Solid-state thermo- and photochromism in N,N′-bis(5-X-salicylidene)diamines (X = H, Br)†
Damir A.
Safin
,
Koen
Robeyns
and
Yann
Garcia
*
Institute of Condensed Matter and Nanosciences, MOST – Inorganic Chemistry, Université Catholique de Louvain, Place L. Pasteur 1, 1348 Louvain-la-Neuve, Belgium. E-mail: yann.garcia@uclouvain.be, damir.safin@ksu.ru; Fax: +32(0) 1047 2330; Tel: +32(0) 1047 2831
First published on 17th October 2012
Abstract
Sixteen N,N′-bis(salicylidene)diamines of common formula (o-OHC6H4C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N–)2Z [Z = o-C6H4 (1a), m-C6H4 (2a), p-C6H4 (3a), Ø (4a), CH2(p-C6H4)2 (5a), O(p-C6H4)2 (6a), S(p-C6H4)2 (7a), SO2(p-C6H4)2 (8a)] and N,N′-bis(5-bromosalicylidene)diamines of common formula (2-OH-(5-Br)C6H3CN–)2Z [Z = o-C6H4 (1b), m-C6H4 (2b), p-C6H4 (3), Ø (4b), CH2(p-C6H4)2 (5b), O(p-C6H4)2 (6b), S(p-C6H4)2 (7b), SO2(p-C6H4)2 (8b)] have been synthesized and characterized by elemental analysis, X-ray powder diffraction, NMR, diffuse reflectance, Raman, IR and fluorescence spectroscopy. Molecular structures of 2a, 6a, 7a, 8a and 4b were elucidated by single crystal X-ray diffraction. Their structures are stabilized by two intramolecular hydrogen bonds and weak intermolecular π⋯π stacking interactions. All molecules are thermochromic, while molecules 2a and 2b exclusively display photochromism upon irradiation at 450 and 365 nm, respectively, which is reversible and exhibits relatively slow thermal relaxation.
N–)2Z [Z = o-C6H4 (1a), m-C6H4 (2a), p-C6H4 (3a), Ø (4a), CH2(p-C6H4)2 (5a), O(p-C6H4)2 (6a), S(p-C6H4)2 (7a), SO2(p-C6H4)2 (8a)] and N,N′-bis(5-bromosalicylidene)diamines of common formula (2-OH-(5-Br)C6H3CN–)2Z [Z = o-C6H4 (1b), m-C6H4 (2b), p-C6H4 (3), Ø (4b), CH2(p-C6H4)2 (5b), O(p-C6H4)2 (6b), S(p-C6H4)2 (7b), SO2(p-C6H4)2 (8b)] have been synthesized and characterized by elemental analysis, X-ray powder diffraction, NMR, diffuse reflectance, Raman, IR and fluorescence spectroscopy. Molecular structures of 2a, 6a, 7a, 8a and 4b were elucidated by single crystal X-ray diffraction. Their structures are stabilized by two intramolecular hydrogen bonds and weak intermolecular π⋯π stacking interactions. All molecules are thermochromic, while molecules 2a and 2b exclusively display photochromism upon irradiation at 450 and 365 nm, respectively, which is reversible and exhibits relatively slow thermal relaxation.
Introduction
Organic photochromism, which is rarely observed in the crystalline state,1 has been optimized for information storage,2,3 molecular machines,4 photo optical switches,5 displays,6 sensors,7 and non-linear optics,8 and therefore still attracts broad interest. Among the molecules that exhibit both thermo- and photochromism in the crystalline state, N-salicylidene aniline derivatives can be highlighted.9–12 This substance class, which has been known for more than a hundred years,13 is attractive from a synthetic point of view since these Schiff bases are straightforwardly synthesized by condensation of the corresponding aldehyde and amine, and readily form complexes with a variety of metals.14 Their optical properties were first systematically studied by Cohen and Schmidt in the sixties.9 Their thermochromism, which corresponds to a temperature-induced reversible colour change,15 results from a tautomeric thermal equilibrium between the uncolored enol form and the yellow cis-keto form. In this instance, the presence of an o-OH group is crucial. Irradiation of the enol and cis-keto forms in the UV and visible wavelengths, respectively, produces the red metastable trans-keto form thanks to cis/trans photoisomerization of the C–N bond (Scheme 1).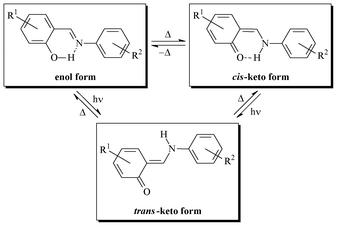 | ||
| Scheme 1 | ||
It was recognized that the solid state thermochromism of N-salicylidene aniline derivatives is evidence of the essential planarity of single molecules with a low dihedral angle between phenolic and benzoic rings (Φ < 25°). These molecules are closely packed in their crystal lattice thanks to π⋯π and/or CH⋯π interactions. Photochromism is caused by a significant rotation (Φ > 25°) and the formation of an open crystal packing, which favour the formation of the trans-keto form.16
Moreover, thermo- and photochromic properties were stated to be mutually exclusive.9 However, a number of examples among N-salicylidene aniline derivatives, showing both thermo- and photochromic properties, contradict the above-mentioned rule.17,18 Hence, it is currently impossible to correctly explain thermo- and photochromism of N-salicylidene aniline derivatives solely on the basis of their crystal structures. Energy differences between the ground and excited states must also be taken into account,19 together with a complete crystal structure determination.
Thermo- and photochromic properties of mono-N-salicylidene aniline derivatives have been thoroughly studied,15–20 but bis(salicylidene)21–26 and tris(salicylidene)27 derivatives have attracted much less attention from an optical properties point of view.
Herein, we discuss eight N,N′-bis(salicylidene)diamines and eight N,N′-bis(p-bromosalicylidene)diamines, each presenting two N-salicylidene imine functions linked by spacers of various natures (Chart 1). The crystal structures of 2a, 6a, 7a, 8a and 4b were elucidated by single crystal X-ray diffraction, and compared with the crystal structures of 1a,283a,21a,b,294a,305a,311b,32and 2b.33 Furthermore, the optical properties of 1–8 were systematically studied by UV-visible diffuse reflectance spectroscopy, fluorometry and discussed in line with their crystal structures.
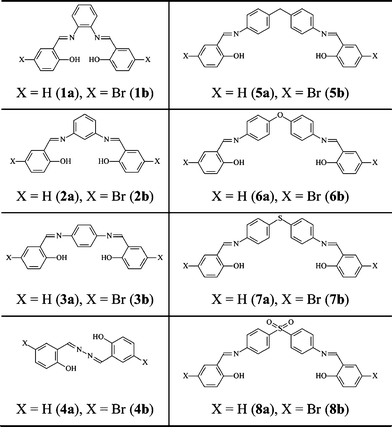 | ||
| Chart 1 | ||
Results and discussion
Synthesis
N,N′-Bis(salicylidene)diamines and N,N′-bis(5-bromosalicylidene)diamines 1–8 were synthesized by reacting the corresponding diamine with salicylaldehyde or 5-bromosalicylaldehyde, respectively, in ethanol. The target molecules are isolated as yellow, yellowish-orange or orange crystalline solids, and fully characterized by CHN analyses and NMR spectroscopy. All compounds are soluble in most polar solvents, including water, and insoluble in n-hexane.1H NMR spectra of 1–7 and 8b in CDCl3 reveal a single set of signals for the C6H3, C6H4, CHN and OH protons at 6.60–8.06, 8.50–8.69 and 11.27–13.37 ppm, respectively. The spectra of 5 also contain a singlet for the CH2 protons at 4.05 ppm. By contrast, the spectrum of 8a contains a double set of slightly different signals: a number of multiplets for the C6H4 protons at 6.64–8.05 ppm, two singlets for the CHN protons at 8.57 and 8.59 ppm, and two singlets for the OH protons at 12.61 and 12.69 ppm. Single crystals of 2a, 6a, 7a, 8a and 4b were obtained by slow evaporation of the solvent from a CH2Cl2–n-hexane solution.
Structural aspects
The presence of different spacers and terminal aryl groups leads to significant changes in the crystal structures, as shown by powder X-ray diffraction patterns of 1–8 (Fig. S1 and S2, ESI†). Thus, the spacer moieties and functional groups dramatically influence the crystal packing, both by steric effects and by the formation of new supramolecular networks (additional H–bonds, π⋯π stacking interactions, etc.)Molecular structures and crystal packing of 2a, 6a, 7a, 8a and 4b are shown in Fig. 1–4, whereas the crystal and structure refinement data are given in the Experimental section.
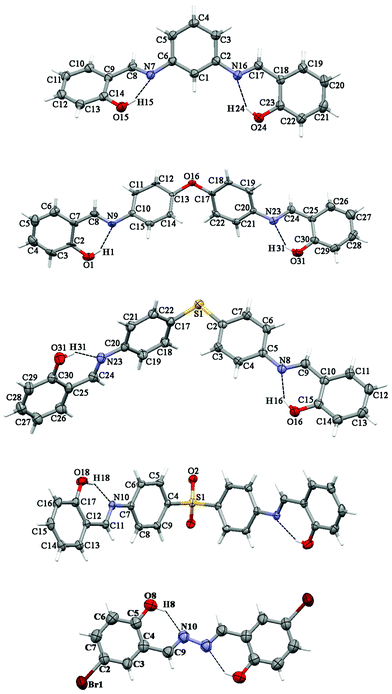 | ||
| Fig. 1 Thermal ellipsoid (50%) plot of 2a, 6a,347a, 8a and 4b (from top to bottom). Dotted lines indicate intramolecular interactions. | ||
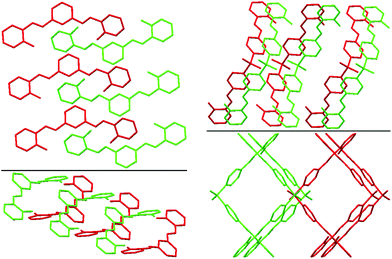 | ||
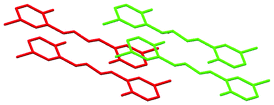
| Fig. 2 Best views of the crystal packing of 2a (left) and 8a (right) in “eye-catching” mode using two colours to better separate the overlapped molecules. Hydrogen atoms were omitted for clarity. | ||
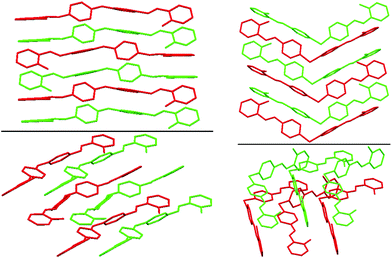 | ||
| Fig. 3 Best views of the crystal packing of 6a (left) and of 7a (right) in “eye-catching” mode using two colours to better separate the overlapped molecules. Hydrogen atoms were omitted for clarity. | ||
 | ||
| Fig. 4 Best view of the crystal packing of 4b in “eye-catching” mode using two colours to better separate the overlapped molecules. Hydrogen atoms were omitted for clarity. | ||
Compounds 2a, 6a, 7a and 8a crystallize in the monoclinic space groups P21/c, P21/c, P21/n and C2/c, respectively, while the compound 4b crystallizes in the triclinic space group P/1. It should be noted that compound 8a has crystallographically-imposed 2-fold symmetry with the central S(1) atom on the twofold axis. Each of the molecules were found in the enolimine form. The bond lengths of C–O are 1.342(3)–1.357(3) Å and those of (N)C–C(aryl) are 1.413(3)–1.456(5) Å, which indicates single bonds, whereas a double bond is revealed from (aryl)CN being 1.259(5)–1.302(3) Å (Table S1, ESI†). Furthermore, the bond angles for C–NC in 2a, 6a, 7a and 8a are in the range 120.0(2)–122.22(19)°, indicating sp2-hybridization of the nitrogen atoms (Table S1, ESI†). The molecules of 6a, 7a and 8a contain two N-salicylidene aniline derivatives linked by an oxygen atom, sulfur atom or sulfone group (Fig. 1) with the dihedral angles for C6H4–O–C6H4, C6H4–S–C6H4 and C6H4–(O)S(O)–C6H4 being 53.72(8), 73.63(10) and 80.66(10)°, respectively (Table S1, ESI†). Furthermore, the aromatic rings of two N-salicylidene aniline residues in 7a were found slightly rotated with respect to each other with dihedral angles of 3.22(11) and 2.13(11)°, while the same dihedral angles are increased in 8a to 14.53(13)°, and are significantly increased in 6a to 50.40(9) and 51.64(9)° (Table S1, ESI†). The geometry of 2a was determined by a significant rotation of one of the salicylideneamine residues around the C(6)–N(7) bond with the torsion angle C(5)–C(6)–N(7)–C(8) being 38.1(3)° (Fig. 1, Table S1, ESI†), whereas the second salicylideneamine residue was found slightly rotated around the C(2)–N(16) bond with the torsion angle C(3)–C(2)–N(16)–C(17) being −9.8(4)° (Fig. 1, Table S1, ESI†). Thus, the corresponding salicylideneamine residue is removed from the least squares plane of all other atoms of the molecule with the dihedral angle between the C6H4 spacer and the (o-OH)C6H4 group being 46.53(12)° (Table S1, ESI†). A similar situation was found in the crystal structure of 1a, where one of the salicylideneamine residues is significantly rotated from the plane of all other atoms of the molecule.28 It should be noted that in the crystal of 4b, the least square planes formed by each molecule are found to be parallel to each other, with the longest dimensions of molecules situated in one direction, while in the crystal structure of the nonsubstituted analogue 4a, the least square planes formed by each molecule are about 65°, rotated with the longest dimensions of molecules crystallized in two directions.30
The crystal structures of 2a, 6a, 7a, 8a and 4b are stabilized by two intramolecular hydrogen bonds of the O–H⋯N type, which are formed between the hydrogen atoms of the OH groups and the nitrogen atoms of the imine groups (Fig. 1, Table S2, ESI†). Furthermore, the crystal structures of 2a and 8a are additionally stabilized by intermolecular hydrogen bonds of the C–H⋯O#1 type (Table S2 and CIF file in ESI†). In the former compound, these H–bonds are formed between the oxygen atom of the OH group and the hydrogen atom of the CH group of the spacer phenylene ring of a further molecule, while in the latter compound intermolecular H–bonds are formed between the oxygen atom of the SO2 group and the hydrogen atom of one of the spacer phenylene rings of a further molecule. As a result of these intermolecular interactions, polymeric chains are formed in the crystal structures of 2a and 8a. Additionally, the crystal structures of 2a, 6a, 7a, 8a and 4b form weak π⋯π stacking interactions between the aryl rings (Table S3, ESI†). Moreover, molecules of 6a, 7a, 8a and 4b are stacked in the crystal (Fig. 2–4). However, in 7a each of the N-salicylidene aniline residues of a molecule are packed almost perpendicularly to two N-salicylidene aniline derivatives of a further molecule (Fig. 3), whereas in 6a, 8a and 4bN-salicylidene aniline residues of a molecule are parallel to a further molecule (Fig. 2–4) similar to that observed in the crystal packing of 5a.31
Optical properties
As shown in Fig. 5, the powders of 1–8 reveal thermochromism from yellow (4a, 5a, 6a, 7a, 6b, 7b), yellowish orange (2a, 3a, 8a, 2b, 3b, 4b, 5b, 8b) or orange (1a, 1b) at room temperature to pale yellow or white on cooling to 77 K. While the orange colour is typical of the trans-keto form, the observed colour change on cooling arises from the cis-keto/enol tautomerisation.15 | ||
| Fig. 5 Photographs showing thermochromism for powders 1–8 at 298 and 77 K. | ||
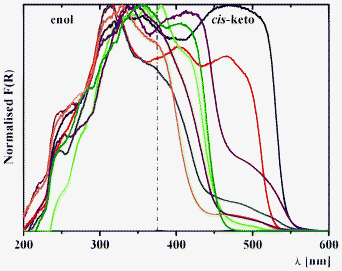 | ||
| Fig. 6 Kubelka–Munk spectra of 1a (black), 2a (red), 3a (blue), 4a (green), 1b (purple), 2b (orange), 3b (brown) and 4b (olive) at 298 K. | ||
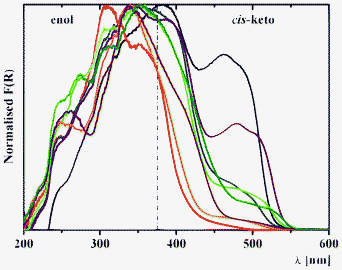 | ||
| Fig. 7 Kubelka–Munk spectra of 5a (black), 6a (red), 7a (blue), 8a (green), 5b (purple), 6b (orange), 7b (brown) and 8b (olive) at 298 K. | ||
Diffuse reflectance spectroscopy of 1–3 allows us to sense the influence of location of the salicylidene groups onto the phenylene ring from ortho-, meta- and para-positions (Fig. 6). The diffuse reflectance spectrum of 3a (Fig. 6), which was reported in the literature data,39 indicate the presence of both enol and keto tautomers, which is also found for 1, 2 and 3b. Interestingly, two bands are identified for the cis-keto form, with maxima at ∼400 and ∼475 nm, which are clearly detected for 2a, and that indicate the presence of two different conformations for the cis-keto form.18c Remarkably, the normalized Kubelka–Munk function F(R) decreases in the order 1 > 2 > 3, and is associated to a bathochromic shift of these bands (Fig. 6). The orange colour observed for 1a and 1b is confirmed by the presence of a band in the trans-keto range. For 4a and 4b, where two chromic groups are directly linked, introduction of the bromine substituent leads to a disappearance of the band at ∼475 nm, corresponding to one of the cis-keto forms (Fig. 6). Since both 4a and 4b are completely planar in the crystal phase, the observed phenomenon might be explained by the fact that in the crystal of 4b, molecules are found to be situated in one direction, while in the crystal structure of the nonsubstituted analogue, 4a molecules are crystallized in two directions (see X-ray description above).
Diffuse reflectance spectroscopy of the series 1–8 allows us to estimate the influence of spacer nature and relative electronegativity (Fig. 7). For the series a, incorporation of a more electronegative fragment on the spacer (ξ(CH2) < ξ(S) < ξ(SO2) < ξ(O)) leads to a relative intensity decrease of the band of the cis-keto form at ∼475 nm in the order 5a (CH2) > 7a (S) > 8a (SO2) > 6a (O) (Fig. 7). However, for the series b a relative intensity decrease is observed in the order 5b (CH2) > 8b (SO2)> 6b (O) ≈ 7b (S) (Fig. 7). This difference in the structures 5b–8b might be explained by the simultaneous influence of withdrawing the bromine substituent as well as by the nature of the spacer.
Diffuse reflectance spectra of 1–8 were also recorded after a 30 min irradiation period at 254, 365 and 450 nm, in order to photo-address the enol and keto forms at room temperature. Photochromic properties of 2a are evident upon irradiation at 450 nm with the rising of a new band above 520 nm attributed to the population of the metastable trans-keto form after photo-isomerization (Fig. 8). This result is in good agreement with the qualitative observation of Kawato.16b,39 Most interestingly, 2b is also found photochromic upon irradiation at 365 nm (Fig. 8). The other title molecules are not photochromic, regardless of the irradiation wavelength and time. Thus, only 2a and 2b exhibit both thermo- and photochromic properties (Fig. 5 and 8).
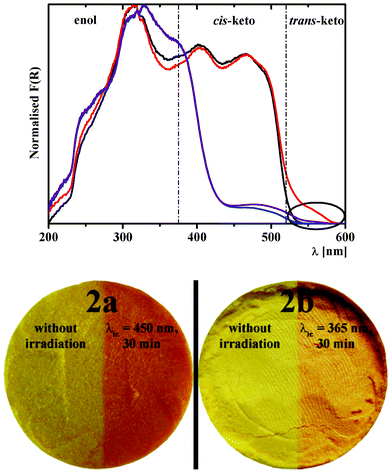 | ||
| Fig. 8 Kubelka–Munk spectra of 2a before (black) and after (red) irradiation at 450 nm for 30 min at 298 K, and of 2b before (blue) and after (purple) irradiation at 365 nm for 30 min at 298 K. The circle highlights the increase of the trans-keto band upon irradiation, which is observed for both 2a and 2b. The photographs show the effect of light irradiation on the colour of powders of 2a and 2b at 298 K, as described above. | ||
The absence of photochromism of 1a, 3a, 4a, 5a and 8a, which was first noticed by Kawato et al.,16b,39 was explained by the formation of a so-called “closed-packed structure” and/or a planar conformation between aromatic rings (Φ < 25°).16b,39 This concept, which could also be applied to explain the non-photochromic character of 6a, 7a, 8a, 1b, 3b, 4b, 5b, 6b, 7b and 8b (see X-ray discussion above) can only be considered in the first approximation, especially for bis(salicylidene) derivatives, where larger structural modifications are expected upon photoisomerization.
In the absence of a crystal structure for 2a, Kawato et al. suggested that having two salicylidene groups out of the plane of the diaminophenyl moiety could be responsible for its photochromic properties. A mechanism pointing out a steric inhibition was even suggested (Scheme 2).16b,39 This thoughtful assumption was made by analysing the crystal structure of the non photochromic molecule 1a, which revealed only one salicylidene group in an out of plane position.
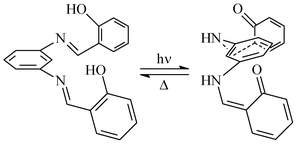 | ||
| Scheme 2 Suggested mechanism for the photoisomerization of 2a.16b,39 | ||
The crystal structure of 2a, which was determined for the first time in the present work, contradicts this scenario, since only one salicylidene group is out of the plane of the diaminophenyl moiety (Fig. 1) and still the molecule is photochromic. Thus, it can be postulated for N,N′-bis(salicylidene)diamines that two conditions are necessary for solid state photochromism: (1) a nonplanar conformation of at least one salicylidene group; (2) sufficient room around the reaction center.26b
The reproducibility of the thermal relaxation of the metastable trans-keto form was studied for 2a and 2b by time dependent diffuse reflectance spectroscopy (Fig. 9, top). Irradiation of 2a and 2b for 30 min at λmax = 450 and 365 nm, respectively, induces a large increase of the Kubelka–Munk function, corresponding to the formation of the trans-keto form as detected in Fig. 8. After stopping irradiation, thermal relaxation is observed for both molecules (Fig. 9, top). The process can be repeated several times as shown for two cycles (Fig. 9, top). Fitting these relaxation data with a first-order exponential equation allowed us to evaluate a kinetic constant of k = 2.6 × 10−5 s−1 for 2b, which is much lower than that of 2a (k = 3.1 × 10−2 s−1).39 The relaxation of 2a appears to be elaborated and contains more than one chemical relaxation pathway, as found for N-salicylidene 4-amino-3,5-bis(pyridine-2-yl)-1,2,4-triazole18b and 5-chloro-N-salicylidene-3-aminomethylpyridine.18c A similar slow back thermal relaxation was observed for 2,4-(OH)2C6H3CHN–(4-COOMe)C6H4 (k = 2.7 × 10−5 s−1).40 The lowest kinetic constants were found for 2-OH-3,5-tBu2C6H2CHN–3-C6H4 (k = 2.0 × 10−7 s−1),41 2-OH–C6H4CHN–3-Py (k = 9.9 × 10−8 s−1)18a and 2-OH-3,5-tBu2C6H2CHN–4-Py (k = 1.8 × 10−8 s−1).40
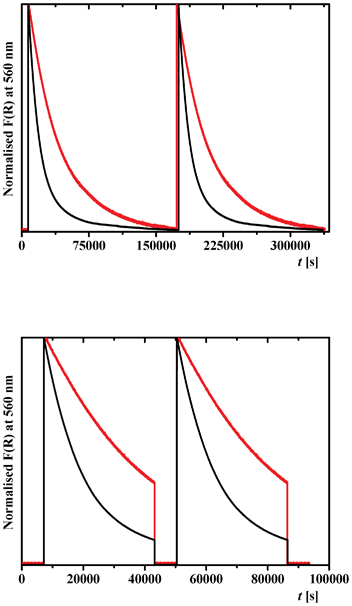 | ||
| Fig. 9 Time dependence of the Kubelka–Munk function F(R) of 2a (black) and 2b (red), at 560 nm showing: (top) a reproducible thermal relaxation of the metastable trans-keto form after irradiation of 2a and 2b at 450 and 365 nm, respectively, for 30 min at 298 K; (bottom) reversibility of photochemical relaxation of the metastable trans-keto form with irradiation cycles at 450 and 546 nm for 2a, and 365 and 546 nm for 2b. Two cycles are shown. | ||
We have examined too the reversibility of the trans/cis-keto isomerization for 2a and 2b by time dependent diffuse reflectance spectroscopy (Fig. 9, bottom). After photoexcitation of 2a and 2b at 450 and 365 nm, respectively, and thermal relaxation for over 10 h, irradiation at 546 nm for 30 min induces a fast return to the initial F(R) value. The cycle can be repeated by irradiation once again at 450 and 365 nm, respectively (Fig. 9, bottom).
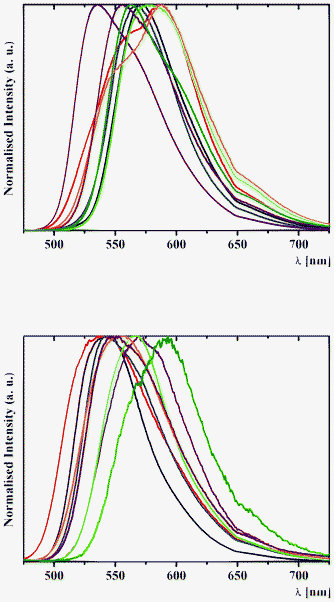 | ||
| Fig. 10 Normalised solid-state emission spectra of (top) 1a (black), 2a (red), 3a (blue), 4a (purple), 1b (green), 2b (orange), 3b (brown) and 4b (olive) and (bottom) 5a (black), 6a (red), 7a (blue), 8a (purple), 5b (green), 6b (orange), 7b (brown) and 8b (olive), with an excitation wavelength of 400 nm at 298 K. | ||
The assignation of bands in the emission spectra of 1–8 was made feasible by the examination of excitation spectra at 620 nm (Fig. 11). The emission band in the spectra of 1 and 3–8 is assigned to the emission of the excited cis-keto* form. Two intense emission bands in the spectra of 2a and 2b are also assigned to the emission of the cis-keto* forms (Fig. 11). However, these cis-keto* forms are slightly different, probably due to the presence of two different salicylidene functions in the structures of 2a and 2b (Fig. 1, see X-ray discussion above). Furthermore, the origin of the band at about 650 nm in the emission spectra of 1–8 (Fig. 11) could be assigned to the radiative de-excitation of either a cis-keto* form higher in energy than the previous one or the excited trans-keto* form.5
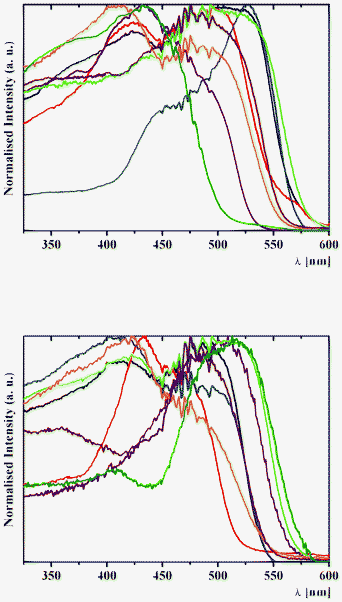 | ||
| Fig. 11 Normalised solid-state excitation spectra of (top) 1a (black), 2a (red), 3a (blue), 4a (purple), 1b (green), 2b (orange), 3b (brown) and 4b (olive) and (bottom) 5a (black), 6a (red), 7a (blue), 8a (purple), 5b (green), 6b (orange), 7b (brown) and 8b (olive) with an emission wavelength of 620 nm at 298 K. | ||
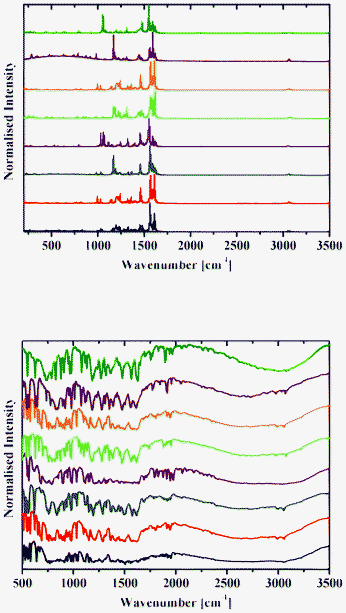 | ||
| Fig. 12 Raman (top) and DRIFT (bottom) spectra of 1a (black), 2a (red), 3a (blue), 4a (purple), 1b (green), 2b (orange), 3b (brown) and 4b (olive) at 298 K. | ||
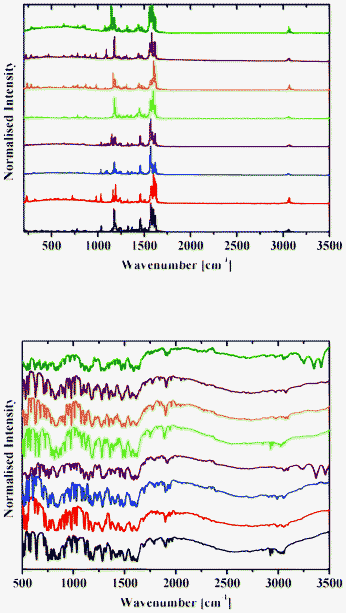 | ||
| Fig. 13 Raman (top) and DRIFT (bottom) spectra of 1b (black), 2b (red), 3b (blue), 4b (purple), 5b (green), 6b (orange), 7b (brown) and 8b (olive) at 298 K. | ||
Raman spectra of 1–8 contain an intense band at about 1600 cm−1 (Fig. 12 and 13), which corresponds to the CN group.42 A number of bands for the OH groups, corresponding to the enol form, are located in the region around 3050 cm−1.
DRIFT spectra were recorded on pure samples of 1–8 to avoid any matrix effect usually observed with KBr and other media used to record IR spectra. They contain a broad absorption band in the range 2250–3200 cm−1, with an intense band with the extremum at around 2900 cm−1 characteristic of the OH and/or NH stretching vibration.13 A band at about 1600 cm−1 corresponds to the CN group (Fig. 12 and 13).42
Conclusions
In summary, we have synthesized a number of N,N′-bis(salicylidene)diamines of the common formula (o-OHC6H4CN–)2Z [Z = o-C6H4 (1a), m-C6H4 (2a), p-C6H4 (3a), Ø (4a), CH2(p-C6H4)2 (5a), O(p-C6H4)2 (6a), S(p-C6H4)2 (7a), SO2(p-C6H4)2 (8a)] and N,N′-bis(5-bromosalicylidene)diamines of the common formula (2-OH-(5-Br)C6H3CN–)2Z [Z = o-C6H4 (1b), m-C6H4 (2b), p-C6H4 (3b), Ø (4b), CH2(p-C6H4)2 (5b), O(p-C6H4)2 (6b), S(p-C6H4)2 (7b), SO2(p-C6H4)2 (8b)] by the condensation of the corresponding diamine and salicylaldehyde or 5-bromosalicylaldehyde, respectively, in the ethanol medium.
According to single crystal X-ray diffraction data, 2a, 6a, 7a, 8a and 4b contain two intramolecular hydrogen bonds of the O–H⋯N type, and are additionally stabilized by weak intermolecular π⋯π stacking interactions.
As concluded from diffuse reflectance spectroscopy data, 1–8 show the presence of a mixture of the dominant enol and cis-keto forms in the solid state at room temperature. All compounds are thermochromic, while 2a and 2b exclusively show photochromism upon irradiation at 450 and 365 nm, respectively. This photochromism exhibits slow back thermal relaxation (e.g. k = 2.6 × 10−5 s−1 for 2b). The main emission properties of 1–8 originate from the cis-keto* to cis-keto relaxation.
Experimental
General procedures
NMR spectra in CDCl3 were obtained on a Bruker AC 300 MHz spectrometer at 25 °C. 1H NMR spectra were recorded at 299.948 MHz. Chemical shifts are reported with reference to SiMe4. Elemental analyses were performed on a Perkin Elmer 2400 CHN microanalyser. X-Ray powder diffractograms were performed with a Siemens D5000 X-ray diffractometer, with a Cu anticathode (λK-α = 1.5418 Å) in Bragg–Brentano geometry (θ/θ mode). The samples were deposited on silicon after calibration with a quartz standard. Diffuse reflectance spectra were obtained with a Varian Cary 5E spectrometer using polytetrafluoroethylene (PTFE) as a reference. Spectra were measured on pure solids to avoid matrix effects. Eventual distortions in the Kubelka–Munk spectra that could result from the study of pure compounds have not been considered, because no comparison with absorption spectra was necessary. Diffuse reflection infrared Fourier-transform (DRIFT) spectra were recorded with a FT-IR Bruker IFS 66/S spectrometer; the resolution was set to 4 cm−1 and the operating range was 500–4000 cm−1. Solid-state emission spectra were obtained with a Fluorolog-3 (Jobin-Yvon-Spex Company) spectrometer. Kubelka–Munk and emissions spectra were normalized to allow meaningful comparisons. Light irradiation was carried out with a LOT-ORIEL 200 W high-pressure mercury Arc lamp (LSN261). Raman spectra in the solid state were obtained with a FTIR Nicolet Magna 860 with Raman unit and Nd:YVO4 (λ = 1064 nm) laser.Syntheses
A solution of salicylaldehyde (10 mmol, 1.22 g) or 5-bromosalicylaldehyde (10 mmol, 2.01 g) dissolved in ethanol (20 mL) was added to solutions of o-phenylenediamine, m-phenylenediamine, p-phenylenediamine, hydrazine monohydrate, 4,4′-diaminodiphenylmethane, 4-aminophenyl ether, 4,4′-diaminodiphenylsulfide, 4-aminophenylsulfone, respectively (5 mmol; 0.54, 0.54, 0.54, 0.25, 0.99, 1.00, 1.08 or 1.24 g) in ethanol (20 mL). The resulting yellow solution was stirred for 30 min and, afterwards, heated at reflux for 2 h. It was then allowed to cool to room temperature to give a fine crystalline yellow, yellowish-orange or orange powder. The product was filtered, washed with ethanol (10 mL) and n-hexane (3 × 50 mL), and dried under vacuum.X-Ray crystallography
Single crystals of 2a, 6a, 7a, 8a and 4b were obtained by slow evaporation of the solvent from a CH2Cl2–n-hexane solution. X-Ray data collection was performed on a Mar345 image plate detector using Mo-Kα radiation (Zr-filter) at ambient temperature. The data were integrated with the crysAlisPro software.43 The implemented empirical absorption correction was applied. The structures were solved by direct methods using the SHELXS-97 program44 and refined by full-matrix least squares on |F2| using SHELXL-97.44 Non-hydrogen atoms were anisotropically refined and the hydrogen atoms were placed on calculated positions in riding mode with temperature factors fixed at 1.2 times Ueq of the parent atoms and 1.5 times Ueq for methyl groups. Figures were generated using the program Mercury.45Acknowledgements
This work was funded by the Fonds National de la Recherche Scientifique-FNRS (FRFC No. 2.4508.08, 2.4537.12, IISN 4.4507.10) and ARC Louvain. We thank the F.R.S.-FNRS (Belgium) for a post-doctoral position allocated to D. A. S.References
- (a) M. Irie, Chem. Rev., 2000, 100, 1683 CrossRef CAS; (b) M. Irie, Photochem. Photobiol. Sci., 2010, 9, 1535 RSC; (c) M. Irie and M. Moritomo, Pure Appl. Chem., 2009, 81, 1655 CrossRef CAS.
- B. L. Feringa, W. F. Jager and B. de Lange, Tetrahedron, 1993, 49, 8267 CrossRef CAS.
- S. Kawata and Y. Kawata, Chem. Rev., 2000, 100, 1777 CrossRef CAS.
- B. L. Feringa, Acc. Chem. Res., 2001, 34, 504 CrossRef CAS.
- (a) M. Irie, in Molecular Switches, ed. B. L. Feringa, Wiley-VCH, Weinheim, 2001, p. 37 Search PubMed; (b) K. Matsuda and M. Irie, in Chemistry of Nano-molecular System – Towards the Realization of Molecular Devices, ed. T. Nakamura, T. Matsumoto, H. Tada and K.-I. Sugiura, Springer, 2002, p. 25 Search PubMed.
- (a) J. Yao, K. Hashimoto and A. Fujishima, Nature, 1992, 355, 624 CrossRef CAS; (b) C. Bechinger, S. Ferrer, A. Zaban, J. Sprague and B. A. Gregg, Nature, 1996, 383, 608 CrossRef CAS.
- V. A. Bren, A. D. Dubonosov, V. I. Minkin, T. N. Gribanova, V. P. Rybalkin, E. N. Shepelenko, A. V. Tsukanov and R. N. Borisenko, Mol. Cryst. Liq. Cryst., 2005, 431, 417 CrossRef CAS.
- (a) K. Nakatani and J. A. Delaire, Chem. Mater., 1997, 9, 2682 CrossRef CAS; (b) J. A. Delaire and K. Nakatani, Chem. Rev., 2000, 100, 1817 CrossRef CAS.
- (a) M. D. Cohen and G. M. J. Schmidt, J. Phys. Chem., 1962, 66, 2442 CrossRef CAS; (b) M. D. Cohen, G. M. J. Schmidt and S. Flavian, J. Chem. Soc., 1964, 2041 RSC; (c) M. D. Cohen, Y. Hirshberg and G. M. J. Schmidt, J. Chem. Soc., 1964, 2051 RSC; (d) M. D. Cohen, Y. Hirshberg and G. M. J. Schmidt, J. Chem. Soc., 1964, 2060 RSC; (e) J. Bregman, L. Leiserowitz and G. M. J. Schmidt, J. Chem. Soc., 1964, 2068 RSC; (f) M. D. Cohen and S. Flavian, J. Chem. Soc. B, 1967, 317 RSC; (g) M. D. Cohen and S. Flavian, J. Chem. Soc. B, 1967, 321 RSC; (h) M. D. Cohen, S. Flavian and L. Leiserowitz, J. Chem. Soc. B, 1967, 329 RSC; (i) M. D. Cohen and S. Flavian, J. Chem. Soc. B, 1967, 334 RSC; (j) M. D. Cohen, J. Chem. Soc. B, 1968, 373 RSC.
- Organic Photochromic and Thermochromic Compounds, ed. J. C. Cran, and R. J. Guglielmetti, vol. 2, Plenum Press, New York, 1999 Search PubMed.
- M. Mikami and S. Nakamura, J. Phys. Chem. B, 2004, 33, 579 Search PubMed.
- E. Hadjoudis, Mol. Eng., 1995, 5, 301 CrossRef CAS.
- (a) A. Senier and F. G. Shepheard, J. Chem. Soc. Trans., 1909, 95, 441 RSC; (b) A. Senier and F. G. Shepheard, J. Chem. Soc. Trans., 1909, 95, 1943 RSC; (c) A. Senier, F. G. Shepheard and R. Clarke, J. Chem. Soc. Trans., 1912, 101, 1950 RSC; (d) A. Senier and R. Clarke, J. Chem. Soc. Trans., 1914, 105, 1917 RSC; (e) A. Senier and R. B. Forster, J. Chem. Soc. Trans., 1914, 105, 2462 RSC.
- For example (a) S. Kumar, D. N. Dhar and P. N. Saxena, J. Sci. Ind. Res., 2009, 68, 181 CAS , and references therein; (b) F. Robert, B. Tinant, R. Clérac, P.-L. Jacquemin and Y. Garcia, Polyhedron, 2010, 29, 2739 CrossRef CAS.
- T. Fujiwara, J. Hadara and O. Keiichiro, J. Phys. Chem. B, 2004, 108, 4035 CrossRef CAS.
- (a) E. Hadjoudis and I. M. Mavridis, Chem. Soc. Rev., 2004, 33, 579 CAS; (b) K. Amimoto and T. Kawato, J. Photochem. Photobiol., C, 2005, 6, 207 CrossRef CAS; (c) T. Haneda, M. Kawano, T. Kojima and M. Fujita, Angew. Chem., Int. Ed., 2007, 46, 6643 CrossRef CAS; (d) Y. Inokuma, M. Kawano and M. Fujita, Nat. Chem., 2011, 3, 349 CrossRef CAS.
- E. Hadjoudis, M. Vitterakis, I. Moustakali and I. Mavridis, Tetrahedron, 1987, 43, 1345 CrossRef CAS.
- (a) F. Robert, A. D. Naik, B. Tinant, R. Robiette and Y. Garcia, Chem.–Eur. J., 2009, 15, 4327 CrossRef CAS; (b) F. Robert, A. D. Naik, F. Hidara, B. Tinant, R. Robiette, J. Wouters and Y. Garcia, Eur. J. Org. Chem., 2010, 621 CrossRef CAS; (c) F. Robert, P.-L. Jacquemin, B. Tinant and Y. Garcia, CrystEngComm, 2012, 14, 4396 RSC; (d) D. A. Safin, K. Robeyns and Y. Garcia, CrystEngComm, 2012, 14, 5523 RSC.
- S. D. Chatziefthimiou, Y. G. Lazarou, E. Hadjoudis, T. Dziembowska and I. M. Mavridis, J. Phys. Chem. B, 2006, 110, 23701 CrossRef CAS.
- E. Hadjoudis, S. D. Chatziefthimiou and I. M. Mavridis, Curr. Org. Chem., 2009, 13, 269 CrossRef CAS.
- (a) N. Hoshino, T. Inabe, T. Mitani and Y. Maruyama, Bull. Chem. Soc. Jpn., 1988, 61, 4207 CrossRef CAS; (b) T. Inabe, N. Hoshino, T. Mitani and Y. Maruyama, Bull. Chem. Soc. Jpn., 1989, 62, 2245 CrossRef CAS; (c) E. Ito, H. Oji, T. Araki, K. Oichi, H. Ishii, Y. Ouchi, T. Ohta, N. Kosugi, Y. Maruyama, T. Naito, T. Inabe and K. Seki, J. Am. Chem. Soc., 1997, 119, 6336 CrossRef CAS; (d) T. Sekikawa, T. Kobayashi and T. Inabe, J. Phys. Chem. B, 1997, 1, 10645 CrossRef; (e) M. Ziólek, J. Kubicki, A. Maciejewski, R. Naskr□cki and A. Grabowska, J. Chem. Phys., 2006, 124, 124518 CrossRef; (f) M. Ziólek, G. Burzinski and J. Karolczak, J. Phys. Chem. A, 2009, 113, 2854 CrossRef.
- Y. Yildiz, Z. Kiliç and T. Hökelek, J. Mol. Struct., 1988, 441, 1 CrossRef.
- Z. Popović, V. Roje, G. Pavlović, D. Matković-Calogović and G. Giester, J. Mol. Struct., 2001, 597, 39 CrossRef.
- E. Hadjoudis, A. Rontoyianni, K. Ambroziak, T. Dziembowska and I. Mavridis, J. Photochem. Photobiol., A, 2004, 162, 521 CrossRef CAS.
- (a) P. Xue, R. Lu, G. Chen, Y. Zhang, H. Nomoto, M. Takafuji and H. Ihara, Chem.–Eur. J., 2007, 13, 8231 CrossRef CAS; (b) P. Chen, R. Lu, P. Xue, T. Xu, G. Chen and Y. Zhao, Langmuir, 2009, 25, 8395 CrossRef CAS.
- (a) M. S. M. Rawat and J. L. Norula, Indian J. Chem., Sect. B: Org. Chem. Incl. Med. Chem., 1987, 26, 232 Search PubMed; (b) M. Taneda, K. Amimoto, H. Koyama and T. Kawato, Org. Biomol. Chem., 2004, 1, 499 RSC; (c) M. Taneda, H. Koyama and T. Kawato, Chem. Lett., 2007, 36, 354 CrossRef CAS; (d) M. Taneda, H. Koyama and T. Kawato, Res. Chem. Intermed., 2009, 35, 643 CrossRef CAS.
- K. Tanaka, R. Shimoura and M. R. Caira, Tetrahedron Lett., 2010, 51, 449 CrossRef CAS.
- (a) N. B. Pahor, M. Calligaris, P. Delise, G. Dodic, G. Nardin and L. Randaccio, J. Chem. Soc., Dalton Trans., 1976, 2478 RSC; (b) C. Subrahmanyam, M. Seshasayee and G. Aravamudan, Cryst. Struct. Commun., 1982, 11, 1719 CAS.
- S. Zhang, S.-J. Ye, T. Zhang and X.-M. Li, Chem. : Res. Chin. Univ., 2004, 20, 562 CAS.
- (a) G. Arcovito, M. Bonamico, A. Domenicano and A. Vaciago, J. Chem. Soc. B, 1969, 733 RSC; (b) X.-X. Xu, X.-Z. You, Z.-F. Sun, X. Wang and H.-X. Liu, Acta Crystallogr., Sect. C: Cryst. Struct. Commun., 1994, 50, 1169 CrossRef; (c) M. Mijanuddin, W. S. Sheldrick, H. Mayer-Figge, M. Ali and N. Chattopadhyay, J. Mol. Struct., 2004, 639, 161 CrossRef; (d) S. Parsons, P. Lovatt, P. Tasker and P. Wood, Private Communication, 2004 Search PubMed; (e) S. M. El-Medani, M. M. Aboaly, H. H. Abdalla and R. M. Ramadan, Spectrosc. Lett., 2004, 37, 619 CrossRef CAS; (f) S.-L. Liu, Y. Chen, J.-F. Dai and H.-W. Liu, Hecheng Huaxue, 2004, 12, 219 CAS; (g) W. Tang, Y. Xiang and A. Tong, J. Org. Chem., 2009, 74, 2163 CrossRef CAS.
- H. Birkedal and P. Pattison, Acta Crystallogr., Sect. C: Cryst. Struct. Commun., 2006, 62, o139 Search PubMed.
- M. Kabak, A. Elmali, Y. Elerman and T. N. Durlu, J. Mol. Struct., 2000, 553, 187 CrossRef CAS.
- K. Ha, Acta Cryst., 2011, 67, o2234 Search PubMed.
- G. H. Shahverdizadeh and E. R. Tiekink, Acta Crystallogr., Sect. E: Struct. Rep. Online, 2011, E67, o798 CAS.
- E. Hadjoudis, V. Verganelakis, C. Trapalis and G. Kordas, Mol. Eng., 1999, 8, 459 CrossRef CAS.
- L. Z. Zhang, Y. Xiang, P. Cheng, G. Q. Tang and D. Z. Liao, Chem. Phys. Lett., 2002, 358, 278 CrossRef CAS.
- S. Sakagami, T. Koga and A. Takase, Mol. Cryst. Liq. Cryst., 2002, 373, 71 CrossRef CAS.
- M. Ziółek and I. Sobczak, J. Inclusion Phenom. Macrocyclic Chem., 2009, 63, 211 CrossRef.
- T. Kawato, H. Kanatomi, H. Koyama and T. Igarashi, J. Photochem., 1986, 33, 199 CrossRef CAS.
- M. Sliwa, S. Létard, I. Malfant, M. Nierlich, P. G. Lacroix, T. Asahi, H. Masuhara, P. Yu and K. Nakatani, Chem. Mater., 2005, 17, 4727 CrossRef CAS.
- T. Kawato, H. Koyama, H. Kanatomi and M. Isshiki, J. Photochem., 1985, 28, 103 CrossRef CAS.
- (a) I. Casades, M. Alvaro, H. Garcia and M. N. Pillai, Eur. J. Org. Chem., 2002, 2074 CrossRef CAS; (b) W. Turbeville and P. K. Dutta, J. Phys. Chem., 1990, 94, 4060 CrossRef CAS.
- Oxford Diffraction Data collection and data reduction, Version 171.34.40.
- G. M. Sheldrick, Acta Crystallogr., Sect. A: Found. Crystallogr., 2008, A64, 112 CrossRef CAS.
- I. J. Bruno, J. C. Cole, P. R. Edgington, M. Kessler, C. F. Macrae, P. McCabe, J. Pearson and R. Taylor, Acta Crystallogr., Sect. B: Struct. Sci., 2002, B58, 389 CrossRef CAS.
Footnote |
| † Electronic Supplementary Information (ESI) available: Fig. S1 and S2, Tables S1–S3. CCDC reference numbers 828843 (2a), 840365 (6a), 828844 (7a), 828845 (8a) and 830149 (4b). For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c2ra21631k |
| This journal is © The Royal Society of Chemistry 2012 |