One novel 3-D vanadoborate with unusual 3-D Na–O–Na network†
Jian
Zhou
*ab,
Xing
Liu
a,
Feilong
Hu
b,
Huahong
Zou
c,
Rong
Li
a and
Xiaojuan
Li
b
aCollege of Chemistry, Chongqing Normal University, Chongqing, 401331, PR China. E-mail: Jianzhou888888@163.com; Jianzhou888888@yahoo.com.cn; Fax: +86-775/2622315; Tel: +86-775/2622315
bCollege of Chemistry and Material, Yulin Normal University, Yulin 537000, PR China
cSchool of Chemistry and Chemical Engineering of Guangxi Normal University, Guilin 541004, PR China
First published on 13th September 2012
Abstract
A new 3-D vanadoborate {Na2B18V12O54(OH)6(H2O)[Na8(H2O)16]}·2H2O (1) has been hydrothermally synthesized and structurally characterized. The crystal of 1 is built up from an unusual 3-D Na–O–Na network and entrapped [V12B18O54(OH)6(H2O)]10− clusters (denoted [V12B18]). Such a Na–O–Na network is constructed by the linkage of rare 1-D helical [Na4(H2O)84+]n chains and [NaO6] octahedra. 1 represents the rare example of extended POMs with 3-D inorganic metal–oxygen networks. Magnetic measurements illustrate that 1 has antiferromagnetic exchange interactions between metal centers.
Introduction
Polyoxometalates (POM), known as an outstanding class of molecules with unmatched structural variety and versatility, have provoked significant interest owing to their potential applications in catalysis, materials science, medicine, nanotechnology, and magneto-chemistry.1 Recently, the design and construction of extended POM materials has made important progress.2 A remarkable approach for designing such extended POMs is the use of the coordination ability of polyoxoanions to combine with different metal cations or metal complexes (MCs). Typical POMs built up from metal cations are a series of mixed-metal framework materials based on the linkages of spherical [V18O42(XO4)] cages and [M(H2O)4]2+ groups (X = V, S; M = Fe, Co, Mn, Zn).3 MCs acting as bridging nodes may also link POM units to form a variety of extended structures.2d,e,4 Notably, MCs mainly include discrete complex cations, 1-D chains and 2-D layers, while 3-D metal–organic networks are very scarce, and appear to be limited to those of [{Fe(tpypor)}3Fe(Mo6O19)2]·xH2O,4h [Ni(4,4′-bpy)2]2[VIV7VV9-O38Cl]·(4,4′-bpy)·6H2O,4e [{Cu2(triazolate)2(H2O)2}Mo4O13],4f and [Cu3(4,4′-bipy)5(MeCN)2]PW12O40·2C6H5CN.4g Moreover, no extended POMs with 3-D inorganic metal–oxygen networks have been documented until now to the best of our knowledge.The use of s-block metal-containing aggregates to dictate the assembly of infinite network materials in a predictable manner is quickly emerging as a highly promising strategy.5 In the case of sodium metal, sodium ions typically have a high affinity for water or oxygen-based ligands to form a diversification of metal-containing aggregates as secondary building units (SBUs), which further link by direct covalent bonding to various extended structures.5c,6 These structures are usually 1-D chains or 2-D layers, but 3-D networks are relatively rare. The only example is {[Na20(Ni8L12)(H2O)28](H2O)13(CH3OH)2}n (L = 4,5-imidazoledicarboxylic acid),5c where all of the sodium ions assemble into a 3-D network with open channels filled by the cubic [Ni8L12] clusters. Herein, we present a new 3-D vanadoborate {Na2B18V12O54(OH)6(H2O)[Na8(H2O)16]}·2H2O (1), which is constructed by the linkages of a new 3-D Na–O–Na network and [V12B18] clusters. 1 is the rare example of extended POMs with 3-D inorganic metal–oxygen networks.
Experimental section
General remarks
All analytical grade chemicals were obtained commercially and used without further purification. The UV/Vis spectra were recorded at room temperature using a computer-controlled PE Lambda 900 UV/Vis spectrometer equipped with an integrating sphere in the wavelength range of 200–800 nm. FT-IR spectra were recorded with a Nicolet Magna-IR 550 spectrometer in dry KBr disks in the 4000–400 cm−1 range. Powder X-ray diffraction (XRD) patterns were collected on a D/MAX-3C diffractometer using graphite-monochromatized Cu Kα radiation (λ = 1.5406 Å). Variable temperature magnetic susceptibility measurements were carried out in the temperature range of 2–300 K with a Quantum Design MPMS-XL5 SQUID magnetometer. The magnetic data were corrected for the diamagnetic contribution of the sample holder and the diamagnetic contribution of the sample by using Pascal constants. Thermogravimetric analyses (TG) were performed using a Mettler TGA/SDTA851 thermal analyzer under N2 atmosphere with a heating rate of 10 °C min−1 in the temperature region of 25–400 °C. The chemical analysis was carried out using inductively coupled plasma (ICP) methods on an ESCALAB2000 analyzer.Synthesis of {Na2B18V12O54(OH)6(H2O)[Na8(H2O)16]}·2H2O (1)
A mixture of Na2SO3 (2 mmol, 0.2516 g), NH4VO3 (1 mmol, 0.1170 g), H3BO3 (3 mmol, 0.1856 g), diaminopropane (dap, 0.25 mL) and water (2.5 mL) was stirred for 0.2 h. Then, the final mixture with a pH of 8.5 was sealed in a 25 mL Teflon®-lined autoclave and heated at 110 °C for 7 days. After cooling to room temperature slowly, brown block crystals were isolated. The crystals were filtered and dried at room temperature (73% yield based on NH4VO3). The experimental and simulated XRD patterns of 1 are shown in Fig. S1, ESI.† The experimental peak positions are in agreement with the simulated XRD pattern, indicating the phase purity of 1. The difference in reflection intensity between experimental and simulated XRD patterns is probably due to the preferred orientation effect of the powder sample during collection of the experimental XRD data. ICP analysis shows that the molar ratio of Na![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :V is 10.11:11.97, consistent with the stoichiometry.
:V is 10.11:11.97, consistent with the stoichiometry.
Crystal structure determination
Single-crystal X-ray diffraction data for 1 were recorded on a Rigaku Mercury CCD diffractometer using a ω-scan method with graphite monochromated Mo Kα radiation (λ = 0.71073 Å) at 296(2) K to a maximum 2θ value (50.20°). Absorption corrections were applied using multi-scan technique. The structure of 1 was solved by Direct Methods (SHELXS-97)7 and refined by full-matrix least-squares techniques using the SHELXL-97 program.8 All atoms were refined with anisotropic displacement parameters. No H atoms associated with water molecules and OH groups were located from the difference Fourier map. Na3, O4w, O8w, and O9w atoms were disordered over two positions with occupation factors of 0.85/0.15, 0.50/0.50, 0.85/0.15 and 0.85/0.15. The O10w and O11w have an occupancy of 0.5. Relevant crystal and collection data parameters and refinement results can be found in Table 1. Additional details of crystal data in CIF format can be found in the Supporting Information.† Ranges of some important bond lengths and angles for 1 are listed in Table S1, ESI†. The CSC reference number is 424870 (1).| 1 | |
|---|---|
| Formula | B18H44Na10O79V12 |
| FW | 2344.08 |
| Crystal system | Monoclinic |
| Space group | P21/n |
| a/Å | 13.589(3) |
| b/Å | 17.012(3) |
| c/Å | 14.339(3) |
| β (°) | 97.84(3) |
| V/Å3 | 3283.9(12) |
| Z | 2 |
| T/K | 296(2) |
| D c/Mg m−3 | 2.370 |
| μ/mm−1 | 1.842 |
| F(000) | 2216 |
| 2θ(max)/° | 50.20 |
| Total reflns collected | 18412 |
| Unique reflns | 5824 |
| No. of parameters | 571 |
| R 1[I > 2σ(I)] | 0.0545 |
| wR2 (all data) | 0.1573 |
Results and discussion
Description of the structure
The dap molecules and SO32− ions in the preparation of 1 take a role as a reductive agent, although they are not incorporated in the final structure. Besides, the dap molecule also acts as an alkaline agent to adjust the pH value of the reaction system. The asymmetric unit of 1 contains 5 Na+ ions, 6 V atoms, 9 B atoms, 30 oxygen atoms and 9.5 water molecules (Fig. 1a). Each V atom coordinates with five O atoms to make a VO5 square pyramid with the V–O distances ranging from 1.6201(10) to 2.0184(8) Å and O–V–O angles from 77.65(4) to 147.00(3)°. Six VO5 square pyramids are held together to form a [V6O18] unit by edge-sharing O atoms (Fig. 1b). The B2/5/8 atoms are triangularly coordinated by three O atoms, while the other boron atoms are tetrahedrally coordinated by four O atoms. The B–O distances are in the range of 1.3546(17)–1.5230(16) Å and O–B–O angles vary from 106.17(8) to 123.39(11)°. Two BO4 tetrahedra and one BO2(OH) triangle are held together to form a cyclohexane-like B3O6(OH)4− unit. Six B3O6(OH)4− units share their corners to give a puckered [B18O36(OH)6]24− ring (Fig. 1c). One [B18O36(OH)6]24− ring is sandwiched between two [V6O18] units to give a [V12B18] cluster (Fig. 1d). The center of the [V12B18] cluster is occupied by one water molecule. These V oxidation states were confirmed by valence bond calculation,9 which shows an average valence of about 4.15; this is close to the 4.17 expected for a VIV:VV ratio of 5:1, so two of the twelve vanadium atoms are VV and ten are VIV. The framework stoichiometry of [V12B18] creates a charge of −10, which can be balanced by ten Na+ ions. The existence of V(IV) accounts for the brown color of the crystal.
![(a) The asymmetric unit of 1; the structures of (b) the [V6O18] unit, (c) [B18O36(OH)6]24− ring, and (d) [V12B18] cluster.](/image/article/2012/RA/c2ra21635c/c2ra21635c-f1.gif) | ||
| Fig. 1 (a) The asymmetric unit of 1; the structures of (b) the [V6O18] unit, (c) [B18O36(OH)6]24− ring, and (d) [V12B18] cluster. | ||
In 1, all sodium atoms are coordinated by six O atoms to give distorted octahedron [NaO6] (Fig. S2, ESI†). The Na–O bond lengths are in the range 2.265(3)–2.8427(13) Å and are in the typical ranges of the corresponding bond lengths reported in the literature.6 Four [NaO6] octahedra are joined together by edge and corner-sharing O atoms to form a [Na4O19] tetramer unit, which is further interconnected by sharing a corner O atom to give a left and right-handed helical [Na4(H2O)84+]n chain parallel to the b axis. The left- and right-handed helices are arranged alternately along the [001] direction (Fig. 2a–e). The period of the helix is 17.0 Å by the centroid of Na+ ions and the dimension of the helical channel is approximately 9.1 × 11.9 Å2. Although a few Na–O–Na chains are observed in the coordination polymers, they are not helical chains.6c,d So the helical chain in 1 represents a new type. The alternate connectivity between the helical chains and the Na(1)O6 octahedra through their vertices generates a novel 3-D Na–O–Na network with 14-ring channels in the [100] direction. The apertures of the 14-ring channels are elliptical in shape with a cross-section of 14.1 × 17.7 Å2 (Fig. S3, ESI†). The [V12B18] clusters act as multidentate ligands, which are fixed within the 14-ring channels as a guest part (Fig. 2f).
![(a) View of the left/right-handed helical [Na4(H2O)84+]n chains parallel in 1 along the [100] direction; (b) The cross section and (c) the profile of a left-handed helical chain; (d) The cross-section and (e) the profile of a right-handed helical chain; (f) perspective view of the 3-D Na–O–Na framework along the [100] direction with the bigger channels occupied by V12B18 clusters.](/image/article/2012/RA/c2ra21635c/c2ra21635c-f2.gif) | ||
| Fig. 2 (a) View of the left/right-handed helical [Na4(H2O)84+]n chains parallel in 1 along the [100] direction; (b) The cross section and (c) the profile of a left-handed helical chain; (d) The cross-section and (e) the profile of a right-handed helical chain; (f) perspective view of the 3-D Na–O–Na framework along the [100] direction with the bigger channels occupied by V12B18 clusters. | ||
So far, a few vanadoborates containing Na+ ions have been reported.10 Their extended frameworks are usually constructed by mono- or dinuclear sodium complexes as bridging nodes. For instance, the organically decorated [V12B16O50(OH)7(en)]7− clusters10a are linked by mononuclear [Na(H2O)2]+ bridges to form a 1-D dual chain, the{(VO)10[B14O30(OH)2]2}{Mn4(C2O4)[B2O4(OH)2]2}20− clusters are connected to form a 2-D layer by mononuclear [Mn(H2O)2]2+ and [Na(H2O)3]+ bridges10b and the [(VO)12O4{B8O17(OH)4}2]12− clusters are connected by unusual binuclear [Na2(H2O)2]2+ bridges into 1-D chain.10c But extended vanadoborates based on multinuclear [Nax(H2O)y]x+ (x ≥ 3) bridges have not been observed until now to the best of our knowledge. Hence, the 3-D extended framework built from the linkage of multinuclear sodium complexes and [V12B18] clusters in 1 represents a new vanadoborate structural motif.
UV/vis and IR spectra
The UV-vis spectrum of 1 displays three intense absorption bands (Fig. S4, ESI†). The higher energy band at 233 (5.32 eV) and 312 nm (3.97 eV) can be assigned to O→V and O→B charge transfers of [V12B18] cluster.11 The weak absorption at 555 nm (2.23 eV) presumably arises from the d–d electronic transition. As shown in Fig. S5, ESI,† there are no bands corresponding to the stretching of –NH2 and –CH2 groups, which confirms the absence of organic amine groups and is in good agreement with the single-crystal structural analyses. An intense peak centered at 3401 cm−1 that is assigned to the O–H stretch of the waters. A lower intensity band at 1649 cm−1 is assigned to the O–H bond of the waters. The strong band at 948 cm−1 is due to terminal V–O stretching. The 789 and 710 cm−1 bands are attributed to symmetrical and asymmetrical V–O–V stretching. The bands at 1417 and 1354 cm−1 are due to the asymmetrical stretch of the [BO3] group. The band at 1058 cm−1 is attributed to the asymmetrical stretch of the [BO4] tetrahedron. Thermogravimetric analysis of 1 was carried out under N2 atmosphere from 25 to 400 °C with a heating rate of 10 °C min−1 (Fig. S6, ESI†). A two-step weight loss occurs in the range of 25–140 °C, which is related to the removal of H2O and the –OH groups (found, 11.6%; calcd, 12.1%).Magnetic properties
The magnetic susceptibility of 1 was measured at 2–300 K. Fig. 3 shows the magnetic behavior of 1 in the form of the product χMT versus temperature, where χM is the molar magnetic susceptibility. The product of χMT decreases smoothly from 3.53 cm3 K mol−1 at 300 K to 0.23 cm3 K mol−1 at 2 K. The χMT value of 3.53 cm3 K mol−1 at 300 K is close to the spin only value of 3.75 cm3 K mol−1 expected for ten uncoupled S = 1/2 V4+ centers considering g = 2, indicating significant antiferromagnetic exchange interactions. Moreover, the plot of χM−1versus T in the range of 300–30 K is well described by the Curie–Weiss law with a Curie constant C = 4.54 cm3 K mol−1 and Weiss constant θ = −81.79 K (Fig. S7, ESI†), which further confirms the occurrence of strong antiferromagnetic exchange interactions between the V4+ ions. Such a strong antiferromagnetic coupling has been observed in most polyoxovanadate clusters.12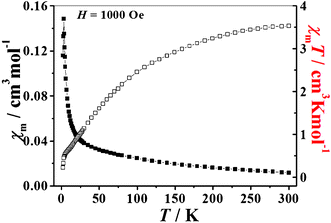 | ||
| Fig. 3 The temperature dependence of the molar magnetic susceptibility χM (■) and the product of the molar magnetic susceptibility and temperature χMT (□) for 1 between 2 and 300 K. | ||
Conclusions
A new 3-D vanadoborate was successfully made under hydrothermal conditions and structurally characterized. It provides a rare example of extended POMs constructed from an unusual 3-D Na–O–Na network and entrapped [V12B18] clusters. The successful isolation of 1 may open up possibilities for synthesizing new extended POMs with particular functions by the linkages of 3-D s-block metal–oxygen network and various POM clusters.Acknowledgements
This work was supported by the NNSF of China (No. 20961011 and 21163022), the NSF of Guangxi Province (No. 2010GXNSFB013017), the NSF of the Education Committee of Guangxi Province (No. 201012MS182), the Key Project of Chinese Ministry of Education (No. 212132) and Program for Excellent Talents in Guangxi Higher Education Institutions. The authors are also grateful to Chongqing Normal University for financial support (12XLB022).References
- (a) J. T. Rhule, C. L. Hill and D. A. Judd, Chem. Rev., 1998, 98, 327 CrossRef CAS; (b) D. E. Katsoulis, Chem. Rev., 1998, 98, 359 CrossRef CAS; (c) M. T. Pope and A. Müller, Angew. Chem., Int. Ed. Engl., 1991, 30, 34 CrossRef; (d) A. Müller, H. Reuter and S. Dillinger, Angew. Chem., Int. Ed. Engl., 1995, 34, 2328 CrossRef; (e) S. Uchida, R. Kawamoto, T. Akatsuka, S. Hikichi and N. Mizuno, Chem. Mater., 2005, 17, 1367 CrossRef CAS; (f) D.-L. Long, C. Streb, Y.-F. Song, S. Mitchell and L. Cronin, J. Am. Chem. Soc., 2008, 130, 1830 CrossRef CAS.
- (a) S.-T. Zheng, J. Zhang and G.-Y. Yang, Angew. Chem., Int. Ed., 2008, 47, 3909 CrossRef CAS; (b) Y. Sawada, W. Kosaka, Y. Hayashi and H. Miyasaka, Inorg. Chem., 2012, 51, 4824 CrossRef CAS; (c) H. E. Moll, A. Dolbecq, J. Marrot, G. Rousseau, M. Haouas, F. Taulelle1, G. Rogez, W. Wernsdorfer, B. Keita and P. Mialane, Chem.–Eur. J., 2012, 18, 3845 CrossRef; (d) J. Zhou, J. Zhang, W.-H. Fang and G.-Y. Yang, Chem.–Eur. J., 2010, 16, 13253 CrossRef CAS; (e) S. Reinoso, Dalton Trans., 2011, 40, 6610 RSC.
- (a) M. I. Khan, J. Solid State Chem., 2000, 152, 105 CrossRef CAS; (b) M. I. Khan, E. Yohannes and D. Powell, Chem. Commun., 1999, 23 RSC.
- (a) J.-W. Zhao, H.-P. Jia, J. Zhang, S.-T. Zheng and G.-Y. Yang, Chem.–Eur. J., 2007, 13, 10030 CrossRef CAS; (b) J.-W. Zhao, B. Li, S.-T. Zheng and G.-Y. Yang, Cryst. Growth Des., 2007, 7, 2658 CrossRef CAS; (c) C.-M. Liu, D.-Q. Zhang, M. Xiong and D.-B. Zhu, Chem. Commun., 2002, 1416 RSC; (d) B.-Z. Lin and S.-X. Liu, Chem. Commun., 2002, 2126 RSC; (e) S. Liu, L. Xie, B. Gao, C. Zhang, C. Sun, D. Li and Z. Su, Chem. Commun., 2005, 5023 RSC; (f) D. Hagrman and J. Zubieta, Chem. Commun., 1998, 2005 RSC; (g) C. Inman, J. M. Knaust and S. W. Keller, Chem. Commun., 2002, 156 RSC; (h) D. Hagrman, P. J. Hagrman and J. Zubieta, Angew. Chem., Int. Ed., 1999, 38, 3165 CrossRef CAS.
- (a) D. J. MacDougall, B. C. Noll and K. W. Henderson, Inorg. Chem., 2005, 44, 1181 CrossRef CAS; (b) D. J. MacDougall, J. J. Morris, B. C. Noll and K. W. Henderson, Chem. Commun., 2005, 456 RSC; (c) R.-Q. Zou, H. Sakurai and Q. Xu, Angew. Chem., Int. Ed., 2006, 45, 2542 CrossRef CAS; (d) K. W. Henderson, A. R. Kennedy, L. Macdonald and D. J. MacDougall, Inorg. Chem., 2003, 42, 2839 CrossRef CAS.
- (a) Z. Wu, Z. Liu, P. Tian, L. Xu, H. Song, X. Bao, X. Liu and X. Liu, Cryst. Res. Technol., 2006, 41, 1049 CrossRef CAS; (b) F. Avecilla, J. A. Peters and C. F. G. C. Geraldes, Eur. J. Inorg. Chem., 2003, 4179 CrossRef CAS; (c) G. Cao, R.C. Haushalter and K. G. Strohmaier, Inorg. Chem., 1993, 32, 127 CrossRef CAS; (d) W. J. Dressick, C. George, S. L. Brandow, T. L. Schull and D. A. Knight, J. Org. Chem., 2000, 65, 5059 CrossRef CAS; (e) L. Wang, Z. Chu, W. Huang and S. Gou, Inorg. Chem. Commun., 2009, 12, 4 CrossRef CAS; (f) K. D. Demadis and P. Baran, J. Solid State Chem., 2004, 177, 4768 CrossRef CAS; (g) S. Samanta, S. Mukhopadhyay, D. Mandal, R. J. Butcher and M. Chaudhury, Inorg. Chem., 2003, 42, 6284 CrossRef CAS; (h) M. C. K. França, J. Eon, M. Fournier, E. Payen and O. Mentré, Solid State Sci., 2005, 7, 1533 CrossRef; (i) M. Lindsjö, A. Fischer and L. Kloo, Z. Anorg. Allg. Chem., 2005, 631, 1497 CrossRef.
- G. M. Sheldrick, SHELXS-97, Program for solution of crystal structures, University of Göttingen, Germany, 1997 CrossRef; G. M. Sheldrick, Acta Crystallogr., Sect. A: Found. Crystallogr., 1990, 46, 467 CrossRef.
- G. M. Sheldrick, SHELXL-97, Program for refinement of crystal structures, University of Göttingen, Germany, 1997 CrossRef; G. M. Sheldrick, Acta Crystallogr., Sect. A: Found. Crystallogr., 2008, 64, 112 CrossRef.
- (a) I. D. Brown and D. Altermatt, Acta Crystallogr., Sect. B: Struct. Sci., 1985, 41, 244 CrossRef; (b) N. E. Brese and M. O'Keeffe, Acta Crystallogr., Sect. B: Struct. Sci., 1991, 47, 192 CrossRef.
- (a) Y.-Q. Sun, G.-M. Li and Y.-P. Chen, Dalton Trans., 2012, 41, 5774 RSC; (b) Y. Cao, H. Zhang, C. Huang, Q. Yang, Y. Chen, R. Sun, F. Zhang and W. Guo, J. Solid State Chem., 2005, 178, 3563 CrossRef CAS; (c) Y. Cao, H. Zhang, C. Huang, Y. Chen, R. Sun and W. Guo, J. Mol. Struct., 2005, 733, 211 CrossRef CAS.
- (a) H. Ma, X. Meng, J. Sha, H. Pang and L. Wu, Solid State Sci., 2011, 13, 850 CrossRef CAS; (b) X. Liu, J. Zhou, Z. Zhou and F. Zhang, J. Cluster Sci., 2011, 22, 65 CrossRef CAS.
- (a) J. Zhou, S.-T. Zheng, W.-H. Fang and G.-Y. Yang, Eur. J. Inorg. Chem., 2009, 5075 CrossRef CAS; (b) Y. Qi, Y. Li, C. Qin, E. Wang, H. Jin, D. Xiao, X. Wang and S. Chang, Inorg. Chem., 2007, 46, 3217 CrossRef CAS; (c) S.-T. Zheng, J. Zhang and G.-Y. Yang, Inorg. Chem., 2005, 44, 2426 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: XRD patterns, TG curve, optical absorption and IR spectra. CCDC reference number 892031. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c2ra21635c |
| This journal is © The Royal Society of Chemistry 2012 |