NMR relaxation and structural elucidation of peptides in the presence and absence of trifluoroethanol illuminates the critical molecular nature of integrin αvβ6 ligand specificity†
Jane L.
Wagstaff
a,
Michelle L.
Rowe
a,
Shu-Ju
Hsieh
ab,
Danielle
DiCara
cd,
John F.
Marshall
c,
Richard A.
Williamson
*a and
Mark J.
Howard
*a
aProtein Science Group, School of Biosciences, University of Kent, Canterbury, Kent CT2 7NJ, UK. E-mail: m.j.howard@kent.ac.uk; r.a.williamson@kent.ac.uk
bInstitute of Biological Chemistry, Academia Sinica, Taipei, Taiwan
cQueen Mary University of London, Barts and The London School of Medicine and Dentistry, Institute of Cancer and CRUK Clinical Centre, John Vane Science Centre, Charterhouse Square, London, EC1M 6BQ, UK
dMRC Laboratory of Molecular Biology, Hills Road, Cambridge, CB2 0QH, UK
First published on 17th September 2012
Abstract
Integrin αvβ6 is an important emerging target for both imaging and therapy of cancer that requires specific ligands based on Arg-Gly-Asp (RGD) peptides. There remains little correlation between integrin-RGD ligand specificity despite studies suggesting an RGD-turn-helix ligand motif is required. Here, we describe the application of 15N NMR relaxation analyses and structure determination of αvβ6 peptide ligands in the presence and absence of trifluoroethanol (TFE) to identify their critical molecular nature that influences specificity, interaction and function. Two linear peptides; one known to demonstrate αvβ6 specificity (FMDV2) and the other based on a natural RGD ligand (LAP2), were compared to two additional peptides based on FMDV2 but cyclised in different positions using a disulphide bond (DBD1 and DBD2). The cyclic adaptation in DBD1 produces a significant alteration in backbone dynamic properties when compared to FMDV2; a potential driver for the loss in αvβ6 specificity by DBD1. The importance of ligand dynamics are highlighted through a comprehensive reduced spectral density and ModelFree analysis of peptide 15N NMR relaxation data and suggest αvβ6 specificity requires the formation of a structurally rigid helix preceded by a RGD motif exhibiting slow internal motion. Additional observations include the effect of TFE/water viscosity on global NMR dynamics and the advantages of using spectral density NMR relaxation data to estimate correlation times and motional time regimes for peptides in solution.
Introduction
Integrins are heterodimeric glycoproteins composed of non-covalently linked α and β subunits1 that dynamically translate extracellular matrix cues into intracellular responses, including mechanical, biochemical, and genetic signals, thereby modulating cell proliferation, survival, migration, and invasion.2 These properties make many integrins key determinants of the survival and spread of cancer and one such integrin, αvβ6, is only expressed on epithelia and then only during processes of tissue remodelling; including wound healing, chronic inflammation, and cancer.3,4 Many investigations have identified αvβ6 as imparting a pro-invasive and aggressive phenotype when overexpressed on cancer cells4–7 that reflects the clinical situation in humans, since survival from cancer of the colon, cervix or from non-small cell lung cancer is reduced significantly if cancers express high levels of αvβ6.8 Integrin αvβ6 is scarce or absent in normal tissue but its presence drives tumour invasion and shortens survival, therefore it represents a major new specific molecular target for many cancers. There are 24 known integrins that cover a variety of signalling functions1 and it is imperative that specific molecular targeting to αvβ6 is achieved to maximise opportunities for tumour imaging and therapy. The challenge for specific molecular recognition of ligands toward αvβ6 is complicated by it being a member of the arginine-glycine-aspartic acid (RGD) receptor class of integrins that includes αvβ3, αvβ5, αvβ8, α8β1, α5β1 and αIIbβ3.Peptide agonists with increased specificity to αvβ6 have been found to recognise an extended motif Asp-Leu-Xxx-Xxx-Leu (DLXXL)9 creating a larger recognition sequence of RGDLXXL. We have previously reported that 20-mer peptide ligands in 30% (v/v) trifluoroethanol (TFE) form structures with RGDLXXL turn-helices where increased helix length correlated with affinity toward αvβ6.10 TFE promotes secondary structure in peptides11 and influences protein structure and function12 therefore the relevance of the TFE induced helix to αvβ6 binding requires confirmation. 1H STD NMR10 and 13C, 1H 2D STD NMR,13 has established αvβ6 peptide ligands bind in a helical conformation that mirrors the TFE induced structure; justifying the approach of analysing the structures of these peptides using TFE/buffer solutions. The highest specificity peptide toward αvβ6 to date is A20-FMDV2, with the primary sequence NAVPNLRGDLQVLAQKVART. This peptide sequence was taken from the surface GH loop motif from foot and mouth disease virus serotype O1 BFS capsid protein VP114 and is a known ligand for integrin αvβ6.10 The FMDV GH loop sequence is very divergent, however the primary recognition motif RGD and to an extent the longer motif RGDLXXL is highly conserved among field isolates.15 A20-FMDV2 has been shown to exhibit extremely high specificity toward αvβ6 over other integrins from in vitro10 and in vivo studies, the latter using mouse models that illustrate clear selectivity of A20-FMDV2 for αvβ6 over αvβ3; thus proving the potential of αvβ6 as a cancer target.8,16
A20-FMDV2 exhibits great promise but, there are advantages to providing either mimetics or modified versions of this peptide to enhance affinity, increase in vivo half-life and reduced clearance for maximum avidity in clinical use. One such method to improve stability is to cyclise the peptide. Cyclised versions of A20-FMDV2 were produced using a disulphide bond but despite an increase in serum stability, this peptide produced little if any increase in αvβ6 affinity and a decrease in αvβ6 specificity. This surprising observation suggests αvβ6 is sensitive to subtle ligand properties and we report in this 15N NMR dynamics study that ligand conformational rigidity correlates with αvβ6 ligand specificity.
Isotopic enrichment provides relatively straightforward access to backbone dynamics over the nanosecond to picosecond timescale as well as affording much-improved precision in the structural elucidation process. NMR backbone dynamics report on the overall tumbling behaviour of the peptide in solution, defined by the molecular correlation time τm, as well as dynamic internal detail via order parameters S2 and internal motion correlation times τe reporting on each 15NH bond vector across the peptide backbone. The order parameter can also be devolved into two separate order parameter processes; Sf2 for fast motions and Ss2 for slow motions where all order parameters are permitted to take values between 0.0 and 1.0; these extremes defining a fully disordered random or rigid ordered system respectively. Another important role of NMR relaxation data is to identify and isolate conformational exchange events through the parameter Rex. NMR solution dynamics have been useful tools in the study of protein internal and global motions related to function but has found very limited use in peptides to date with few examples existing beyond the utility of natural abundance 13C NMR relaxation to study nascent structure,17,18 observing flexibility differences between oxidised and reduced peptide forms19 or in membrane peptide analysis.20,21 Our study is the first to observe dynamic changes in the presence and absence of TFE using 15N NMR peptide relaxation with combined ModelFree and spectral density methods that verify this approach for peptides within TFE-buffer systems. Four peptides were used in this study:
FMDV2 (NAVPNLRGDLQVLAQKVART-Hsl)
DBD1 (EKCPNLRGDLQVLAQKVCRT-Hsl)
DBD2 (CYVPNLRGDLQVLAQKVAKC-Hsl)
LAP2 (GFTTGRRGDLATIHGLNRPF-Hsl)
All peptides contain a C-terminal modification of a methionine to a homoserine lactone (Hsl) that occurs from cyanogen bromide cleavage of the recombinant peptide from its fusion partner; hence the removal of ‘A20’ in peptide names used in previous studies; FMDV2 is A20-FMDV2-Hsl and provides a benchmark for a peptide with high αvβ6 affinity. DBD1 and DBD2 are modified FMDV2 peptides, each with a different intramolecular disulphide bond linkage that was designed to support the original A20-FMDV2 structure.10 The disulphide bond serves two purposes; (a) to force stabilisation of the turn-helix topology and (b) to provide in vivo serum stability for clinical use; cyclising peptides has been a successful approach to stabilise peptides in the radiopharmaceutical field for many years. Additional residues Glu1-Lys2 in DBD1 and Tyr2 in DBD2 were inserted to enable in vivo imaging labels to be easily attached to the peptides. LAP2 is a natural αvβ6 ligand based upon the latency associated peptide (LAP) of a transforming growth factor (TGFβ1) with a single mutation M16L to allow cleavage of the peptide from the recombinant fusion partner by cyanogen bromide.22,23 Unlike FMDV2, LAP and LAP2 have high affinities for integrins other than αvβ6 and provide comparison data with respect to this wider spectrum of targets.
This NMR based structural and dynamic study of these four 21-mer αvβ6 peptide ligands illustrates that differences in peptide design, through disulphide bond cyclisation, dramatically alters the fundamental properties of the ligand that in turn influence their affinity and specificity toward αvβ6. Our study informs on the critical molecular nature of integrin ligands that defines their interaction and function; optimal ligands for αvβ6 can be designed in the future. These results also illustrate that the design of drugs or agents toward integrins requires careful consideration of structure and dynamic ligand properties.
Results
Peptide production and purification
Production and purification of all peptides was completed with comparable yields by following our published protocol23 with one exception: post-translation formylation was observed with LAP2 peptide that was subsequently circumvented by solubilising the peptide for CNBr cleavage in trifluoroacetic acid.Peptide NMR assignments and structural elucidation
All peptides were assigned in the presence and absence of TFE and the structures of the peptides in the presence of TFE solved using 2D and 3D 15N edited NOESY datasets. Assigned 15N–1H HSQC datasets for each peptide and all peptide NMR assignments are available (Fig. S1 and Table S1, ESI†). The structure closest to the mean for each peptide is shown in Fig. 1 and structural ensembles and statistics shown in Fig. S2 and Table S2 in ESI†. All ensembles show variation due to low numbers of structural restraints beyond the helical regions but disorder is better identified using NMR relaxation dynamics. Structural limits for each peptide are also defined in Fig. 1 that includes a schematic of contacts and restraints. Fig. 1 confirms all peptides conform to the RGD-turn helix motif as reported previously10 and additional structural information is reported in the ESI†.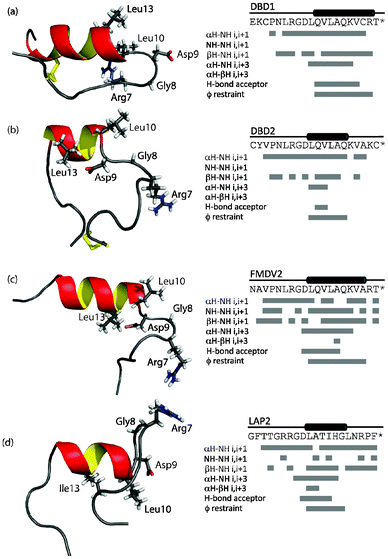 | ||
| Fig. 1 Structures closest to the mean for each peptide and NOE contacts, hydrogen bond restraints assigned for DBD1(a), DBD2 (b), FMDV2 (c) and LAP (d) in the presence of 30% (v/v) TFE. Key residues of the RGDLXXL/I motif are shown as sticks on each structure. | ||
15N NMR relaxation studies ± TFE
Relaxation parameters were obtained for individual amino acids in each peptide in the absence of TFE (Fig. S3, ESI†) and mean values for each parameter within its respective peptide are summarised in Table 1(a). In the absence of TFE, relaxation parameter sets distinguish disulphide bonded cyclic peptides from free peptides. FMDV2 and LAP2 free peptides exhibit variability across T1 (longitudinal relaxation time), T2 (transverse relaxation time) and η (heteronuclear NOE) that defines both termini from larger T1 and T2 values and highly negative η values. DBD1 and DBD2 values are significantly different and confirm a compact topology is present for these peptides even in the absence of TFE. The global correlation time τm, in the absence of TFE, was estimated for DBD1, DBD2, FMDV2 and LAP2 to be 2.4 ± 0.1, 3.8 ± 0.4, 1.4 ± 0.4 and 3.3 ± 0.2 ns using dipolar T1 and T2 equations to correlate average observed to theoretical T1/T2 ratios for each peptide. Despite using S2 = 0.2 in the theoretical treatment (to account for unstructured random coil), these results confirm that T1/T2 ratios place emphasis on the spin–spin relaxation time (T2) for the estimation of τm. As peptides provide a small population of relaxation data, estimated τm values need additional supporting validation if subsequent ModelFree analysis is required.| (a) No TFE | |||
|---|---|---|---|
| 15N T1 (ms) | 15N T2 (ms) | η | |
| DBD1 | 558 ± 5 | 394 ± 5 | −0.70 |
| DBD2 | 551 ± 5 | 298 ± 5 | −0.71 |
| FMDV2 | 696 ± 5 | 592 ± 5 | −1.20 |
| LAP2 | 768 ± 5 | 461 ± 5 | −1.20 |
| (b) 30% (v/v) TFE | |||
|---|---|---|---|
| 15N T1 (ms) | 15N T2 (ms) | η | |
| DBD1 | 582 ± 31 | 114 ± 12 | −0.54 |
| DBD2 | 523 ± 6 | 157 ± 3 | −0.40 |
| FMDV2 | 572 ± 15 | 169 ± 6 | −0.27 |
| LAP2 | 768 ± 6 | 229 ± 5 | −0.59 |
Relaxation parameters were obtained for individual amino acids in each peptide in the presence of TFE (Fig. S4, ESI†) and mean values for each parameter within its respective peptide are summarised in Table 1(b). FMDV2 and LAP2 illustrate a reduction of both T2 and η across the structurally defined helical regions for each peptide to confirm the formation of a rigid secondary structure element in the presence of TFE validating the structural limits as outlined in Fig. 1. Relaxation data of DBD1 and DBD2 support TFE promoting helix formation despite these peptides being constrained through the disulphide bond. Using an average S2 of 0.86 representing a structured state, the global correlation time τm for DBD1, DBD2, FMDV2 and LAP2 were estimated to be 6.6 ± 0.7, 4.9 ± 0.1, 4.9 ± 0.2 and 3.7 ± 0.1 ns respectively in the presence of TFE; also exhibiting variation influenced by T2 when using T1/T2 ratio estimates. This is most noticeable with DBD1 where the average 15N T2 in TFE is depressed compared to other peptides and suggests exchange line broadening is present in the data. Discounting DBD1, all estimations of τm in TFE are approximately double that expected based on back extrapolation of protein data using residue number24 that predicts 21 residue structures at 10 °C to have a τm of 2.8 ns. The 30%(v/v) TFE/buffer mixture is reported to have double the viscosity of water at 10 °C25 which explains this increase in correlation time. This also rationalizes the practical advantage of using NOE over ROE for TFE based peptide structure determination. Equally, knowledge of this viscosity effect further supports our use of low levels of TFE to complete the co-operative transition of helix formation as devised from previous far-UV CD observations.10 These relaxation data must be considered in context with advanced analysis methods below.
Reduced spectral density mapping
15N relaxation parameters report on the motional properties of each peptide and such motions are translated through spectral density functions that, being defined from the autocorrelation function, describe the limit and amplitude of frequencies of motions experienced from the reorientations of 15N–1H bond vectors in each peptide. 15N relaxation parameters (Fig. S3 and S4, ESI†) were used to determine reduced spectral density values that makes no assumptions on timescales of motion in the molecule under investigation; a clear advantage with small datasets. Spectral densities were estimated at three frequencies: zero, ωN and 0.87ωH where we have adopted the notation that describes each spectral density as J(0), J(ωN) and J(ωh).26 Timescales of motion of the 15N–1H bond vector are expressed by each spectral density estimate where J(0) describes millisecond to nanosecond motion, J(ωN) nanosecond motion and J(ωh) nanosecond to picosecond motion that are qualitatively described as slow, intermediate and fast time regimes respectively. Plots of spectral densities with residue number for each peptide in the absence and presence of TFE are available (Fig. S5, ESI†). Spectral density values can be interrogated graphically in order to appreciate the motional time regimes within proteins under investigation27–29 and Fig. 2 and Fig. 3 illustrate this approach for peptide data in the absence and presence of TFE. The solid curved lines in each figure are an aid to interpretation and represent the simple Lorentzian model of spectral density calculated for a range of correlation times; equivalent to an order parameter S2 = 1.0 for an isotropic system. Specific motional time points for these curves are shown in Fig. S6, ESI†. The scatter of data within each curve is due to global and local motion that can be explained using ModelFree order parameters S2, Sf2 and Ss2 as well as exchange line broadening contributions, Rex. Linear curve fitting of the data defines line segments that intercept the solid Lorentzian line at specific time points to provide estimates of the timescales of motion for each peptide in the absence and presence of TFE. Data was further utilised to understand specific motional behaviour of both RGD-turn (blue data points) and helix motifs (red data points) as defined from the structural data.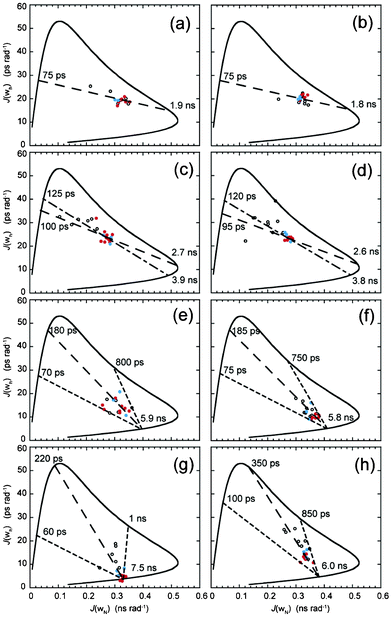 | ||
| Fig. 2 Parametric curves showing the dependence of J(ωN) on J(ωh) for individual 15N nuclei from (a) DBD1-TFE, (b) DBD2-TFE, (c) FMDV2-TFE, (d) LAP2-TFE, (e) DBD1+TFE, (f) DBD2+TFE, (g) FMDV2+TFE and (h) LAP2+TFE. Defined structure regions from Fig. 1 are coloured blue for RGD-turn and red for the helix region for each peptide. The continuous curved line represents the dependence of theoretical Lorentzian spectral density functions with variable correlation time. The large-dashed line (– – – –) is a least-squared fit to all the data points in (a–h), the small-large-dashed line (- – - –) is a fit to only RGD-helix data points in (a–d) and the small-dashed line (- - - -) is the motional extremes as defined from the data. Motional times defined as the intercept of each dashed line with the theoretical curve are shown on individual plots. | ||
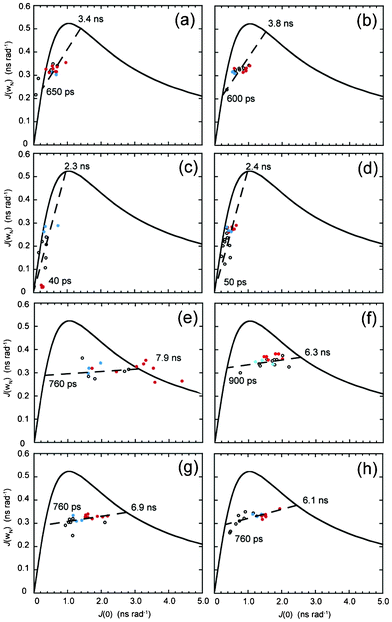 | ||
| Fig. 3 Parametric curves showing the dependence of J(ωh) on J(0) for individual 15N nuclei from (a) DBD1-TFE, (b) DBD2-TFE, (c) FMDV2-TFE, (d) LAP2-TFE, (e) DBD1+TFE, (f) DBD2+TFE, (g) FMDV2+TFE and (h) LAP2+TFE. Defined structure regions from Fig. 1 are coloured blue for RGD-turn and red for the helix region for each peptide. The continuous curved line represents the dependence of theoretical Lorentzian spectral density functions with variable correlation time. The large-dashed line (– – – –) represents a least-squared fit to all the data points in (a–h). Motional times defined as the intercept of each dashed line with the theoretical curve are shown on individual plots. | ||
Fig. 2 (a–d) displays J(ωh) vs. J(ωN) plots for each peptide in the absence of TFE. DBD1 (Fig. 2a) and DBD2 (Fig. 2b) can be analysed using a single fitted dashed line with virtually all 15N–1H bond vectors across the peptides clustering regardless of position in the sequence or presence in the RGD-turn or helix motif. Intercept points for both DBD1 and DBD2 provide similar estimates of the motional times τ of 1.9 ns/75 ps and 1.8 ns/75 ps (global/local motion); supporting both disulphide bonded peptides tumbling in concert over the entire sequence. In contrast, non-cyclic peptides FMDV2 (Fig. 2c) and LAP2 (Fig. 2d) display a larger distribution of their spectral density data in the absence of TFE and linear line fitting is different when applied to the entire dataset or to data from the RGD-helix motif. For FMDV2, motional times τ of 2.7 ns/100 ps (all values) and 3.9 ns/125 ps (RGD-helix) are obtained with LAP2 providing 2.6 ns/95 ps (all values) and 3.8 ns/120 ps (RGD-helix). As with DBD1 and DBD2, data is comparable between FMDV2 and LAP2 for all residues suggesting similar motional properties but increases in τ for the RGD-helix support the presence of nascent structure and short-term structural order. The position of data on each fitted line is influenced by its effective order parameter S2 that can be quantitatively determined as shown in the original manuscript.27 However, it is sufficient here to notice FMDV2 and LAP2 data is distributed to the left of the fitted line and closer to the J(ωN) axis supporting overall S2 values for FMDV2 and LAP2 as lower than those for DBD1 and DBD2 in the absence of TFE. Equally, RGD and helix motif data for FMDV2 and LAP2 occurs further from the J(ωN) axis and supports an increased order parameter S2 compared to data outside these motif regions. This is underpinned by the larger motional time values obtained for these motif regions and we conclude that this method of data handling is useful for discerning structural propensity and relative order in poorly defined regions. Furthermore, these estimates of τ proved useful for ModelFree analysis shown below.
Fig. 2(e–h) displays J(ωh) vs. J(ωN) plots for each peptide in the presence of TFE with data average fitted dashed lines that define overall and RGD-helix motional limits converging to a point of common overall correlation time. Fig. 2(e) and 2(f) show the cyclic peptides DBD1 and DBD2 continue to exhibit similar J(ωh) and J(ωN) in the presence of TFE with average motional times, τ, estimated to be 5.9 ns/180 ps and 5.8 ns/185 ps respectively; even the overall limits estimated from the curves are comparable at 70 ps/800 ps and 75 ps/750 ps respectively. Despite significant differences in structure, the overall dynamic nature of DBD1 and DBD2 are similar on both J(ωh) and J(ωN) time scales and such comparable motional properties must be driven primarily by the cyclic nature rather than defined structural regions. Differences are apparent upon comparison of RGD and helix regions with DBD2+TFE helix data being closer to the intercept point and so having higher order parameter S2 values than the RGD-turn. In contrast, DBD1+TFE provides a diffuse data pattern with the helix data specifically not forming a compact arrangement close to the intercept. This is, in part, due to DBD1+TFE data being unique and the only dataset to be described by a single order parameter S2 together with a internal correlation time τe. However, residual broadening was also observed in Fig. 3(e). In contrast, non-cyclic peptides demonstrate more diverse J(ωh) and J(ωN) defined motional properties that are reflective of the different structural regions promoted by the co-solvent. Fig. 2(g) and 2(h) provide average motional times for FMDV2+TFE and LAP2+TFE of 7.5 ns/220 ps and 6.0 ns/350 ps respectively and motional limits of 60 ps/1 ns and 100 ps/850 ps. Motional values are similar but FMDV2 has a global correlation time 1.5 ns slower in addition to a lower average internal correlation time but with broader limits. The most important observation is the compact nature of FMDV2 helix data points (Fig. 2(g)) supporting order parameter S2 values approaching 1.0 for these NH vectors. The RGD-turn region data are distributed just beyond the helix and unstructured terminal residues define the extreme limits in Fig. 2(g). LAP2+TFE data has a broad distribution along the linear average line suggesting less order overall than that observed for FMDV2. Again, these estimates of correlation time and motional time regimes proved useful for ModelFree analysis.
Fig. 3(a–h) demonstrate the use of J(ωN) vs. J(0) for all peptides in the absence or presence of TFE to evaluate data for chemical exchange contributions that manifest as line broadening and generate larger values of J(0). This is clearly demonstrated in Fig. 3(e) for DBD1+TFE where helix data fall outside the theoretical Lorentzian line. These data can also be interrogated via a linear line that acts as a barometer for the order parameter S2 and the dotted-Lorentzian line intercept signifies S2 is zero on the left intercept and is 1.0 on the right intercept. Fig. 3(a–d) illustrate the difficulty in producing linear fits for data in the absence of TFE due to the narrow range of J(0) obtained, that in turn was expected from systems tumbling with low correlation times and low effective order parameters. In contrast, Fig. 3(e–h), for peptides in the presence of TFE, provided correlation time estimates more comparable to those obtained from Fig. 2(e–h). The value of 7.9 ns for DBD1+TFE is 2.0 ns longer than that derived from Fig. 3(e) but the NH vectors in the helix of this peptide are clearly in chemical exchange. Fig. 3(f–h) provide correlation time estimates for DBD2+TFE, FMDV2+TFE and LAP2+TFE of 6.3 ns, 6.9 ns and 6.1 ns which compare favourably with Fig. 2(f–h) estimates of 5.8 ns, 7.5 ns and 6.0 ns.
Lipari-Szabo ModelFree analysis of NMR relaxation parameters
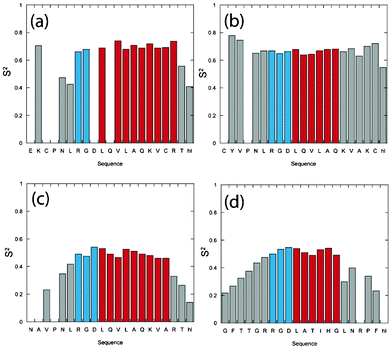
Fig. 4 displays the ModelFree parameters obtained for all peptides in the presence of TFE where each appropriate model was identified using a common selection strategy30 together with global correlation time data obtained from extended spectral density analysis. This approach provided lower chi-squared fitting values than using correlation times obtained directly from NMR relaxation parameters. The overall order parameter S2 for each peptide supports increased order across the entirety of both disulphide bonded cyclic peptides unlike FMDV2 and LAP2 where S2 values <0.5 are observed at the termini.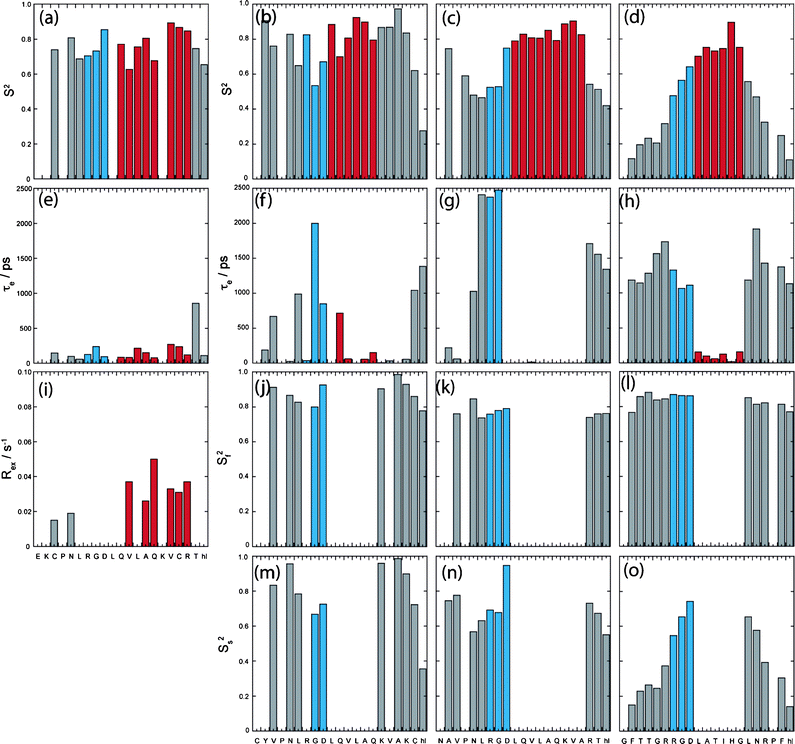 | ||
| Fig. 4 Column plots describing the results of model-free Lipari-Szabo analysis of (a), (e) and (i) DBD1+TFE, (b), (f), (j) and (m) DBD2+TFE, (c), (g), (k) and (n) FMDV2+TFE and (d), (h), (l) and (o) LAP2+TFE with amino acid sequence. Data reporting within TFE defined structure regions from Fig. 1 are coloured blue for RGD-turn and red for the helix region for each peptide. | ||
DBD1+TFE relaxation data was unique in being the only dataset to be described using a single order parameter S2 together with the correlation time for internal motion τe. However, residues Cys3, Asn5, Val12, Ala14, Gln15, Val17, Cys18, Arg19 and Thr20 all required contributions to account for additional line broadening due to chemical exchange, characterised by Rex as identified in Fig. 4(i); confirming the validity of data points to the right of Lorentzian boundary in Fig. 3(e). The unique nature of DBD1+TFE data is also seen with concomitant τe contributions <500 ps (with the exception of Thr20) that are smaller than those within the remaining peptides. This is most apparent for the RGD region that displays an average τe across the three amino acids of ca. 0.15 ns compared to 0.96 ns, 1.60 ns and 1.15 ns for DBD2+TFE, FMDV2+TFE and LAP2+TFE respectively. Rex and τe contributions label DBD1+TFE as having unique properties among the peptides studied.
Helical regions of DBD2+TFE, FMDV2+TFE and LAP2+TFE provided optimal ModelFree chi-squared values when using a single order parameter and internal correlation time. However, data for most residues outside the helical region did not provide satisfactory fits from a S2/τe model and optimal fits were obtained using two internal motion processes covering fast and slow nanosecond timescale events, as described by their own specific order parameters Sf2 and Ss2. Sufficient data redundancy was maintained by estimating only τe in addition to Sf2 and Ss2 and in these cases Sf2·Ss2 = S2. Unlike DBD1+TFE, DBD2+TFE exhibits slow and fast motion contributions although S2 values remain greater than 0.7 across the peptide with the exception of Cys20 and Hsl21 (Fig. 4b); suggesting a reasonably rigid backbone as expected. These differences compared to DBD1+TFE are most likely due to the open structural arrangement of DBD2+TFE (Fig. 1b). The RGD region of DBD2+TFE displays internal motion of 0.04/2.00/0.85 ns for Arg7/Gly8/Asp9 and is slower compared to DBD1+TFE (Fig. 4e) but not as slow as observed for Arg7/Gly8 of FMDV2+TFE (see below).
ModelFree data for FMDV2+TFE and LAP2+TFE highlight different overall S2 order parameter in Fig. 4c and 4d. Helical regions provide average S2 of 0.83 ± 0.04 and 0.76 ± 0.07 for FMDV2+TFE and LAP2+TFE respectively. The helix in FMDV2+TFE displays the highest overall rigidity of all peptides studied and despite not having a disulphide-bond to hold the peptide in a cyclic conformation, FMDV2+TFE has average termini S2 values of 0.54 ± 0.11 compared to 0.28 ± 0.14 for LAP2+TFE suggesting increased rigidity in the termini of FMDV2+TFE. Fig. 5g and 5h highlight significant slow internal motions in both termini of LAP2+TFE that manifest in the C-terminus of FMDV2+TFE but are not as prevalent within its N-terminus. This is supported by NOEs observed in FMDV2+TFE between N-terminal residues and the turn-helix. In contrast, LAP2+TFE displays mobile termini where Ss2 < Sf2 that dominates as the low overall order parameters in Fig. 5d. The RGD region of FMDV2+TFE suggests Asp9 has similar dynamic characteristics as the helix with Ss2 = 0.95 but Arg7 and Gly8 have significant slow internal motion values of 2.38 ns and 2.48 ns respectively. In contrast, the internal correlation times of Arg7/Gly8 of LAP2+TFE are more comparable to Gly8 in DBD2+TFE. FMDV2+TFE has the most ordered helical region and termini of the non-cyclic peptide pair but also exhibits comparatively slow internal motions in the Arg-Gly region of the RGD motif when compared to the other peptides. LAP2+TFE exhibits very flexible termini making this peptide unique.
 | ||
| Fig. 5 Column plots describing the order parameter S2 results of model-free Lipari-Szabo analysis of (a) DBD1-TFE, (b) DBD2-TFE, (c) FMDV2-TFE and (d) LAP2-TFE with amino acid sequence. Data reporting within TFE defined structure regions from Fig. 1 are coloured blue for RGD-turn and red for the helix region for each peptide. | ||
A ModelFree investigation of relaxation data in the absence of TFE using correlation times derived from Fig. 2(a–d) is shown in Fig. 5. Data fits support both cyclic peptides having rigidity across their respective sequences, as suggested above. Order parameter S2 remains fairly constant across the RGD-helix regions of both DBD1-TFE and DBD2-TFE; supporting the disulphide bonds promoting interaction between the N- and C-terminal regions of each peptide. In contrast both FMDV2-TFE and LAP2-TFE provide similar S2 profiles that would be expected from peptides with no defined structure in solution although increased order parameter toward the centre of the peptide acknowledges a reduction in the backbone degrees of freedom. Close inspection of Fig. 5c and 5d indicate that the helix forming regions have increased order, as illustrated in Fig. 5c where Val17 has an S2 of 0.45 but the equivalent residue 5 amino acids from the N-terminus, Asn4, has an S2 value of 0.35. This provides evidence for the S2 maximum is at Leu13 in FMDV2-TFE unlike the sequence centre at Gln11. The asymmetry observed in the order parameter of FMDV2 in the absence of TFE suggests the presence of nascent structure in this peptide; these subtle but significant changes are supported by the spectral density data in Fig. 2c and 2d; this verifies the use of NMR relaxation parameters in the study nascent structure. The equivalent asymmetry in the order parameter profile of LAP2-TFE is less obvious due to the smaller helix placing the largest S2 values in the middle of the peptide.
Implications of peptide structure and NMR relaxation dynamics for integrin recognition and specificity
Specificity of biotinylated peptides for αvβ6 was assessed using the paired cell lines A375P.Beta6.puro and A375P.puro.10 Both cell lines express integrins αvβ8, αvβ5, αvβ3 and α5β1 (data not shown) but only A375P.Beta6.puro expresses αvβ6. Biotinylated A20FMDV2, DBD1 and DBD2 bind to A375P.Beta6.puro at nanomolar concentrations but only at micromolar concentrations to A375P.puro (Fig. S7, ESI†), showing a high degree of both affinity and specificity for αvβ6. DBD1 shows the highest degree of binding to the αvβ6-negative A375P.puro, followed by DBD2, and finally A20FMDV2, which has an extremely low level of binding even at 10 μM (Fig. S7, ESI†). A20LAP has previously been shown to have a much lower affinity for αvβ610 and in similar experiments also bound to A375P.puro at micromolar concentrations (data not shown).Discussion
Our previous structure-function studies of RGD peptides toward integrin αvβ6 highlighted a clear correlation between helical length and affinity toward the integrin.10,13 These investigations supported the proposition that TFE-generated helical peptide structures are adopted upon binding the integrin. From a functional viewpoint, FACS analysis of each peptide confirms FMDV2 as the most αvβ6 specific peptide and both flow cytometry (Fig. S7, ESI†) and solid-phase binding assays (data not shown) demonstrate that cyclisation via a disulphide bond does not detrimentally change affinity toward αvβ6 in DBD1 and DBD2. However like LAP2, DBD1 and DBD2 also bind to integrin αvβ6-negative cells that express integrins αvβ3, αvβ5, αvβ8 and α5β1; the unique specificity of FMDV2 to integrin αvβ6 is lost due to the addition of the disulphide bond even though DBD1 and DBD2 are sequence related to FMDV2.Fig. 1 illustrates that each peptide adopts a specific helical conformation in TFE; a potential basis for both affinity and specificity differences. Structural differences between DBD1 and FMDV2 originate from the disulphide bond, as proven by the similarities between 15N HSQC data for reduced DBD1 and FMDV2 (Fig. S8, ESI†). The DBD1 helix is considerably different in that Leu10 is not within the helical section and this peptide forms an RGDL-helix motif in contrast to the RGD-helix motif that DBD2, FMDV2 and LAP2 exhibit. However, DBD1 displays a higher affinity than FMDV2 for the integrin although it is not αvβ6 specific; suggesting the change in structural motif is not detrimental to αvβ6 recognition. Our previous study ranked FMDV2 with higher affinity than LAP based on helix length10 where length of the helix in TFE followed as (DBD1 > FMDV2 > DBD2 > LAP2) which correlates with αvβ6 affinity (DBD1 > A20FMDV2 ≥ DBD2 > A20LAP). Affinity to integrins other than αvβ6 expressed on A375P.puro cells follows DBD1 > DBD2 > FMDV2, such that specificity to other integrins does not correlate with helix length.
LAP2+TFE is the most dynamically flexible peptide with lower S2 values suggesting significant motion at both termini, in contrast to that observed for FMDV2+TFE. These dynamic differences clearly separate LAP2 and FMDV2 as distinct from each other and may account for differences in affinity and specificity for αvβ6. Also, slow internal motions observed in the Arg-Gly of the RGD region of FMDV2 separate it from the other three peptides studied. These observations suggest the hypothesis that αvβ6 specificity is dynamically driven by a suitable peptide ligand creating a rigid helix with a high order parameter and an RGD motif with slow internal motions; properties that FMDV2 demonstrates. It is worth remembering that LAP2 contains the M16L mutation which could be argued as sufficiently different to the natural substrate (LAP) to question these conclusions, but 15N HSQC overlays of LAP and LAP2 (Fig. S9, ESI†) show the similarity between these two peptides. 15N-labelled LAP could never be produced in sufficient quantities for NMR relaxation studies; necessitating the use of LAP2 instead.
Critical observations involve the role of the disulphide bonds in both DBD1 and DBD2 that create two peptides with dramatically different dynamic properties upon helix formation. The disulphide bond promotes helix formation by destabilising the unfolded state but it also enforces a reduction in specificity toward αvβ6. This makes DBD1 and DBD2 less useful as starting points for the design of mimetics for cancer therapy or imaging. DBD2 displays dynamic properties most similar to FMDV2 and it follows that DBD2's specificity to αvβ6 is closest to FMDV2; an observation that further supports the hypothesis that ligand dynamics drives αvβ6 specificity. Equally, the dynamics of DBD2 are sufficiently diverse from FMDV2 to differentiate their respective affinities and specificities toward αvβ6. This suggests specificity and affinity are finely balanced for ligands towards αvβ6 and increased affinity will sacrifice specificity and vice versa. This concept is supported by the dynamic and exchange active peptide DBD1 having the highest affinity toward αvβ6 at the cost of being a peptide that recognises other integrins, an observation that equally suggests integrins other than αvβ6 interact with conformationally labile ligands. Ultimately, NMR relaxation dynamics support FMDV2 forming a rigid RGD-helix motif, as shown by the tight compact nature of data in Fig. 2g, and that this rigid nature supports an optimal fit to the αvβ6 ligand-binding site. In addition, this rigidity is not conducive to binding to other integrins, including others from the αv class and supports the β6 subunit as the key ingredient for αvβ6 recognition. As a result, the β-unit must define specificity across the integrin classes and can sense the sequence, structure and dynamics of potential ligands.
Conclusions
Isotopically enriched peptides have allowed a precise structure and dynamic study of ligands with different affinities and specificities toward integrin αvβ6. This approach supports our initial hypothesis that peptide ligand helix formation is linked to affinity and specificity but has equally identified the dynamic nature of peptides as important. Our results suggest that integrin αvβ6 specific peptides require the formation of a structurally rigid helix directly following the RGD motif that exhibits slow internal motion. In combination, specificity and affinity toward αvβ6 appear to be dictated by both the dynamic nature and RGD-turn helix length in the peptide ligand; FMDV2 is optimised with respect to both properties. As a cautionary observation, our data also confirms that cyclising peptides can change their dynamic behaviour in a manner that alters their affinity and specificity toward αvβ6. This observation could hold true for many different protein-target systems investigated in vitro or within the field of drug screening and discovery.Finally, from a technological viewpoint, this study has shown that 15N NMR relaxation for peptides can be achieved using the same theoretical and practical solution dynamics as used over the last 20+ years in protein NMR. The combined use of reduced spectral density and ModelFree formalism has created a unique insight into ligand dynamic properties that can explain subtle differences in ligand affinity and specificity for integrin αvβ6. We have also confirmed that trifluroethanol-water systems radically affect the viscosity of solutions that manifest in longer correlation times for peptides.
Experimental
Plasmid preparation and fusion protein expression and purification
The production and purification of recombinant, isotopically enriched peptides was completed as previously described23 and is also reported in detail within the ESI†.NMR sample preparation
Lyophilised peptide was prepared for NMR analysis to a final concentration of 1 mM as determined by sample weight. Peptides were dissolved in NMR buffer of 20 mM Na2HPO4, 100 mM NaCl, pH 6.5 with 10% (v/v) D2O (Goss scientific Ltd). Samples had a final volume of 330 μL and transferred to a Shigemi (BMS-005V) NMR tube for NMR analysis. Peptides analyzed in the presence of TFE were prepared in 20 mM Na2HPO4, 100 mM NaCl, pH 6.5 with 30% (v/v) d3-TFE to a final volume of 330 μL. 30%(v/v) TFE was chosen to complement our previous studies where far-UV circular dichroism confirmed integrin peptides complete the co-operative transition of helix formation below this TFE ratio; typically between 15–25% TFE(v/v).10 The disulphide bonded peptides DBD1 and DBD2 were oxidized in the presence of a 10× molar excess of oxidised glutathione before analysis.NMR data acquisition, processing and analysis
Standard NMR experiments29 were carried out at 10 °C on a Varian UnityINOVA with RT-HCN probe and Bruker AV3 spectrometer with QCI-F cryoprobe operating at 14.1 Tesla (1H resonance frequency of 600 MHz). Specific experiments used for peptide structure elucidation are described in the ESI†. Each 15N T1, T2 and heteronuclear NOE experiment was acquired with the same spectral resolution as HSQC experiments. 10 15N T1 experiments were completed for each peptide and solvent with differing relaxation delays of 64, 128, 256, 384, 512, 640, 768 and 896 ms with 250 and 640 ms delays repeated. 15N T2 data were collected with 11 different experiments with relaxation delays of 40, 60, 80, 100, 120, 140, 160, 180 and 200 ms with 80 and 160 ms delays repeated. Heteronuclear NOE experiments were acquired with a transient cycle of 5.5 s both with and without saturation of amide protons. All NMR data were processed using NMRpipe31 on Linux PCs and all NMR spectra were assigned and relaxation data analysed using the software package CCPN Analysis.32Structure calculation and analysis
NOE distance restraints for each peptide were assigned from 2D and 3D NOESY datasets with a constant bin size of 1.2 to 5.5 Å that is optimal for peptide structures.12 CNS version 1.133 was used to calculate an initial set of 20 structures for each peptide with torsion angle/velocity initialized – simulated annealing from each linear peptide and a total of 10 cycles of energy minimization were completed, each with 200 cooling steps. Iterations of visualization and violation analysis were completed until no van der Waals or NOE violation were seen greater than 0.2 Å in line with previous structural studies of integrin peptides.10 Dihedral angle restraints were applied only to residues with α-helical conformations as determined by direct analysis of structures created from NOE restraints. Following dihedral angle refinement, hydrogen bond donor acceptor pairs were identified from these intermediate structures and used for final structural refinement. All restraints were added to a final calculation of 100 structures, of which the 50 with the lowest reported energy were further water refined using YASARA (AMBER96 force field) and became the final ensemble for each peptide. The precision of each ensemble of 50 structures was assessed using the program PROCHECK-NMR34 to score occupation of favourable, allowed or disallowed regions in the Ramachandran plot. All images of the peptides and ensembles were made using Pymol.35 Limits of the helix for each peptide ensemble were defined by a combination of dihedral angles (confirmed by PROCHECK), NOEs and hydrogen bonds.NMR relaxation data analysis
15N T1 and T2 relaxation data for each nucleus was fitted to a monoexponential decay within CCPN Analysis and heteronuclear NOE data was obtained as either (I − Io)/Io (η) or I/Io (η − 1) for general or ModelFree analysis respectively. Reduced spectral density and theoretical dipolar 15N T1, T2 calculations (for correlation time estimation) were made using the equations published by Kay and co-workers26 and include chemical shift anisotropy contributions where appropriate. The simple Lorentzian model for spectral density as used in Fig. 2, 3 and S9† was taken to be J(ω) = 2/5 [τm/(1 + ω2τm2)]. Linear curve fitting of spectral density data was achieved using KaleidaGraph software. ModelFree 4.030,36–39 was used to provide order parameters, internal motions and exchange broadening analysis. Specific details regarding these analyses are given in the results and discussion section of the text.Determination of peptide integrin αvβ6 specificity by FACS analysis
Cell lines A375P.Beta6.puro and A375P.puro10 were washed with 20 mM Tris pH 7.5, 50 mM NaCl, 20 mM EDTA, 0.1% BSA, 0.1% sodium azide and with 20 mM Tris pH 7.5, 50 mM NaCl, 0.5 mM MgCl2, 1 mM CaCl2, 0.1% BSA, 0.1% sodium azide (MGCA 0.1/0.1), then incubated with biotinylated peptides. Cells were washed with MGCA 0.1/0.1 and bound peptide detected with anti-biotin antibody (Jackson ImmunoResearch) followed by Alexa-488 anti-mouse (Molecular Probes). Cells were analysed by flow cytometry (Beckton Dickinson LSR-1 with CellQuest software) and signal normalised to 10D5 (anti-alphaVbeta6 monoclonal antibody) binding to A375P.Beta6.puro.Acknowledgements
We are gratefully acknowledge Antonio Saha for assistance with peptide flow cytometry experiments. We also thank the BBSRC for funding JLW through a doctoral training account (BB/D526461/1) and The Wellcome Trust for Equipment Grant 091163/Z/10/Z (MJH and RAW).References
- R. O. Hynes, Cell, 2002, 110, 673–687 CrossRef CAS.
- C. A. Morton, I. Campbell and R. M. MacKie, Br. J. Dermatol., 1996, 135, 853–855 CrossRef CAS.
- D. Kohda, C. J. Morton, A. A. Parkar, H. Hatanaka, F. M. Inagaki, I. D. Campbell and A. J. Day, Cell, 1996, 86, 767–775 CrossRef CAS.
- C. J. Morton, D. J. Pugh, E. L. Brown, J. D. Kahmann, D. A. Renzoni and I. D. Campbell, Structure, 1996, 4, 705–714 CrossRef CAS.
- G. J. Thomas, M. P. Lewis, I. R. Hart, J. F. Marshall and P. M. Speight, Int. J. Cancer, 2001, 92, 641–650 CrossRef CAS.
- G. J. Thomas, M. P. Lewis, S. A. Whawell, A. Russell, D. Sheppard, I. R. Hart, P. M. Speight and J. F. Marshall, J. Invest. Dermatol., 2001, 117, 67–73 CrossRef CAS.
- G. J. Thomas, M. L. Nystrom and J. F. Marshall, J. Oral Pathol. Med., 2006, 35, 1–10 CrossRef CAS.
- B. Dalhus, M. Saarinen, U. H. Sauer, P. Eklund, K. Johansson, A. Karlsson, S. Ramaswamy, A. Bjork, B. Synstad, K. Naterstad, R. Sirevag and H. Eklund, J. Mol. Biol., 2002, 318, 707–721 CrossRef CAS.
- S. Kraft, B. Diefenbach, R. Mehta, A. Jonczyk, G. A. Luckenbach and S. L. Goodman, J. Biol. Chem., 1999, 274, 1979–1985 CrossRef CAS.
- D. Dicara, C. Rapisarda, J. L. Sutcliffe, S. M. Violette, P. H. Weinreb, I. R. Hart, M. J. Howard and J. F. Marshall, J. Biol. Chem., 2007, 282, 9657–9665 CrossRef CAS.
- M. Buck, Q. Rev. Biophys., 1998, 31, 297–355 CrossRef CAS.
- J. F. Povey, C. M. Smales, S. J. Hassard and M. J. Howard, J. Struct. Biol., 2007, 157, 329–338 CrossRef CAS.
- J. L. Wagstaff, S. Vallath, J. F. Marshall, R. A. Williamson and M. J. Howard, Chem. Commun., 2010, 46, 7533–7535 RSC.
- D. Logan, R. Abu-Ghazaleh, W. Blakemore, S. Curry, T. Jackson, A. King, S. Lea, R. Lewis, J. Newman and N. Parry, Nature, 1993, 362, 566–568 CrossRef CAS.
- P. Wittung-Stafshede, Acc. Chem. Res., 2002, 35, 201–208 CrossRef CAS.
- S. H. Hausner, D. DiCara, J. Marik, J. F. Marshall and J. L. Sutcliffe, Cancer Res., 2007, 67, 7833–7840 CrossRef CAS.
- M. Campbell and P. Humphries, Methods Mol. Biol., 2011, 763, 355–367 CAS.
- M. M. Humphries, P. F. Kenna, M. Campbell, L. C. Tam, A. T. Nguyen, G. J. Farrar, M. Botto, A. S. Kiang and P. Humphries, Eur. J. Hum. Genet., 2012, 20, 64–68 CAS.
- M. Campbell, M. M. Humphries, A. S. Kiang, A. T. Nguyen, O. L. Gobbo, L. C. Tam, M. Suzuki, F. Hanrahan, E. Ozaki, G. J. Farrar, P. F. Kenna and P. Humphries, EMBO Mol. Med., 2011, 3, 235–245 CrossRef CAS.
- K. E. Caroccia, R. Estephan, L. S. Cohen, B. Arshava, M. Hauser, O. Zerbe, J. M. Becker and F. Naider, Biopolymers, 2011, 96, 757–771 CrossRef CAS.
- D. S. Libich and G. Harauz, Eur. Biophys. J., 2008, 37, 1015–1029 CrossRef CAS.
- J. S. Munger, X. Z. Huang, H. Kawakatsu, M. J. D. Griffiths, S. L. Dalton, J. F. Wu, J. F. Pittet, N. Kaminski, C. Garat, M. A. Matthay, D. B. Rifkin and D. Sheppard, Cell, 1999, 96, 319–328 CrossRef CAS.
- J. L. Wagstaff, M. J. Howard and R. A. Williamson, Mol. BioSyst., 2010, 6, 2380–2385 RSC.
- V. A. Daragan and K. H. Mayo, Prog. Nucl. Magn. Reson. Spectrosc., 1997, 31, 63 CrossRef CAS.
- Y. Takada, X. Ye and S. Simon, GenomeBiology, 2007, 8, 215 CrossRef.
- N. A. Farrow, O. Zhang, A. Szabo, D. A. Torchia and L. E. Kay, J. Biomol. NMR, 1995, 6, 153–162 CrossRef CAS.
- M. Andrec, G. T. Montelione and R. M. Levy, J. Biomol. NMR, 2000, 18, 83–100 CrossRef CAS.
- M. L. Maguire, G. Guler-Gane, D. Nietlispach, A. R. Raine, A. M. Zorn, N. Standart and R. W. Broadhurst, J. Mol. Biol., 2005, 348, 265–279 CrossRef CAS.
- P. Barthe, L. Chiche, N. Declerck, M. A. Delsuc, J. F. Lefevre, T. Malliavin, J. Mispelter, M. H. Stern, J. M. Lhoste and C. Roumestand, J. Biomol. NMR, 1999, 15, 271–288 CrossRef CAS.
- A. M. Mandel, M. Akke and A. G. Palmer, J. Cell. Biochem., 1995, 29–29 Search PubMed.
- F. Delaglio, S. Grzesiek, G. W. Vuister, G. Zhu, J. Pfeifer and A. Bax, J. Biomol. NMR, 1995, 6, 277–293 CrossRef CAS.
- W. F. Vranken, W. Boucher, T. J. Stevens, R. H. Fogh, A. Pajon, M. Llinas, E. L. Ulrich, J. L. Markley, J. Ionides and E. D. Laue, Proteins: Struct., Funct., Bioinf., 2005, 59, 687–696 CrossRef CAS.
- A. T. Brunger, P. D. Adams, G. M. Clore, W. L. DeLano, P. Gros, R. W. Grosse-Kunstleve, J. S. Jiang, J. Kuszewski, M. Nilges, N. S. Pannu, R. J. Read, L. M. Rice, T. Simonson and G. L. Warren, Acta Crystallogr., Sect. D: Biol. Crystallogr., 1998, 54, 905–921 CrossRef CAS.
- R. A. Laskowski, J. A. Rullmannn, M. W. MacArthur, R. Kaptein and J. M. Thornton, J. Biomol. NMR, 1996, 8, 477–486 CrossRef CAS.
- The PyMOL Molecular Graphics System, Version 1.5.0.4 Schrödinger, LLC.
- G. Lipari and A. Szabo, J. Am. Chem. Soc., 1982, 104, 4546–4559 CrossRef CAS.
- G. Lipari and A. Szabo, J. Am. Chem. Soc., 1982, 104, 4559–4570 CrossRef CAS.
- A. M. Mandel, M. Akke and A. G. Palmer, J. Mol. Biol., 1995, 246, 144–163 CrossRef CAS.
- A. G. Palmer, M. Rance and P. E. Wright, J. Am. Chem. Soc., 1991, 113, 4371–4380 CrossRef CAS.
Footnote |
| † Electronic Supplementary Information (ESI) available: See DOI: 10.1039/c2ra21655h |
| This journal is © The Royal Society of Chemistry 2012 |