Enhanced visible photocatalytic activity of hybrid Pt/α-Fe2O3 nanorods†
Long
Chen
a,
Feng
Li
a,
Binbin
Ni
a,
Jiao
Xu
a,
Zhengping
Fu
*a and
Yalin
Lu
*abc
aCAS Key Laboratory of Materials for Energy Conversion, Department of Materials Science and Engineering, University of Science and Technology of China, Hefei, 230026, P. R. China. E-mail: fuzp@ustc.edu.cn; Tel: 86 0551 3607330
bNational Laboratory for Physical Sciences at the Microscale, Hefei, 230026, P. R. China. E-mail: yllu@ustc.edu.cn; Tel: 86 0551 3603004
cLaser Optics Research Center, US Air Force Academy, CO, 80840, USA
First published on 24th August 2012
Abstract
α-Fe2O3 nanorods with highly defective surfaces were used as a support to further synthesize hybrid Pt/α-Fe2O3 nanorods. Transmission electron microscopy analyses suggest that Pt nanoparticles are selectively deposited on the surface defects such as edge-steps. Pt nanoparticles in metallic and electron deficient states, as well as the correlation between chemisorbed oxygen species and the peripheral Pt area, were revealed by the X-ray photoelectron spectroscopy spectra. The hybrid nanorods with an optimized amount of Pt nanoparticles and a large peripheral Pt area exhibit far better visible light photocatalytic activity for degrading methylene blue than α-Fe2O3.
Introduction
Binary hybrid nanocrystals which combine two disparate materials into one (such as semiconductor–semiconductor,1,2 magnet–semiconductor,3 metal–semiconductor,1,4–10 and metal–magnet11,12 systems) have attracted considerable attention in terms of their novel properties arising from their controllable topologies and geometries.13,14 Particularly, the metal–semiconductor hybrid system can become a classic example to optimize the activity of photocatalysts. In such metal–semiconductor nanostructures, semiconductors usually serve as the light harvesting component, absorbing photons and creating excitons: electrons can readily move through the heterointerface, whereas the holes localize in the energetically lower semiconductors due to the Schottky contact between semiconductor and metal nanoparticles with large work function (i.e. high electronegativity).15,16 Thus, the redox reactions based on metal–semiconductor photocatalysis systems can be facilitated by this interfacial charge transferring and separation. α-Fe2O3 has long been regarded as a promising photocatalyst due to its abundance in nature, non-toxicity, good corrosion resistance, low processing cost and substantial visible light absorption.17 However, its performance is greatly limited by the poor conductivity and high electron-hole recombination loss.18 Past efforts have been focusing on architecture controlling and heteroatom doping. For example, one-dimensional nanostructures, particularly nanorods, offer several advantages as compared to spherical nanoparticles, like higher surface-to-volume ratio and the effective photogenerated charge carrier separation through a designed path (i.e. the oriented rods).19,20 Moreover, heteroatom doping, especially combined with noble metal nanoparticles, can enhance the properties of α-Fe2O3. Wei and co-workers have fabricated hybrid Pd/α-Fe2O3 nanoparticulate catalysts, which can act as efficient photocatalysts for metal salts reduction.21 Pradhan and Parida have reported that the substitutions of Fe3+ of α-Fe2O3 nanorods by Pt4+ can improved the performance towards the decolorization of methylene blue (MB).22 Recently, Au-functionalized hematite hybrid nanospindles exhibit much higher activity for applications in both gas sensing and CO oxidation when comparing to the pristine hematite nanospindles, and Pt/α-Fe2O3 hybrid nanospindles are also cursorily mentioned.23 However, both of the references22,23 have not provided more detailed investigations on Pt/α-Fe2O3 hybrids. The associated metal growth mechanism is still unclear and the photocatalytic application exploration on α-Fe2O3 based metal–semiconductor hybrids is still limited, thus further research is undoubtedly needed. Herein, Pt/α-Fe2O3 hybrid nanorods are rationally designed with the Pt nanoparticles in a well-dispersed state on the as-synthesized defective surface of hematite nanorods, which present efficient photocatalytic activity for organic pollutant degradation. The X-ray photoelectron spectra reveals that the Pt nanoparticles are in metallic and electron deficient states, which will enhance the charge separation and improve the photocatalytic property. The degradation efficiency is correlated to the amount of Pt nanoparticles and the peripheral Pt area, and the maximal degradation efficiency is obtained by optimizing both of them.Experimental
Materials preparation
The Pt/α-Fe2O3 hybrids were prepared in two steps. First, α-Fe2O3 nanorods were synthesized following the previously reported method.24 Typically, an aqueous salt solution at pH 1.5 (set by HCl) containing 0.15 M FeCl3 and 1.0 M NaNO3 was kept at 95 °C for 10 h. The obtained yellow product was rinsed with water, dried at 60 °C and subsequently heated in air at 450 °C for 1 h to allow a complete crystal phase transition from β-FeOOH to α-Fe2O3. Second, polyol reduction method was used to deposit Pt onto α-Fe2O3 nanorods.25 0.1 g α-Fe2O3 powder was immersed in a mixed solution containing 2 mL H2PtCl6 aqueous solution (19.3 mM L−1) and 8 mL ethanol, and then the slurry was dried at 60 °C after ultrasonication for 30 min. Ethylene glycol (40 mL) was added to the above mentioned dry powder followed by stirring for 10 min to afford a homogenous suspension. The suspension was kept at various reaction temperatures (Tr) and durations with continuous stirring and protected from light during the whole process. The Pt/α-Fe2O3 hybrids were collected by centrifugation, washed with distilled water and dried at 60 °C, and denoted as PHH1 (Tr = 85 °C, 4 h), PHH2 (Tr = 90 °C, 4 h), PHH3 (Tr = 95 °C, 3 h), PHH4 (Tr = 95 °C, 4 h), PHH5 (Tr = 100 °C, 4 h), respectively.Photocatalytic tests
The photocatalytic activity and the degradation mechanism were investigated by photo-degradation of MB under the irradiation of 20 W fluorescent lamp with a wavelength range of 400–760 nm for a series of time. In the photocatalytic activity tests, 20 mg of each Pt/α-Fe2O3 hybrid catalyst was suspended in 100 mL of 10 mg L−1 MB solution. For comparison, the photocatalysis properties of pure α-Fe2O3 nanorods were also tested under the same conditions. In the investigation of photocatalytic mechanism tests, 3 mL aqueous solution containing 0.5 mg Pt/α-Fe2O3 hybrid and 0.02 mg MB with/without ethanol as sacrificial hole scavenger were employed. All the photocatalytic measurements were carried after reaching adsorption equilibrium in 30 min. The concentration of reduced MB after the irradiation in each sample was estimated using its maximum absorbance at 664 nm from UV–vis absorption spectrum.Characterization
X-Ray powder diffraction (XRD) patterns were recorded on a Rigaku-TTR III X-ray diffractometer with Cu-Kα radiation. The morphologies of the powders were observed by field emission scanning electron microscopy (FESEM, JSM-6700F) and transmission electron microscopy (TEM, JEOL-2010 and JEM-2011). The chemical composition of the samples was analyzed by X-ray photoelectron spectroscopy (XPS) (Thermo Electron ESCALAB-250). All the binding energy data were calibrated by C 1s component at 284.8 eV.Result and discussion
The yellow product of the hydrothermal treatment is β-FeOOH, which is then transformed to red brown α-Fe2O3 after thermal treatment, which are verified by XRD analysis (ESI, Fig. S1†). Fig. 1a and 1b show the FESEM images of β-FeOOH nanorods and α-Fe2O3 nanorods, respectively. The β-FeOOH nanorods and α-Fe2O3 nanorods with 40–50 nm diameter and 1–1.2 μm length (aspect ratio ∼ 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :20) are self-assembled as bundles, most probably driven by the tendency to minimize the interfacial energy among those primary nanorods with the same crystallographic orientation. The XRD (ESI, Fig. S1b†) and SEAD patterns (inset in Fig. 1e) show that the as prepared α-Fe2O3 nanorods possess high crystallinity. However, TEM image in Fig. 1b shows that cavity-like and ditch-like pores have formed on the α-Fe2O3 nanorods surfaces after both thermal and ultrasonic treatment. The pores are possibly formed by removal of the residual Cl− ions held in the tunnels of β-FeOOH crystal lattice26 and H2O in bundle-like nanorods20 during the thermal treatment process. The ultrasonic treatment can also split the bundled nanorods into separate nanorods with the destruction of their joints of the bundled nanorods, which will generate residual pores. Such rough surfaces expose highly active planes and sites, which will serve as the preferred positions for depositing metals,4,6–8 and be the key factor for getting highly dispersed Pt nanoparticles in this study. Fig. 1c shows the overview TEM image of prepared Pt/α-Fe2O3 hybrid (PHH4). It is clear that the bundle-like structures, formed by weak physical interactions between single nanorods, have been split by ultrasonic treatment during the Pt decoration step. Meanwhile, there also exists incomplete separation and damage, which results in the bundle-like and fractal shapes (ESI, Fig. S2†). However, both Fig. 1c and Fig. S2† exhibit homo-dispersed Pt nanoparticles deposited on the exposed surfaces of α-Fe2O3.
:20) are self-assembled as bundles, most probably driven by the tendency to minimize the interfacial energy among those primary nanorods with the same crystallographic orientation. The XRD (ESI, Fig. S1b†) and SEAD patterns (inset in Fig. 1e) show that the as prepared α-Fe2O3 nanorods possess high crystallinity. However, TEM image in Fig. 1b shows that cavity-like and ditch-like pores have formed on the α-Fe2O3 nanorods surfaces after both thermal and ultrasonic treatment. The pores are possibly formed by removal of the residual Cl− ions held in the tunnels of β-FeOOH crystal lattice26 and H2O in bundle-like nanorods20 during the thermal treatment process. The ultrasonic treatment can also split the bundled nanorods into separate nanorods with the destruction of their joints of the bundled nanorods, which will generate residual pores. Such rough surfaces expose highly active planes and sites, which will serve as the preferred positions for depositing metals,4,6–8 and be the key factor for getting highly dispersed Pt nanoparticles in this study. Fig. 1c shows the overview TEM image of prepared Pt/α-Fe2O3 hybrid (PHH4). It is clear that the bundle-like structures, formed by weak physical interactions between single nanorods, have been split by ultrasonic treatment during the Pt decoration step. Meanwhile, there also exists incomplete separation and damage, which results in the bundle-like and fractal shapes (ESI, Fig. S2†). However, both Fig. 1c and Fig. S2† exhibit homo-dispersed Pt nanoparticles deposited on the exposed surfaces of α-Fe2O3.
 | ||
| Fig. 1 FESEM images of β-FeOOH nanorods (a) and α-Fe2O3 nanorods (b), respectively. Inset of (b) show TEM image of the α-Fe2O3 nanorods after ultrasonic treatment; (c) overview TEM image of Pt/α-Fe2O3 hybrid (PHH4); (d) magnified TEM image of Pt/α-Fe2O3 hybrid (PHH4); (e) and (f) High Resolution TEM images of Pt/α-Fe2O3 hybrid (PHH4). Inset in (e) show Selected Area Electron Diffraction of α-Fe2O3. | ||
The average size of Pt nanoparticles on PHH4 has been statistically analysed to be around 3.9 nm. The magnified image of PHH4 (Fig. 1d) shows that the Pt nanoparticles preferred to deposit on two defective positions of the α-Fe2O3 surfaces, i.e. the edges of ditches (marked by dash-line circle) and the inside of ditches (marked by ellipse). The distinct lattice fringe throughout the α-Fe2O3 nanorods terminated by consecutive edge-steps can be clearly observed in Fig. 1e. Thus, it is reasonable that the nanorods with rough surfaces are enriched with edge-steps, and these highly active edge steps will act as the preferred nucleation sites for Pt, which can be inferred from the two Pt particles shown in Fig. 1d and Fig. 1e. The distance between two lattice-fringes in α-Fe2O3 nanorods measured from the Fig. 1e image are 0.275 nm and 0.420 nm, agree well with the (104) and (01–1) plane spacing (0.272 and 0.416 nm, JCPDS 33-0664, respectively) of the hexagonal structured hematite. The high resolution TEM was used to study the connection between Pt and α-Fe2O3 nanorods and the results are shown in Fig. 1f. The 0.23 nm crystal plane spacing in the Pt nanoparticles matches well to the spacing of (111) planes of face centred cubic platinum (0.229 nm, JCPDS 88-2343).
The TEM images of Pt/α-Fe2O3 hybrids prepared at different conditions and the corresponding Pt nanoparticles size distribution histograms are displayed in Fig. 2 ( PHH1 is shown in Fig. S3, ESI†). The primary particle sizes of the deposited Pt nanoparticles in different Pt/α-Fe2O3 hybrids are highly dispersed, although larger secondary particles are observed in PHH5. Evidently, both size and surface amount of Pt nanoparticles increase when the reaction time is prolonged and the temperature raised, and the average diameters of the Pt nanoparticles range from 1.5 to 5.1 nm under variable growth conditions. An interesting phenomenon in Fig. 2b is that some Pt nanoparticles align themselves in rows, as marked in the frame region, and this phenomenon disappeared in PHH4 and PHH5. As shown previously, there exist plenty of edge-steps on the surface of α-Fe2O3 nanorods, which will serve as the preferred sites for Pt deposition. It is rational to assume that the Pt nanoparticles deposited on the same edge-step will align in a row. Pt nanoparticles in PHH4 and PHH5 present a random distribution due to the growth of the nuclei. The absence of freestanding (unattached) Pt nanoparticles, which form thermally from the direct reaction between Pt precursor and ethylene glycol, indicates that α-Fe2O3 is indispensable for the growth of Pt particles. According to the nucleation theory, the overall energy barrier to nucleation will decrease when the nucleation takes place on defective sites, and the density of stable nuclei will increase while the radius of the nuclei will decrease. Subsequently, the nanoparticles growth will be accompanied by the nucleation process. However, Maeda et al. have reported that the Au particles grew large and dominantly covered steps even at room temperature on TiO2 (1 × 1) surfaces, and smaller Au particles were highly dispersed on the terraces of the cross-linked (1 × 2) surfaces.27 These results indicate that the growth rates of metallic particles at different positions of rough surfaces are different. Based on the above statements, the deposition process of Pt nanoparticles on α-Fe2O3 surface is most probably related to the following scenario. At the initial stage, the Pt precursor adsorbed onto the α-Fe2O3 surface in the ultrasonication and drying process, and defective sites which are rich in dangling bonds would serve as electron (supplied by ethylene glycol, a reducing agent) traps for reducing Pt ions from solution. in the following stage, the initially formed Pt cluster will attract more electrons and grow faster. The asynchronous nucleation and different growth rates of Pt will ensure a well-dispersed Pt state with a specific size range. In addition, comparison of the PHHs presented in Fig. 2 shows that the Pt nanoparticles number significantly increase from PHH2 to PHH4 and decreases in PHH5 with increasing particle size. This phenomenon can be attributed to the Ostwald ripening process, which typically leads to a decreasing Pt nanoparticle density in a long time reaction.
 | ||
| Fig. 2 TEM images of Pt/α-Fe2O3 hybrids. (a) PHH2 (90 °C, 4 h); (b) PHH3 (95 °C, 3 h); (c) PHH4 (95 °C, 4 h); (d) PHH5 (100 °C, 4 h). Insets show Pt nanoparticle size distribution histogram of the corresponding products. The surface Pt atomic ratios (at%) are 0.48% for PHH2, 0.63% for PHH2, 1.79% for PHH4, and 1.95% for PHH5. PHH1 is shown in Fig. S3.† | ||
XPS spectra have been used to provide insights into the electronic properties of the Pt/α-Fe2O3 hybrids. The Fe 2p spectra (ESI, Fig. S4†) reveal that iron exists predominately as Fe3+, with a 2p3/2 peak position of 710.9 ± 0.3 eV and shake-up satellite peak at 718 eV.28 The O 1s XPS spectra (Fig. 3) can be fitted with a main peak located at 529.9 ± 0.2 eV, which is assigned to the lattice oxygen of α-Fe2O3, and a shoulder peak at 531.5 ± 0.3 eV, which can be attributed to the chemisorbed oxygen species on the α-Fe2O3 and the PHHs surface.29 Due to the complexity of the oxygen containing species, we do not ascribe the shoulder peak to distinct species (e.g., O−, surface OH groups etc). The chemisorbed oxygen species on the α-Fe2O3 surface may be due to the destruction of surface Fe–O–Fe by ultrasonic treatment and the following formation of coordinatively unsaturated Fe–O– and Fe– as binding sites for adsorbing chemisorbed oxygen species in polyol reduction process.30 However, Rso, defined as the area ratio of shoulder peak to the main peak of O 1s, increases from 0.23 in α-Fe2O3 to 1.21 in PHH4 and drops to 0.42 in PHH5. This means that a larger amount of chemisorbed oxygen species have formed on the surface of the Pt/α-Fe2O3 hybrids than on the α-Fe2O3 bundle-like nanorods. Moreover, the abnormal change of Rso in PHH5 reveals the amount of surface chemisorbed oxygen species are not exclusively thermal-dependent but have relation to surface Pt as well.
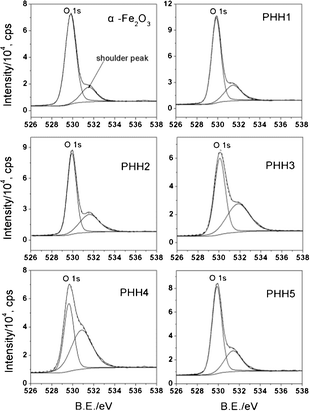 | ||
| Fig. 3 O 1s region XPS spectra registered for α-Fe2O3 and Pt/α-Fe2O3 hybrids. All the spectra can be fitted with a main peak (lattice oxygen of α-Fe2O3) and a shoulder peak (chemisorbed oxygen species). The peak area ratios of shoulder peak to O 1s (Rso) are 0.23 for α-Fe2O3, 0.32 for PHH1, 0.48 for PHH2, 0.77 for PHH3, 1.21 for PHH4, and 0.42 for PHH5. | ||
To elucidate the chemisorbed oxygen species, similar polyol reduction experiments were employed on α-Fe2O3 powder. Briefly, 0.1 g as-prepared α-Fe2O3 powder was processed by the same experimental procedures as PHH2, PHH4 and PHH5 without adding H2PtCl6. The details of the treatment are provided in the ESI.† As revealed in Fig. S5,† the surface chemisorbed oxygen species amount remain unchanged (Rso ∼0.47) even when the reaction temperature is raised to 100 °C, which is convincing evidence that the surface adsorption process of chemisorbed oxygen species (adsorbed at unsaturated Fe–O– and Fe– binding sites) reaches equilibrium and saturation at 90 °C for 4 h. However, the Rso of PHH3 and PHH4 show higher values than that of PHH2 when Pt nanoparticles are deposited on α-Fe2O3 surface. These results strongly suggest that Pt nanoparticles can increase the surface chemisorbed oxygen species amount. Additionally, the reduced surface chemisorbed oxygen species amount in PHH5 implies that the surface chemisorbed oxygen species may be correlated to the surface Pt amount as well as the particular adsorption sites of Pt nanoparticles. The aforementioned TEM images (Fig. 2) exhibit an obvious increasing of surface Pt amounts. However, the Pt nanoparticles number increase from PHH2 to PHH4 and begin to decrease because of the aggregation of initial Pt nanoparticles (the Ostwald ripening process) in PHH5. To realize whether there is a connection between surface chemisorbed oxygen species amount and Pt nanoparticles number, we defined and quantitatively analyzed the specific periphery density (SPD)31 (see ESI†). Here, it should be noted that the concept of periphery area consists of both Pt and hematite surface near the junction of Pt and α-Fe2O3. The relevant data are summarized in Table 1. Based on these results, we believe that the oxygen species are chemisorbed at both α-Fe2O3 surface (unsaturated Fe–O– and Fe– as binding sites) and periphery areas of Pt particles. Remarkably, the variation tendency of Rso is well consistent with the change of obtained SPD (Table 1), we assume that the oxygen species are predominantly chemisorbed around the periphery areas of Pt particles, same as the case of Au/MOx system.32
| α-Fe2O3 | PHH1 | PHH2 | PHH3 | PHH4 | PHH5 | |
|---|---|---|---|---|---|---|
| a The SPD of PHH1 is omitted for its low surface Pt amount. | ||||||
| R so | 0.23 | 0.32 | 0.48 | 0.77 | 1.21 | 0.42 |
| SPD | — | — | 0.051 | 0.117 | 0.187 | 0.12 |
| Pt 4f7/2 B.E. (eV) | — | 72.22 | 71.82 | 71.65 | 71.38 | 71.2 |
| Surface Pt at% | — | 0.14 | 0.48 | 0.63 | 1.79 | 1.95 |
| Average Pt size/nm | — | <1 | 1.5 | 2.8 | 3.9 | 5.1 |
XPS spectra in Pt 4f core-level region for Pt/α-Fe2O3 hybrids are shown in Fig. 4, and the binding energies (B.E.) of Pt 4f7/2 are also listed in Table 1. The B.E. of the Pt 4f7/2 core-level displayed a tendency of negative shift from PHH1 to PHH5, indicating an elevated electron density in the platinum nanoparticles. Moreover, the B.E. of Pt 4f7/2 decreases with the increase of Pt atomic ratio, and reaches 71.2 eV for PHH5 (the value of the metallic Pt33,34). Previous research has reported that the chemical shifts of the Pt 4f7/2 levels achieved ∼1.2 eV per one oxidation state unit as compared to the metallic platinum.35 Since all the Pt 4f7/2 B.E. values deviate greatly from the binding energies of Pt2+ (∼73.6 eV) or Pt4+ (∼76 eV) oxidation state, these should not be chemical shifts but arise from the electron-deficient Pt state (Ptδ+, 0 < δ < 2). As described above, the O 1s XPS spectra suggest that chemisorbed oxygen species formed during the polyol reduction process is most probably coming from the surface unsaturated bonds. We have also shown that Pt will preferentially deposit at defective sites on the hematite surface, and the defective sites such as step edges are characterized with less coordinative saturated bonds. Therefore, the Pt4+ precursor, firstly adsorbed on the α-Fe2O3 surface, will probably be incompletely reduced by ethylene glycol in the initial reduction process with the formation of Fe–Oδ−–Ptδ+. On the other hand, the so-called strong metal-support interaction (SMSI) between the metal and reduced (suboxide) support leads to the electron-rich metal, which is reversed in oxidative environments.36 Therefore, the surface oxidative Fe3+ (revealed in Fig. S4†) can also have an effect on the electronic structure of Pt. Thus, nascent small Pt nanoparticles will be in the electronic-deficient state (Ptδ+, 0 < δ < 2) and cause an obvious positive B.E. shift. The Pt 4f7/2 B.E. from PHH1 to PHH5 decreases with increasing surface Pt amount, which can be ascribed to the increasing concentration of valence electron concentration. The decrease of Pt 4f B.E. should be the result of a transformation from electron-deficient Pt (Ptδ+, 0 < δ < 2) to zero-valent metallic Pt (Pt0). The monotonic shifts can be interpreted by size effect and electron transfer process from α-Fe2O3 to Pt, which is analogous of Jiang's work of Au 4f B.E. in Au/TiO2.37 The small Pt nanoparticles in PHH1 are mostly in an electron-deficient state, and the electronic structure of nascent deposited Pt nanoparticles will be strongly affected by the deposition sites, thereby the Pt 4f B.E. shift is more obvious than other hybrids. As the deposition process continued, the electron-deficient state of larger Pt nanoparticles will be alleviated due to the decreasing unsaturated surface bonds with the enlarged surface Pt nanoparticles size, and the charge transfer from α-Fe2O3 to Pt in the photoemission process will increase the concentration of valence electron concentration in Pt nanoparticles. As a consequence, the change of the surface Pt atomic ratio results in a comprehensive effect, i.e., the step-down shifts of Pt 4f B.E. from PHH1 to PHH5. Clearly, the electronic-deficient state will always exist in PHHs because nascent incomplete reduced Pt always exists; it can be overshadowed by the metallic state of Pt and charge transfer process when Pt nanoparticles grow larger.
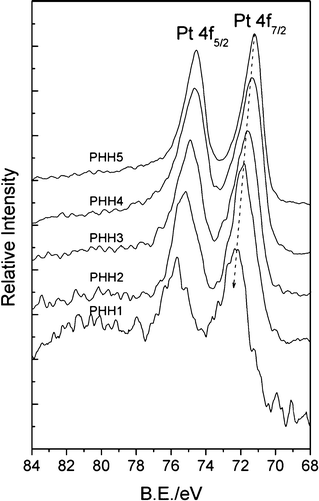 | ||
| Fig. 4 XPS spectra of Pt 4f region registered for Pt/α-Fe2O3 hybrids. The peak positions of Pt 4f7/2 are 72.22 eV for PHH1, 71.82 eV for PHH2, 71.65 eV for PHH3, 71.38 eV for PHH4, and 71.2 eV for PHH5. | ||
Size effects are very important both for catalytic activity38 and for electronic properties. Here, controlled photocatalytic measurements were employed to study the effects of PHHs while MB degradation was used as a test reaction.
Fig. 5a shows a normalized time courses for MB degradation (C/C0, where C0 is the MB concentration after adsorption equilibrium). The degradation curves can be described as a quasi-zero-order limiting step rate expression of the Langmuir–Hinshelwood kinetic model.33 Good crystallinity usually leads to significant enhancement of catalytic properties, whereas lattice defects behaved as recombination centers for the photogenerated electron/hole pairs.16 However, the photocatalytic activity of α-Fe2O3 nanorods is negligible, probably because of the short diffusion length of the minority charge carriers (2–4 nm) and the high electron-hole recombination loss.18,39 The PHH1 exhibited similar photocatalysis performance to that of the α-Fe2O3 nanorods, this result is mainly ascribed to the low Pt amount (at% = 0.14%). The photocatalytic abilities of other Pt/α-Fe2O3 hybrids are remarkably enhanced compared to α-Fe2O3 nanorods, which probably arise from the increase in the charge-separation efficiency due to the electron transfer from α-Fe2O3 nanorods to Pt. The degradability of PHH4 achieves a maximum 86% after irradiation for 150 min, far exceeding that of the single component (α-Fe2O3 bundle-like nanorods), strongly indicate that higher surface Pt atomic ratio can facilitate photocatalytic MB degradation process. However, PHH5 shows a lower catalytic activity compared to PHH4, though PHH5 has a higher surface Pt atomic ratio than PHH4.
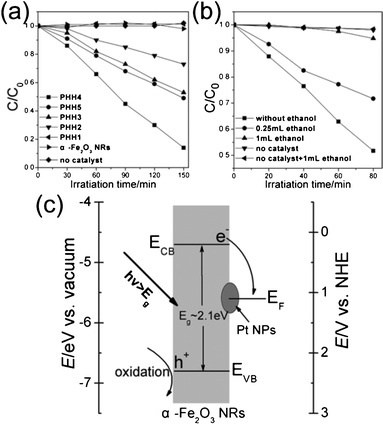 | ||
| Fig. 5 Time courses for photocatalytic degradation of MB using different samples listed in Table 1 (a) and photoexperiments for investigation of degradation mechanism (b); (c) simplified energy band diagram scheme of the Pt/α-Fe2O3 hybrid versus vacuum and NHE. ECB and EVB represent the conduction and valence band edges of α-Fe2O3 nanorods, respectively, and EF is the Fermi level of Pt. | ||
To clarify this phenomenon, we took ethanol as the sacrificial hole scavenger, and the MB degradability of the PHH4 were performed with/without ethanol under 20 W fluorescent lamp irradiation for a definite time. Fig. 5b shows that PHH4 exhibits good degradation performance over the control experiment with 0.25 mL ethanol added. Furthermore, PHH4 degraded only 5% MB if 1 mL ethanol was added, approaching that of the blank control (2%). These results effectively suggest that photoinduced holes rather than electrons play a vital role in MB degradation process, which is opposite to the MB degradation mechanism in Pt/CdSe and Au/CdSe hybrid systems, though the conduction and valence band edge position of CdSe is similar with α-Fe2O3.8,9Fig. 5c shows the simplified energy band diagram scheme of the PHH versus the vacuum level and the normal hydrogen electrode (NHE) level in aqueous solutions, respectively. Both the electron-deficient and metallic Pt (work function = 5.6 eV) can promote the photogenerated electrons to transfer from α-Fe2O3 nanorods and then be trapped by dissolved molecular oxygen,40 while the holes left on the α-Fe2O3 nanorods surface will trapped by hydroxyl ions, which facilitate the separation of hole/electron excitation pairs and inhibit the recombination of photogenerated carriers. Moreover, the UV–vis absorbance spectra of α-Fe2O3 bundle-like nanorods and Pt/α-Fe2O3 hybrids (Fig. S6†) show that the absorption peak of Pt/α-Fe2O3 hybrids remain almost unchanged compared to that of α-Fe2O3 bundle-like nanorods and the light absorption efficiencies are rarely improved. Therefore, in a certain scope, the photocatalytic activity increases with increasing surface Pt amount because of the increasing efficiency of the separation of the hole/electron excitation pairs, and more separated photoexcited holes will take part in MB degradation process. Additionally, as discussed before, the surface chemisorbed oxygen species at the specific periphery areas of Pt nanoparticles increase from PHH1 to PHH4 and decrease in PHH5, which is analogous to the degradation activity tendency. Bao and co-workers have reported that the interface-confined coordinatively unsaturated ferrous sites, mainly at the periphery of the two-dimensional (2D) FeO nanoislands, together with the metal supports, are active for dioxygen.31 Moreover, a general ‘periphery’ mechanism has been proposed to describe the oxidation process of carbon monoxide by Au/support catalysts.32 Therefore, the specific periphery areas of Pt nanoparticles in Pt/α-Fe2O3 are also considered as active centers for MB degradation. The chemisorbed oxygen species, particularly surface OH groups, will participate in the MB photo-degradation process. When the specific periphery areas of Pt nanoparticles decrease, the photocatalytic activity falls. On the other hand, the primary role of photogenerated holes left on the surface of α-Fe2O3 nanorods is to oxidize the hydroxyl ion or serve as the direct oxide for MB degradation. Nevertheless, previous reports show that photogenerated holes can possibly migrate to Pt, indicating that some Pt particles kept trapping the holes and possibly acted as recombination sites.41,42 Therefore, excess Pt nanoparticles on the α-Fe2O3 will act as recombination centers for hole/electron excitation pairs, thus decrease the separation efficiency of the photogenerated carriers. Consequently, an optimum surface amount of Pt is important to the photocatalytic activity.
Conclusion
In summary, Pt/α-Fe2O3 hybrids have been prepared by two facile steps. The Pt precursor is preferentially reduced on defective surface sites such as edge-steps, and then the nascent deposited Pt will grow larger. The XPS spectra analysis on α-Fe2O3 and Pt/α-Fe2O3 hybrids revealed chemical state information: iron exists predominately as Fe3+ and a larger amount of chemisorbed oxygen species have formed on the surface of the Pt/α-Fe2O3 hybrids after ultrasonic treatment and the polyol reduction process. The specific periphery areas of Pt nanoparticles can increase the amount the surface chemisorbed oxygen species. Meanwhile, the unsaturated surface bonds lead to an incomplete reduced Pt precursor and the so-called strong metal-support interaction (SMSI), and finally result in a positive Pt 4f B.E. shift. The general step-down tendency of Pt 4f B.E. shifts in Pt/α-Fe2O3 hybrids result from an increasing valence electron concentration of Pt nanoparticles, which can be ascribed to the size effect and the charge transfer process. The combined analysis of XPS, the simplified energy band diagram scheme and UV–vis absorbance spectra of the Pt/α-Fe2O3 hybrids reveal that the increasing efficiency of the separation of the hole/electron excitation pairs can improve the photocatalytic activity. Moreover, the photocatalytic activity can be optimized by varying the surface amount Pt to obtain a large peripheral Pt area, which act as active centers for absorbing oxygen species.Acknowledgements
This work was supported by the National Basic Research Program of China (2012CB922000) and the Fundamental Research Funds for the central Universities. Prof. Lu also thanks the support from the US Air Force Office of Scientific of Research (AFOSR).References
- D. V. Talapin, H. Yu, E. V. Shevchenko, A. Lobo and C. B. Murray, J. Phys. Chem. C, 2007, 111, 14049–14054 CAS.
- H. Tada, Q. Jin, H. Nishijima, H. Yamamoto, M. Fujishima, S. Okuoka, T. Hattori, Y. Sumida and H. Kobayashi, Angew. Chem., Int. Ed., 2011, 50, 3501–3505 CrossRef CAS.
- K. W. Kwon and M. Shim, J. Am. Chem. Soc., 2005, 127, 10269–10275 CrossRef CAS.
- T. Mokari, E. Rothenberg, I. Popov, R. Costi and U. Banin, Science, 2004, 304, 1787–1790 CrossRef CAS.
- C. Pacholski, A. Kornowski and H. Weller, Angew. Chem., Int. Ed., 2004, 43, 4774–4777 CrossRef CAS.
- A. E. Saunders, I. Popov and U. Banin, J. Phys. Chem. B, 2006, 110, 25421–25429 CrossRef CAS.
- G. Dukovic, M. G. Merkle, J. H. Nelson, S. M. Hughes and A. P. Alivisatos, Adv. Mater., 2008, 20, 4306–4311 CrossRef CAS.
- E. Elmalem, A. E. Saunders, R. Costi, A. Salant and U. Banin, Adv. Mater., 2008, 20, 4312–4317 CrossRef CAS.
- R. Costi, A. E. Saunders, E. Elmalem, A. Salant and U. Banin, Nano Lett., 2008, 8, 637–641 CrossRef CAS.
- V. Subramanian, E. Wolf and P. V. Kamat, J. Phys. Chem. B, 2001, 105, 11439–11446 CrossRef CAS.
- H. Yu, M. Chen, P. M. Rice, S. X. Wang, R. L. White and S. H. Sun, Nano Lett., 2005, 5, 379–382 CrossRef CAS.
- T. Pellegrino, A. Fiore, E. Carlino, C. Giannini, P. D. Cozzoli, G. Ciccarella, M. Respaud, L. Palmirotta, R. Cingolani and L. Manna, J. Am. Chem. Soc., 2006, 128, 6690–6698 CrossRef CAS.
- W. L. Shi, H. Zeng, Y. Sahoo, T. Y. Ohulchanskyy, Y. Ding, Z. L. Wang, M. Swihart and P. N. Prasad, Nano Lett., 2006, 6, 875–881 CrossRef CAS.
- A. Vaneski, A. S. Susha, J. Rodriguez-Fernandez, M. Berr, F. Jackel, J. Feldmann and A. L. Rogach, Adv. Funct. Mater., 2011, 21, 1547–1556 CrossRef CAS.
- A. Wood, M. Giersig and P. Mulvaney, J. Phys. Chem. B, 2001, 105, 8810–8815 CrossRef CAS.
- X. B. Chen, S. H. Shen, L. J. Guo and S. S. Mao, Chem. Rev., 2010, 110, 6503–6570 CrossRef CAS.
- C. M. Eggleston, Science, 2008, 320, 184–185 CrossRef CAS.
- K. Sivula, F. Le Formal and M. Gratzel, ChemSusChem, 2011, 4, 432–449 CrossRef CAS.
- L. Vayssieres, C. Sathe, S. M. Butorin, D. K. Shuh, J. Nordgren and J. H. Guo, Adv. Mater., 2005, 17, 2320–2323 CrossRef CAS.
- H. G. Cha, S. J. Kim, K. J. Lee, M. H. Jung and Y. S. Kang, J. Phys. Chem. C, 2011, 115, 19129–19135 CAS.
- Y. H. Wei, S. B. Han, D. A. Walker, S. C. Warren and B. A. Grzybowski, Chem. Sci., 2012, 3, 1090–1094 RSC.
- G. K. Pradhan and K. M. Parida, ACS Appl. Mater. Interfaces, 2011, 3, 317–323 CAS.
- J. Zhang, X. H. Liu, L. W. Wang, T. L. Yang, X. Z. Guo, S. H. Wu, S. R. Wang and S. M. Zhang, J. Phys. Chem. C, 2011, 115, 5352–5357 CAS.
- L. Vayssieres, N. Beermann, S. E. Lindquist and A. Hagfeldt, Chem. Mater., 2001, 13, 233–235 CrossRef CAS.
- Z. Guo, Y. T. Chen, L. S. Li, X. M. Wang, G. L. Haller and Y. H. Yang, J. Catal., 2010, 276, 314–326 CrossRef CAS.
- C. Z. Wu, P. Yin, X. Zhu, C. Z. Ouyang and Y. Xie, J. Phys. Chem. B, 2006, 110, 17806–17812 CrossRef CAS.
- Y. Maeda, T. Fujitani, S. Tsubota and M. Haruta, Surf. Sci., 2004, 562, 1–6 CrossRef CAS.
- T. Fujii, F. M. F. de Groot, G. A. Sawatzky, F. C. Voogt, T. Hibma and K. Okada, Phys. Rev. B: Condens. Matter, 1999, 59, 3195–3202 CrossRef CAS.
- T. Kawabe, K. Tabata, E. Suzuki, Y. Yamaguchi and Y. Nagasawa, J. Phys. Chem. B, 2001, 105, 4239–4244 CrossRef CAS.
- J. G. Yu, X. X. Yu, B. B. Huang, X. Y. Zhang and Y. Dai, Cryst. Growth Des., 2009, 9, 1474–1480 CAS.
- Q. Fu, W. X. Li, Y. X. Yao, H. Y. Liu, H. Y. Su, D. Ma, X. K. Gu, L. M. Chen, Z. Wang, H. Zhang, B. Wang and X. H. Bao, Science, 2010, 328, 1141–1144 CrossRef CAS.
- G. C. Bond and D. T. Thompson, Gold Bull., 2000, 33, 41–51 CrossRef CAS.
- V. Iliev, D. Tomova, L. Bilyarska, A. Eliyas and L. Petrov, Appl. Catal., B, 2006, 63, 266–271 CrossRef CAS.
- F. Sen and G. Gokagac, J. Phys. Chem. C, 2007, 111, 5715–5720 CAS.
- A. Karpov, M. Konuma and M. Jansen, Chem. Commun., 2006, 838–840 RSC.
- D. W. Goodman, Catal. Lett., 2005, 99, 1–4 CrossRef CAS.
- Z. Q. Jiang, W. H. Zhang, L. Jin, X. Yang, F. Q. Xu, J. F. Zhu and W. X. Huang, J. Phys. Chem. C, 2007, 111, 12434–12439 CAS.
- M. Arenz, K. J. J. Mayrhofer, V. Stamenkovic, B. B. Blizanac, T. Tomoyuki, P. N. Ross and N. M. Markovic, J. Am. Chem. Soc., 2005, 127, 6819–6829 CrossRef CAS.
- J. H. Kennedy and K. W. Frese, J. Electrochem. Soc., 1978, 125, 709–714 CrossRef CAS.
- K. Maeda, M. Higashi, D. L. Lu, R. Abe and K. Domen, J. Am. Chem. Soc., 2010, 132, 5858–5868 CrossRef CAS.
- N. Z. Bao, L. M. Shen, T. Takata and K. Domen, Chem. Mater., 2008, 20, 110–117 CrossRef CAS.
- M. Yoshida, A. Yamakata, K. Takanabe, J. Kubota, M. Osawa and K. Domen, J. Am. Chem. Soc., 2009, 131, 13218–13219 CrossRef CAS.
Footnote |
| † Electronic Supplementary Information (ESI) available. See DOI: 10.1039/c2ra21897f |
| This journal is © The Royal Society of Chemistry 2012 |