PEO surface-decorated silica nanocapsules and their application in in vivo imaging of zebrafish†
Benedict You Wei
Hsu
a,
Cathleen
Teh
b,
Happy
Tan
a,
Siew Yee
Wong
c,
Yu
Zhang
d,
Vladimir
Korzh
b,
Xu
Li
*c and
John
Wang
*ad
aNUS Graduate School for Integrative Sciences and Engineering (NGS), National University of Singapore, Centre for Life Sciences (CeLS) #05-01, 28 Medical Drive, 117456, Singapore. E-mail: msewangj@nus.edu.sg
bLaboratory of Fish Developmental Biology, Institute of Molecular and Cell Biology (IMCB), Agency for Science, Technology and Research (A*STAR), 61 Biopolis Drive, 138673, Singapore
cInstitute of Materials Research and Engineering (IMRE), Agency for Science, Technology and Research (A*STAR), 3 Research Link, 117602, Singapore. E-mail: x-li@imre.a-star.edu.sg
dDepartment of Materials Science & Engineering, National University of Singapore, Blk EA, #03-09, 9 Engineering Drive 1, 117576, Singapore
First published on 12th October 2012
Abstract
To develop a class of robust and colloidally stable silica nanocapsules for bioimaging, we have investigated the interfacial hydrolysis and condensation of silicon alkoxides confined between the PPO core and PEO corona of self-assembled Pluronic® polymeric micelles. The interfacial hydrolysis and condensation, which are completed under the benign conditions of near-neutral pH and at room temperature, give rise to the decoration of silica nanocapsules with a layer of PEO chains dangling on their surface. This feature enables them to be intrinsically stable in aqueous and physiological environments. To demonstrate the silica nanocapsules as effective nanocarriers in bioimaging as well as their biocompatibility, fluorescent conjugated polymer MEH-PPV is encapsulated into the silica nanocapsules and used for in vivo studies with zebrafish as the vertebrate model. When microinjected into the transgenic zebrafish line TG(fli1:EGFP), the cellular internalization of these fluorescent silica nanocapsules does not appear to interfere with larval development nor affect vessel growth. Their biostability is verified by the continued presence of fluorescent-labeled cells in the zebrafish larvae over a long period after the microinjection of fluorescent silica nanocapsules.
Introduction
For the past decade, surface-decorated silica nanocapsules have emerged as one of the most promising candidates for nanocarriers in biomedical fields for diagnosis, controlled delivery and cellular imaging, as shown by their capabilities of encapsulating various therapeutic payloads, functional inorganic nanoparticles and imaging tags.1–17 They are structurally more stable than conventional polymer-based nanocarriers18 while their excellent biocompatibility is affirmed by the classification of silica as “generally recognized as safe” (GRAS) by the US Food and Drug Administration (FDA).19 Indeed, surface-decorated silica nanocapsules are commonly used for specific cellular targeting, where both in vitro as well as in vivo experiments have been conducted.4–10,15,16In order to prepare surface-decorated silica nanocapsules for biomedical applications, several different types of template-assisted approaches have been adopted. These templates can be broadly classified into two major categories: hard and soft templates. Hard (solid) templates include iron oxide nanocrystals,1–6 monodispersed silica7–9 or even gold nanoparticles10,11 while soft templates comprise primarily of microemulsion droplets13–16 and polymeric micelles.20–23 Despite the considerable differences in the detailed synthesis procedures, these template-assisted approaches usually consist of four major processing steps: (i) preparation of an appropriate template; (ii) formation of an outer silica shell on the template surface through controlled chemical precipitation of silica precursors in the solution; (iii) template removal to generate the desired hollow structure and (iv) surface decoration by introducing appropriate functional groups through chemical reaction.
With regard to the hard templating technique, removal of the template typically involves calcination at high temperatures2,6 or immersion in a strongly acidic environment,1,4,10 both of which are potentially detrimental to biologically active compounds.24 Consequently, this technique does not offer the option of an in situ encapsulation of these functional molecules during the synthesis of silica nanocapsules. To overcome such a drawback, several researchers have employed microemulsion droplets, which can contain functional molecules, as the template for silica shell formation.13–16 However, due to droplet coalescence during the synthesis process,24 the resultant silica nanocapsules tend to exhibit a large overall size with diameters > 200 nm.13 This reduces their blood circulation time because of an increased susceptibility to rapid splenic uptake.25 Moreover, cetyltrimethylammonium bromide (CTAB), which is commonly used to stabilize the oil/water interface in the microemulsion system,15 is highly toxic and incomplete removal of this surfactant can impede the use of silica nanocapsules for biomedical applications.
Polymeric micelles can function simultaneously as both an encapsulant and templating agent too.20–23 Polymeric micelle-templated silica nanocapsules typically range from 50–100 nm, which is an ideal dimension for prolonged blood circulation and an enhanced level of drug delivery.25 For example, non-ionic PEO-based block copolymers have been used as templating agents due to their affinity with silica.21,22 However, a highly acidic environment is typically employed to protonate the PEO segments so that silica precursors can readily adsorb onto them through electrostatic interactions. Such harsh synthesis conditions would preclude the in situ encapsulation of many pH-sensitive compounds (e.g. iron oxide nanocrystals and DNA molecules). This limits the versatility of silica nanocapsules as nanocarriers for biomedical applications. Therefore, particular emphasis is placed on the development of synthesis methodologies to fabricate silica nanocapsules under benign conditions. Yuan and coworkers, for example, demonstrated one of such methods by synthesizing cationic block copolymers that were able not only to confine silica shell formation, but also catalyze the hydrolysis and condensation of silica precursors at neutral pH and room temperature.26 However, the procedure involves several organic synthesis steps and therefore careful control is needed in order to develop the most desirable hollow nanostructure.
We have developed an alternative but facile route of synthesizing silica nanocapsules via an interfacial condensation scheme, wherein silica condensation is confined to the interface between the core and corona of polymeric micelles while the hydrolysis and condensation reactions are completed under benign conditions of near-neutral pH and at room temperature.27 Unlike the conventional approach of adding the silica precursor after the polymeric micelles have been formed,21–23 the novelty of our method stems from the fact that the silica precursor (e.g. TMOS) and block copolymer are introduced simultaneously into the aqueous solution. Therefore, during the micellization process of amphiphilic block copolymers, the silica precursor is first sequestrated into the core of the polymeric micelles. Pluronic® block copolymers, which consist of hydrophilic polyethylene oxide (PEO) and hydrophobic polypropylene oxide (PPO) segments arranged in a triblock PEO–PPO–PEO structure, are selected as templating agents to direct the formation of a robust silica shell. As a result, as-synthesized silica nanocapsules are surface-decorated with a layer of PEO chains, which enable them to be intrinsically stable in aqueous environments, exhibit good antifouling behaviour, have a prolonged blood circulation half-life and provide further functionality to which receptor-specific ligands can be attached to. This negates the need for further PEGylation, which is an otherwise conventional technique to improve the pharmacokinetic properties of silica nanocapsules.28 Pluronic® also inhibits P-glycoprotein efflux pumps to suppress the drug resistance of multidrug resistant (MDR) cancer cells and enhances the transgene expression when used for gene therapy applications.29,30 Given the desirable properties of Pluronic® for applications in various biomedical fields, the block copolymer template is opted to be retained after silica shell formation.
In this present study, the sizes and morphologies of PEO surface-decorated silica nanocapsules (PEOSN) are effectively tuned by varying several key synthesis conditions such as polymer block length, solvent type and reactant concentrations. This enables us to elicit a synergistic combination of merits derived from both hollow silica nanocapsules and Pluronic® micelles. Fluorescent PEOSN are then prepared by sequestering the fluorescent conjugated polymer MEH-PPV into the core of Pluronic® micelles, followed by subsequent interfacial silica condensation. Moreover, their application as effective fluorescent probes for bioimaging is conclusively demonstrated by using the transgenic zebrafish line TG(fli1:EGFP), wherein the zebrafish Fli1 promoter is used to drive the expression of enhanced green fluorescent protein (EGFP) in the vessels and highlight all vascularized tissues with green fluorescence.31,32 This provides a strong contrast against the red fluorescent PEOSN in the confocal images. Besides, the zebrafish development is relatively fast and stereotypical such that most organs are vascularized and functional by 5 days post fertilization.32 The manipulated larvae can also be mounted alive in low melting agarose and are small enough for non-invasive, whole animal imaging. Therefore, TG(fli1:EGFP) is an excellent vertebrate model to ascertain the biocompatibility and in vivo behavior of the fluorescent PEOSN.
Experimental section
Materials
Block copolymer Pluronic® L121 (PEO5PPO70PEO5), P123 (PEO20PPO70PEO20), F127 (PEO106PPO70PEO106), poly(2-methoxy-5-(2-ethyl-hexyloxy)-1,4-phenylenevinylene) (MEH-PPV), tetramethylorthosilicate (TMOS; 98%), tetrahydrofuran (THF) and dimethylformamide (DMF) were purchased from Sigma Aldrich and used without further purification.Synthesis of PEOSN
PEOSN was prepared using a procedure reported previously.27 In a typical synthesis, a polymer solution of the desired concentration was prepared by dissolving the respective polymers (Pluronic® L121, P123 or F127) into 900 μL of THF. To each of these polymer solutions, 65 μL of TMOS was then added. The mixture solution was injected into 10 g of deionized water immersed in a water-bath under ultra sonication in 3 min. The solution was sonicated for 10 more minutes, followed by stirring at room temperature for four days to evaporate off THF and ensure a complete hydrolysis and condensation of TMOS at the interface between the core and corona of the copolymer micelles. The amount of block copolymer and TMOS used were subsequently varied systematically to study their influence on the formation of PEOSN. Mixtures of THF and DMF in various ratios are also used to investigate their effect on the core-shell morphology. (Total volume of solvents used before injecting into water is kept at 900 μL for all experiments)Synthesis of fluorescent PEOSN
Fluorescent conjugated polymer MEH-PPV was first dissolved in THF to form a stock solution of 1.0 mg mL−1. 75 μL of this stock solution was then mixed with 75 mg of F127 dissolved in 825 μL of THF. Next, 65 μL of TMOS was added and the resultant mixture solution was subsequently injected into 10 g of deionized water while being sonicated in a water bath for 3 min. The solution was sonicated for another 10 min, followed by stirring at room temperature for four days.Characterizations
Transmission electron microscopy (TEM, JEM-2010F, 200 kV) was employed to study the morphology of the synthesized PEOSN. To prepare the sample for TEM analysis, 40 μL of an aqueous suspension of PEOSN was diluted with 200 μL of deionized water. A small drop of this diluted solution was pipetted directly onto a 200-mesh carbon-coated copper grid. After 2 min, the excess solution was removed by touching the grid edge using a Kimwipe delicate wipe. The sample was finally dried at room temperature in preparation for TEM analysis. Dynamic light scattering (DLS) was performed with Malvern Zetasizer Nano-S using a HeNe laser (633 nm) to measure the nanocapsule size and size distribution. The spectrometer was calibrated by using a standard polystyrene suspension of 60 nm. All measurements were conducted using water as the dispersant in a glass cuvette. 29Si NMR characterization was carried out with a Bruker Avance 400 (DRX400) NMR spectrometer at 400 MHz at room temperature. The sample was loaded into 4.0 mm Zirconia PENCIL™ rotors and capped tightly. The 29Si spectrum was recorded at 79.489 MHz with a repetition delay of 10 s. A 35-ms acquisition time, spin rate of 10,000 rps and 3000 number of scans were employed. Chemical shifts of 29Si NMR were referenced to tetramethylsilane as 0 ppm.Zebrafish care, microinjection and confocal fluorescence imaging
The transgenic zebrafish line, TG(fli1:EGFP), was used as the vertebrate model for the microinjection of fluorescent PEOSN. This transgenic line expresses the green fluorescent protein in the heart and all blood vessels of the embryo.31 TG(fli1:EGFP) was maintained in the Institute of Molecular and Cell Biology (IMCB) zebrafish facility. The embryos were obtained and experiments were carried out in accordance to IACUC rules (the Biopolis IACUC application #090430) and established methods.33 To prevent the formation of melanin and ensure optical translucence of transgenic larvae for confocal imaging, 1-phenyl-2-thiourea (PTU) was added to zebrafish embryos at 22 hpf. For the introduction of silica nanocapsules into the zebrafish circulation, 3 days old TG(fli1:EGFP) larvae were mounted ventral side up in 1% UltraPure™ low melting point agarose (Invitrogen) and fluorescent PEOSN were microinjected into the sinus venosus. For monitoring of localized uptake of silica nanocapsules, 3 days old TG(fli1:EGFP) larvae were mounted dorsal side up in 1% UltraPure™ low melting point agarose and fluorescent PEOSN were microinjected into the brain ventricle. Confocal microscopy images were acquired with an upright (Zeiss Axiovert200 M) laser scanning microscope (LSM Meta 510, Carl Zeiss) using two laser lines (30 mW Argon and 1 mW HeNe) as the excitation source and visualized using the 505/530 nm and 560/615 nm emission band-pass filters respectively. Images were acquired under the same setting for all microinjected larvae.Results and discussion
Synthesis and optimization of PEOSN
As illustrated in Scheme 1, our strategy of preparing PEOSN lies in the sequestration of tetramethylorthosilicate (TMOS) to the hydrophobic micellar core during the self-assembly of Pluronic® block copolymers micelles. Subsequent contact with water molecules at the core-corona interface will result in the hydrolysis and condensation of TMOS to form a thin silica shell. In this regard, we propose that the condensation reaction frontline is on the outer circumference of the interfacial shell. Hydrolyzed TMOS will diffuse through the silica layer and condense on the outer shell surface. Hence, upon initiation at the core-corona interface, the silica shell will extend radially outwards into the hydrophilic PEO corona as the reaction time progresses.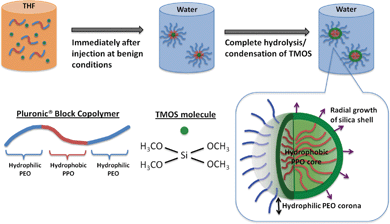 | ||
| Scheme 1 Formation mechanism of PEO surface-decorated silica nanocapsules (PEOSN) | ||
In lieu of our hypothesis for the formation mechanism of PEOSN, the silica shell should ideally be sufficiently thin so that the PEO corona will not be completely enveloped by a thick silica layer at the interfacial region. Instead, free PEO chains can extend from the surface to function as steric stabilizers and prevent the silica nanocapsules from agglomeration. The PEO chains will also offer the opportunity to conjugate other functional groups, such as folate acid, for targeted diagnosis and delivery of payloads.34 Meanwhile, a minimum thickness of the silica shell must still be maintained so as to enhance the mechanical integrity of the Pluronic® micelles and ensure that they remain stable even when subjected to high dilution conditions in physiological environments. Thus, PEOSN will exhibit a synergistic improvement in biomechanical properties to a level which cannot be achieved if the silica nanocapsules and Pluronic® micelles are applied separately. Development of such a robust and colloidally stable nanocarrier can be facilely achieved by our interfacial templating scheme, wherein various synthesis parameters will be varied to confine silica formation at the interfacial region efficiently, thereby controlling the size and core-shell morphology of PEOSN for the development of an optimized nanocapsule structure.
To investigate the effect of PEO block length on the resultant silica nanocapsules, L121 (PEO5PPO70PEO5), P123 (PEO20PPO70PEO20) and F127 (PEO106PPO70PEO106) are selected as templating agents because there is a significant variation in the PEO block length while the PPO block length remains the same. For comparison purpose, the physicochemical properties of these block copolymers are listed in Table 1.29 Employing L121 as a templating agent results in the formation of silica aggregates without a typical core-shell morphology (Fig. 1a). The inability of L121 to self-assemble into polymeric micelles for templated formation of silica nanocapsules is evidenced by DLS analysis too, which indicates the largest hydrodynamic diameter of 650 nm for silica particles arising from L121 as a template (Fig. 1d). This suggests the presence of extensive particle aggregation. Macroscopically, precipitation is observed and this confirms the colloidal instability of L121 mixture solution. These findings could be attributed to the short PEO block length of L121, which limits the aqueous solubility of the block copolymer and its tendency to form micelles. Such characteristics are also supported by the low hydrophilic–lipophilic balance (HLB) and cloud point of L121, as listed in Table 1. As a result, no silica nanocapsules are formed when L121 is selected as a templating agent.
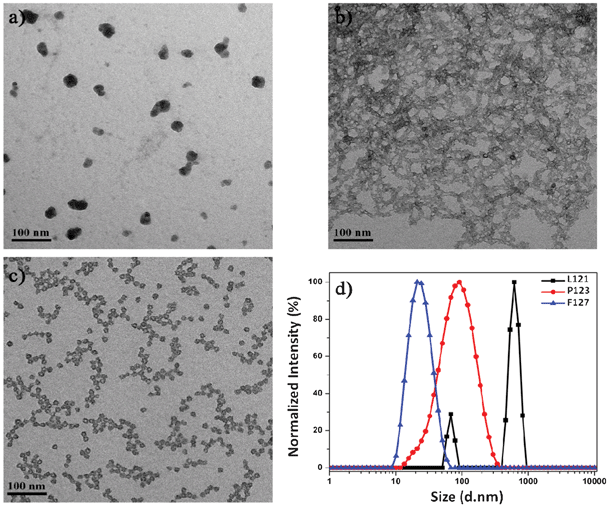 | ||
| Fig. 1 TEM images of the PEOSN derived from (a) L121, (b) P123, and (c) F127 block copolymers and (d) their corresponding DLS result. | ||
| Chemical Formula | Trade name | Molecular weight | PEO wt (%) | Cloud pointa/°C | HLBb |
|---|---|---|---|---|---|
| a Cloud point is determined for 1% aqueous solution and indicates the temperature at which the copolymer solution starts to separate. b HLB measures the degree to which the amphiphilic polymer is hydrophilic or lipophilic. A higher HLB value indicates a greater tendency to form micelles. | |||||
| PEO5PPO70PEO5 | L121 | 4400 | 10 | 14 | 1–7 |
| PEO20PPO70PEO20 | P123 | 5750 | 30 | 90 | 7–12 |
| PEO106PPO70PEO106 | F127 | 12![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 600 600 |
70 | >100 | 22 |
In comparison, P123 and F127 block copolymers have larger HLB values and higher cloud points. Thus, they form micelles readily at room temperature to direct the formation of PEOSN, as indicated by the distinct core-shell morphology of PEOSN derived from P123 and F127 as templates (Fig. 1b and 1c). Nevertheless, while agglomeration is observed with P123-templated PEOSN (Fig. 1b), F127-templated PEOSN has a relatively uniform dispersion in aqueous solution (Fig. 1c). DLS analysis further indicates that F127-templated PEOSN exhibit the smallest hydrodynamic diameter of 25 nm (Fig. 1d). These observations demonstrate the importance of sufficiently long PEO block segments in overcoming the susceptibility of silica nanocapsules towards agglomeration. With progressing time, growth of the silica shell will proceed radially outwards from the core-corona interface. Hence, due to the gradual encroachment of the silica network into the hydrophilic PEO corona, the shorter PEO chains of P123 are unable to provide effective steric stabilization to keep the silica nanocapsules from agglomerating together. As such, F127 is determined to be the preferred choice of templating agent in the synthesis of colloidally stable PEOSN due to its favorable block copolymer composition.
F127 is known to undergo a sol–gel phase transition at sufficiently high concentrations,29,35 during which the F127 micelles are closely packed together and the system becomes highly viscous. Thus, low F127 concentration regimes of < 1.5 wt% were maintained for all synthesis conditions in order to ensure the formation of well-separated F127 micelles.29 The morphological evolution of PEOSN with varying amount of F127 from 25 to 125 mg (i.e. 0.25 to 1.25 wt%) is then investigated by TEM (Fig. S1†) and DLS (Fig. S2†). The core size of PEOSN refers to the inner diameter of the silica shell whereas the outer diameter of PEOSN includes the contribution from the silica shell thickness (Fig. 2).
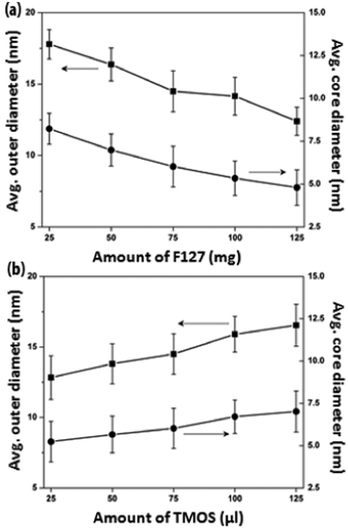 | ||
| Fig. 2 The average outer and core diameters of PEOSN synthesized at varying amount of (a) F127 and (b) TMOS, as measured from TEM. | ||
At a fixed TMOS concentration, a decreasing amount of F127 used will cause more TMOS molecules to be partitioned within each micellar core. Hence, F127 polymeric micelles are expected to undergo a higher degree of core swelling, and this accounts for the increase in average core size and outer diameter of PEOSN from 4.8 to 8.0 nm and 12.7 to 17.8 nm respectively when the amount of F127 used is reduced from 125 to 25 mg (Fig. 2a). In addition, when the ratio of F127 to TMOS decreases, more silica is expected to condense in the PEO corona region and this reduces the effective length of the free PEO chains projecting from the nanocapsule surface. Therefore, agglomeration is more likely to take place, as suggested from the marked increase in hydrodynamic diameter of PEOSN at lower the amount of F127 (i.e. 25–50 mg) in Fig. S2†. Conversely, at a fixed F127 concentration, increasing the amount of TMOS used from 25 to 125 μL brings about an enlargement of the average core size and outer diameter of PEOSN from 5.5 to 7.0 nm and 12.8 to 16.5 nm respectively (Fig. 2b). Formation of a thicker silica shell at higher TMOS concentrations also results in a more well defined core-shell morphology (Fig. S3†). As such, a balance must be struck since a high F127 to TMOS ratio will hamper the formation of a well-defined core-shell morphology while a low F127 to TMOS ratio can cause a greater susceptibility towards particle agglomeration. This is indicated by the gradual cross-linking between PEOSN to form “pearl-necklace” aggregates (Fig. S3e†) and a considerable increase in the hydrodynamic diameter of PEOSN when 125 μL of TMOS is used (Fig. S4†). Despite the variations in our synthesis conditions, the hydrodynamic diameter of PEOSN is consistently larger than its corresponding outer diameter as determined from TEM analysis. This can be ascribed to the PEO chains extending from the outer surface of the silica shell, which exhibit no contrast in the TEM images due to their low electron densities,27 but are reflected in the DLS size measurement. This also confirms the interfacial templating nature of our method whereby the silica shell is formed between the PPO core and PEO corona of the F127 micelles.
In order to elucidate the influence of solvent type on PEOSN formation, the size and morphology of PEOSN synthesized in a mixture of THF and DMF at various mixed solvent ratios of THF/DMF are investigated by TEM (Fig. S5†). As shown in Fig. 3, when the volume fraction of DMF increases, both the core size and outer diameter of the F127-templated PEOSN increases. This is likely attributed to the slower evaporation rate of DMF (boiling point of 155 °C) than that of THF (boiling point of 65 °C). As a result, at larger volume fractions of DMF, the higher content of DMF retained in the micellar core will lead to core swelling and the subsequent formation of an enlarged silica shell. Besides, the addition of DMF to enhance the silicate condensation degree of PEOSN is also demonstrated by 29Si NMR spectroscopy. In the 29Si NMR spectra of Fig. 4a, the three peaks at approximately −92, −102 and −112 ppm can be assigned to the Q2, Q3 and Q4 species respectively. Since the Q4 species denotes the greatest extent of silicate polymerisation, the condensation degree of PEOSN can thus be estimated by determining the area ratio of Q4/(Q2 + Q3). As such, our study suggests the formation of a more condensed silica shell at higher volume fractions of DMF, as illustrated in Fig. 4b. This has positive implications for drug delivery and controlled release applications because the permeability of the silica shell can then be effectively tuned by varying the amount of DMF used.
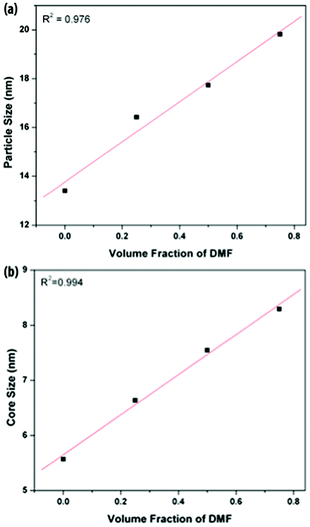 | ||
| Fig. 3 The average (a) outer and (b) core diameters of PEOSN synthesized at different mixed solvent ratios (THF/DMF), as measured from TEM. | ||
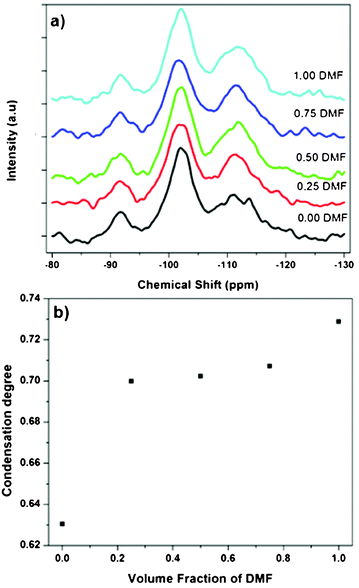 | ||
| Fig. 4 (a) 29Si NMR spectra of PEOSN synthesized at different mixed solvent ratios (THF/DMF), and (b) the corresponding condensation degree of PEOSN as determined by the area ratio of Q4/(Q2 + Q3). | ||
In vivo imaging of fluorescent PEOSN in zebrafish
To demonstrate the application of PEOSN as effective nanocarriers for bioimaging, fluorescent PEOSN are prepared by encapsulating conjugated polymer MEH-PPV, which emits red fluorescence, into the interior of silica nanocapsules. The transgenic zebrafish line TG(fli1:EGFP) is then selected as the vertebrate model for in vivo study with standard laser scanning microscopic technique. After injecting the fluorescent PEOSN into the developing zebrafish larvae, the latter is re-iteratively imaged over many days to assess the biocompatibility, track spatial temporal biodistribution and evaluate the biostability of the injected fluorescent probes.Biocompatibility is assessed by the impact of the fluorescent PEOSN on organ/larvae growth, as well as the gain in vascular network complexity of the zebrafish larvae at different time point of post-injection. Confocal imaging immediately after injection into the cardiac cavity shows that the fluorescent PEOSN are well dispersed in vivo because both the pericardium (the viscous fluid around the heart) and the internal content of the heart fluoresce a uniform red (Fig. 5a). Importantly, the injected larvae remain viable because the heart continues to grow in size and complexity even after 3 days post-exposure to fluorescent PEOSN (Fig. 5b). This confirms the biocompatibility of PEOSN as a synthetic dye carrier. Cellular uptake of fluorescent PEOSN is also demonstrated by the emergence of red fluorescent cells in the cardiac cavity (Fig. 5b). To further assess the extent of cellular uptake by various cells in the zebrafish larvae, the biodistribution of fluorescent PEOSN is examined in the whole larvae, 1-day post injection (Fig. 6). It is observed that red fluorescent cells are predominantly detected close to the green fluorescent (GFP-positive) vessels. Fig. 6(d–f) illustrate some vessels that are imaged at higher magnifications.
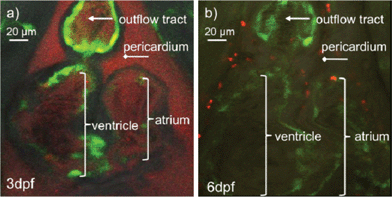 | ||
| Fig. 5 (a) Confocal images of zebrafish heart immediately after injection of fluorescent PEOSN (b) and the same injected larvae imaged 3 days later. | ||
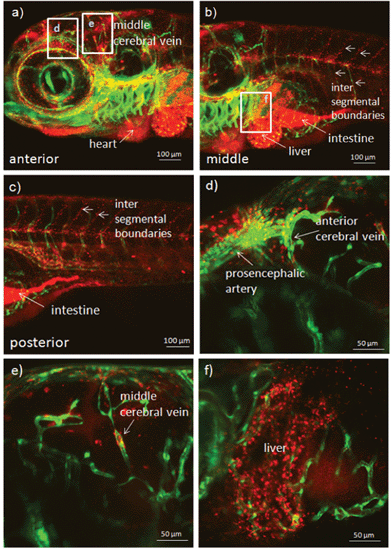 | ||
| Fig. 6 Confocal images of transgenic zebrafish larvae illustrating the biodistribution and uptake of fluorescent PEOSN after 1 day post injection into the heart. Selected areas are enlarged in (d), (e) and (f). | ||
The biostability of fluorescent PEOSN is assessed by investigating the extent to which the red fluorescent signal could be retained after cellular internalization of the silica nanocapsules. This is achieved by direct microinjection of fluorescent PEOSN into the brain ventricle. It is important to note that this procedure is relatively benign when performed on young zebrafish larvae and allows one to address the extent of cellular uptake after localized delivery of fluorescent PEOSN. The brain ventricle, when imaged immediately after microinjection, is highlighted by red fluorescence that is contributed by the injected fluorescent PEOSN (Fig. 7a). Confocal imaging of the same injected larva, 7 days later, still indicates the presence of red fluorescent-labeled cells that are predominantly lining around the site of injection (Fig. 7b). This illustrates the biostability of fluorescent PEOSN. More interestingly, the in vivo biocompatibility of fluorescent PEOSN is strongly supported by the continued brain vascularization in the same injected larva by comparing the vascular network in Fig. 7a with that 7 days later in Fig. 7b. Therefore, the studies conducted in zebrafish in our present work strongly support the use of fluorescent PEOSN as a biocompatible and effective tool for labeling cells in vivo.
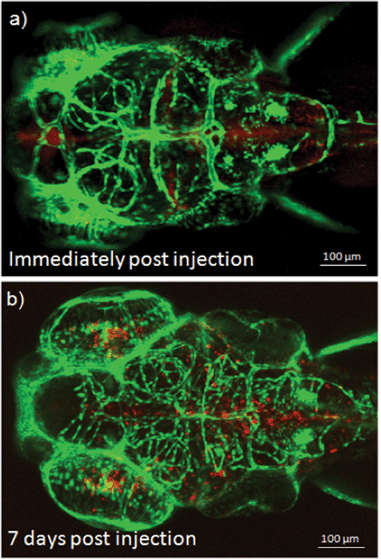 | ||
| Fig. 7 (a) Confocal images of transgenic zebrafish larvae illustrating the cellular uptake and biostability of fluorescent PEOSN immediately and (b) 7 days post injection into the brain ventricle. | ||
Conclusions
We have successfully developed a class of robust silica-based nanocapsules via a novel interfacial hydrolysis and condensation scheme, confined at the interface between the PPO core and PEO corona of the self-assembled F127 polymeric micelles. The silica nanocapsules are surface-decorated with a layer of PEO chains and therefore they are stable in aqueous and physiological environments. Our study demonstrates the precise tuning of the structure and morphology of PEOSN by manipulating several synthesis parameters such as the PEO block length, concentration of block copolymers and TMOS, as well as the type and ratio of mixed solvents. To demonstrate the application of PEOSN for bioimaging, we uploaded fluorescent conjugated polymer MEH-PPV into the interior of PEOSN and conducted in vivo studies with the transgenic zebrafish line TG(fli1:EGFP). The biocompatibility is demonstrated by cellular internalization of these fluorescent PEOSN, which does not interfere with larval development nor affect vessel growth, while the biostability of fluorescent PEOSN is further supported by the presence of fluorescent-labeled cells 7 days post-exposure to the microinjected silica nanocarriers.Acknowledgements
This work is supported by NMRC, Ministry of Health, Singapore (Grant: EDG10may036), and BMRC-SERC Joint Grant (Grant 112 1480002).References
- C. Hui, C. Shen, J. Tian, L. Bao, H. Ding, C. Li, Y. Tian, X. Shi and H. -J. Gao, Nanoscale, 2011, 3, 701 RSC.
- J. Yang, J. Lee, J. Kang, K. Lee, J. -S. Suh, H. -G. Yoon, Y. -M. Huh and S. Haam, Langmuir, 2008, 24, 3417 CrossRef CAS.
- F. -H. Chen, L. -M. Zhang, Q. -T. Chen, Y. Zhang and Z. -J. Zhang, Chem. Commun., 2010, 46, 8633 RSC.
- X. Zhai, M. Yu, Z. Cheng, Z. Hou, P. Ma, D. Yang, X. Kang, Y. Dai, D. Wang and J. Lin, Dalton Trans., 2011, 40, 12818 RSC.
- M. Ma, H. Chen, Y. Chen, X. Wang, F. Chen, X. Cui and J. Shi, Biomaterials, 2012, 33, 989 CrossRef CAS.
- X. Kang, Z. Cheng, D. Yang, P. Ma, M. Shang, C. Peng, Y. Dai and J. Lin, Adv. Funct. Mater., 2012, 22, 1470 CrossRef CAS.
- Y. Chen, H. Chen, L. Guo, Q. He, F. Chen, J. Zhou, J. Feng and J. Shi, ACS Nano, 2010, 4, 529 CrossRef CAS.
- Y. Chen, H. Chen, Y. Sun, Y. Zheng, D. Zeng, F. Li, S. Zhang, X. Wang, K. Zhang, M. Ma, Q. He, L. Zhang and J. Shi, Angew. Chem., 2011, 123, 12713 CrossRef; Angew. Chem. Int. Ed., 2011, 50, 12505 Search PubMed.
- Y. Chen, C. Chu, Y. Zhou, Y. Ru, H. Chen, F. Chen, Q. He, Y. Zhang, L. Zhang and J. Shi, Small, 2011, 7, 2935 CrossRef CAS.
- T.-T. Wang, F. Chai, C.-G. Wang, L. Li, H.-Y. Liu, L.-Y. Zhang, Z.-M. Su and Y. Liao, J. Colloid Interface Sci., 2011, 358, 109 CrossRef CAS.
- L. Zhu, H. Wang, X. Shen, L. Chen, Y. Wang and H. Chen, Small, 2012, 8, 1857 CrossRef CAS.
- Z. Cai, Z. Ye, X. Yang, Y. Chang, H. Wang, Y. Liu and A. Cao, Nanoscale, 2011, 3, 1974 RSC.
- C. I. Zoldesi, C. A. van Walree and A. Imhof, Langmuir, 2006, 22, 4343 CrossRef CAS.
- X. Zhang, L. Clime, H. Roberge, F. Normandin, L'H. Yahia, E. Sacher and T. Veres, J. Phys. Chem. C, 2011, 115, 1436 CAS.
- T. Kim, E. Momin, J. Choi, K. Yuan, H. Zaidi, J. Kim, M. Park, N. Lee, M. T. McMahon, A. Q. Hinojosa, J. W. M. Bulte, T. Hyeon and A. A. Gilad, J. Am. Chem. Soc., 2011, 133, 2955 CrossRef CAS.
- H. Xing, W. Bu, S. Zhang, X. Zheng, M. Li, F. Chen, Q. He, L. Zhou, W. Peng, Y. Hua and J. Shi, Biomaterials, 2012, 33, 1079 CrossRef CAS.
- Z. Xu, C. Li, P. Ma, Z. Hou, D. Yang, X. Kang and J. Lin, Nanoscale, 2011, 3, 661 RSC.
- L. Wang, K. Wang, S. Santra, X. Zhao, L. R. Hilliard, J. E. Smith, Y. Wu and W. Tan, Anal. Chem., 2006, 78, 646 CrossRef.
- Y. Piao, A. Burns, J. Kim, U. Wiesner and T. Hyeon, Adv. Funct. Mater., 2008, 18, 1 Search PubMed.
- H. Tan, J. M. Xue, B. Shuter, X. Li and J. Wang, Adv. Funct. Mater., 2010, 20, 722 CrossRef CAS.
- M. Mandal and M. Kruk, Chem. Mater., 2012, 24, 123 CrossRef CAS.
- F. Chi, Y. N. Guo, J. Liu, Y. Liu and Q. Huo, J. Phys. Chem. C, 2010, 114, 2519 CAS.
- D. Niu, X. Liu, Y. Li, Z. Ma, W. Dong, S. Chang, W. Zhao, J. Gu, S. Zhang and J. Shi, J. Mater. Chem., 2011, 21, 13825 RSC.
- X. W. Lou, L. A. Archer and Z. Yang, Adv. Mater., 2008, 20, 3987 CrossRef CAS.
- T. D. Schladt, K. Schneider, H. Schild and W. Tremel, Dalton Trans., 2011, 40, 6315 RSC.
- J.-J. Yuan, O. O. Mykhaylyk, A. J. Ryan and S. P. Armes, J. Am. Chem. Soc., 2007, 129, 1717 CrossRef CAS.
- H. Tan, N. S. Liu, B. He, S. Y. Wong, Z. K. Chen, X. Li and J. Wang, Chem. Commun., 2009, 41, 6240 RSC.
- P. Petrov, M. Bozukov and C. B. Tsvetanov, J. Mater. Chem., 2005, 15, 1481 RSC.
- M. L. Adams, A. Lavasanifar and G. S. Kwon, J. Pharm. Sci., 2003, 92, 1343 CrossRef CAS.
- S. Fusco, A. Borzacchiello and P. A. Netti, J. Bioact. Compat. Polym., 2006, 21, 149 CrossRef CAS.
- N. D. Lawson and B. M. Weinstein, Dev. Biol., 2002, 248, 307 CrossRef CAS.
- B. M. Weinstein, Trends Cell Biol., 2002, 12, 439 CrossRef CAS.
- M. Westerfield, The Zebrafish Book. A Guide for the Laboratory Use of Zebrafish (Danio rerio), 4th ed., Univ. of Oregon Press, Eugene, 2000 Search PubMed.
- K. H. Bae, Y. Lee and T. G. Park, Biomacromolecules, 2007, 8, 650 CrossRef CAS.
- S. Y. Lee, Y. Lee, J. E. Kim, T. G. Park and C.-H. Ahn, J. Mater. Chem., 2009, 19, 8198 RSC.
Footnote |
| † Electronic Supplementary Information (ESI) available: Supplementary Fig. S1–S5. See DOI: 10.1039/c2ra22472k |
| This journal is © The Royal Society of Chemistry 2012 |