C(sp3)–F reductive elimination from alkylgold(III) fluoride complexes†
Neal P.
Mankad
and
F. Dean
Toste
*
Department of Chemistry, University of California-Berkeley, Berkeley, CA 94720, USA. E-mail: fdtoste@berkeley.edu; Fax: +1 510-666-2504; Tel: +1 510-642-2850
First published on 13th October 2011
Abstract
Rare examples of C(sp3)–F reductive elimination were observed from several cis-F2Au(R)(IPr) intermediates generated by oxidation of (IPr)AuR complexes with XeF2. For R groups bearing β-hydrogens, β-hydride elimination was competitive with C(sp3)–F reductive elimination. For strained cyclic R groups and most acyclic R groups lacking β-hydrogens, carbocation-like rearrangements occurred prior to C(sp3)–F reductive elimination. Kinetics of the decay of one cis-F2Au(R)(IPr) species, stereochemical analysis of reductive elimination with a chiral R group, and DFT analysis collectively suggest C(sp3)–F reductive elimination proceeding through transient cationic [(IPr)Au(F)(R)]+ intermediates with significant ionization of the Au–alkyl bonds.
Introduction
Installation of C–F bonds by transition metal catalysis is particularly fascinating both because of significant interest in fluorocarbon products for important applications,1 but also because of the fundamental challenges posed by designing C–F bond-forming catalytic cycles.2 One main difficulty involves mediating C–F reductive elimination, which is likely to be the key turnover step in many such catalytic cycles. C–F reductive elimination remains rare compared to other types of C–X reductive elimination (X = e.g. C, N, O, Cl, Br, I) despite the large thermodynamic driving force provided by formation of strong C–F bonds (H3C–F BDE = 110 kcal mol−1).3 Key advances in stoichiometric C(sp2)–F reductive elimination4 as well as fluorination of transition metal π-complexes5 have led directly to recent discoveries of corresponding C(sp2)–F and C(allyl)–F bond-forming catalysts.2,4d,5,6 On the other hand, stoichiometric C(sp3)–F reductive elimination from discrete σ-organometallic intermediates has not been extensively studied7 despite a number of reports of stable, high-valent alkyl–metal fluoride complexes.8 Moreover, the reverse process, C(sp3)–F oxidative addition, has only been observed directly in a single recent report.9 Identification of systems that allow for C(sp3)–F reductive elimination and careful study of the principles that govern this process promise to guide the design of future C(sp3)–F bond-forming catalysts.10Results and discussion
Fluorination of Au–C(sp3) bonds
Alkylgold(III) intermediates are well known to undergo facile C–X reductive elimination (X = O, Cl, Br, or I). Recently a mechanistic study was conducted on carbon–halogen reductive elimination from X2Au(Me)(IPr) complexes (IPr = 1,3-bis(2,6-diisopropylphenyl)imidazol-2-ylidene; X = Cl, Br, or I), which produce CH3X even at low temperature.11 In contrast, we recently reported the oxidation of (IPr)AuMe (1) with XeF2 to yield the difluoride analogue cis-F2Au(Me)(IPr) (1-F2), which is remarkably robust towards thermal decomposition by CH3F elimination.12We reasoned that ground-state destabilization of F2Au(R)(IPr) complexes might lead to successful C(sp3)–F bond formation. It is well known that steric pressure facilitates reductive elimination processes in this way.13 Indeed, during the course of our ongoing studies on the role of gold(III) fluorides in catalysis,12,14 we sought to oxidize the more hindered complexes (IPr)Au(tBu) (2) and (IPr)AuCH2CH2tBu (3) with XeF2 in order to study the reactivity of the intermediates cis-F2Au(tBu)(IPr) (2-F2) and cis-F2Au(CH2CH2tBu)(IPr) (3-F2) with arylboronic acid coupling partners. During those studies we found that, if starved of coupling partners, these intermediates decomposed to yield product mixtures containing the gold(I) fluoride complex (IPr)AuF, as well as alkenes resulting from formal β-H elimination and fluoroalkanes presumably derived from C(sp3)–F reductive elimination (Table 1).15 Reaction of (IPr)AuCH2CD2tBu (4), in which the β position is perdeuterated, with XeF2 led to a product mixture only slightly perturbed from that of 3 (Table 1) and in which β-H(D) elimination still outcompeted C–F elimination.
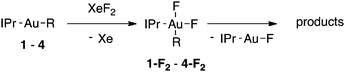
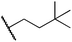
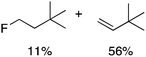
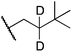
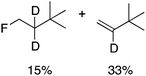
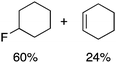
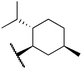
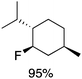
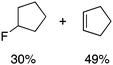
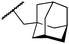
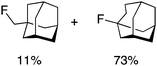
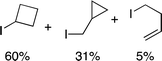
We next examined complexes bearing cyclic alkyl groups in order to disfavor β-H elimination (Table 2). For cyclohexyl derivative 5, C–F reductive elimination indeed outcompeted β-H elimination by a ratio of ∼5![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2. The bulkier (−)-menthyl derivative 6 produced (−)-menthyl fluoride as the exclusive product with perfect stereoretention and with no evidence of alkene products.16 Surprisingly, β-H elimination outcompeted C–F reductive elimination for cyclopentyl derivative 7. For the more strained cycloalkyl derivatives 8 and 9, β-H elimination pathways were marginalized but carbocation-like rearrangement processes were observed as minor pathways relative to direct C–F reductive elimination. Specifically, cyclopropylmethyl fluoride was observed as a minor product from fluorination of cyclobutyl derivative 8, and allyl fluoride was observed as a minor product from fluorination of cyclopropyl derivative 9 (Table 2).
:2. The bulkier (−)-menthyl derivative 6 produced (−)-menthyl fluoride as the exclusive product with perfect stereoretention and with no evidence of alkene products.16 Surprisingly, β-H elimination outcompeted C–F reductive elimination for cyclopentyl derivative 7. For the more strained cycloalkyl derivatives 8 and 9, β-H elimination pathways were marginalized but carbocation-like rearrangement processes were observed as minor pathways relative to direct C–F reductive elimination. Specifically, cyclopropylmethyl fluoride was observed as a minor product from fluorination of cyclobutyl derivative 8, and allyl fluoride was observed as a minor product from fluorination of cyclopropyl derivative 9 (Table 2).
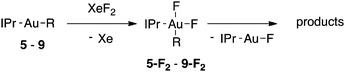
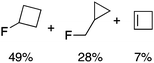
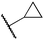
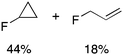
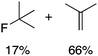
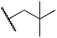
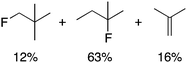
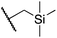
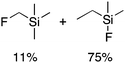
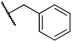
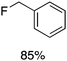
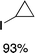
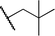
We next studied complexes lacking β-hydrogens (Table 3). Reaction of the neo-pentyl complex 10 with XeF2 also led to carbocation-like behavior, with the rearranged tertiary alkyl fluoride product (tert-pentyl fluoride) dominating over the expected primary alkyl fluoride product (neo-pentyl fluoride). This apparent methyl migration was also observed for the trimethylsilylmethyl derivative 11, with the rearrangement pathway outcompeting the direct C–F reductive elimination pathway to yield ethylfluorodimethylsilane as the major product (Table 3). Alkyl migration even occurred for the 1-adamantylmethyl complex 12 upon reaction with XeF2, further highlighting the preference for AuIII-mediated C(sp3)–F reductive elimination to produce tertiary rather than primary fluoroalkanes when possible. By contrast, reaction of (IPr)AuCH2Ph (13) with XeF2 yielded FCH2Ph in good yield (Table 3).
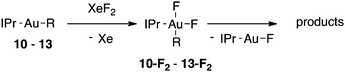
The (IPr)AuF byproduct of these reductive elimination processes was accessed on preparative scale by reaction of 13 with XeF2 and studied by X-ray crystallography. The solid-state structure of (IPr)AuF·2CH2Cl2 revealed an extended hydrogen-bonding network, with the CH2Cl2 solvent molecules acting as hydrogen bond donors towards the terminal Au–F moiety (Fig. 1). Similar hydrogen bonding interactions exist for (SIPr)AuF·2CH2Cl2 (SIPr = 1,3-bis(2,6-diisopropylphenyl) imidazolin-2-ylidene).17 The Au–F distance in (IPr)AuF (2.071(2) Å) is slightly shorter than that in (SIPr)AuF (2.082(2) Å).
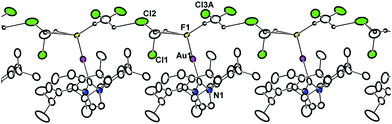 | ||
| Fig. 1 Extended solid-state structure of (IPr)AuF·2CH2Cl2, as 50% ellipsoids. Only solvent hydrogen atoms (in calculated positions) are shown. Selected bond lengths (Å) and angles (°): Au–F, 2.071(2); Au–C, 1.954(5); C–Au–F, 177.65(15). | ||
Kinetics of the decay of an alkylgold(III) difluoride intermediate
For several of the fluorination reactions the cis-F2Au(R)(IPr) intermediates were easily identified by their characteristic 19F NMR properties (Table S1†),12 and it was thus established that (IPr)AuF and the organic products depicted in Tables 1–3 appeared over time as these cis-F2Au(R)(IPr) complexes disappeared. One such intermediate, 11-F2, was chosen for kinetics studies. The disappearance of intermediate 11-F2 obeyed first-order kinetics (Figure S1†). An Eyring plot (Figure S2†) revealed activation parameters of ΔH‡ = 12.7 kcal mol−1 and ΔS‡ = −30 e.u., and thus an activation free energy of ΔG‡ = 21.7 kcal mol−1 at 298 K. When samples were spiked with various concentrations of added tetrabutylammonium fluoride (TBAF), the decay of 11-F2 exhibited an approximately inverse first-order relationship with fluoride concentration (Fig. 2 and S3†).![Dependence of the rate of decay of cis-F2Au(CH2SiMe3)(IPr) (11-F2) on F− concentration, as determined by the method of initial rates. kobs = (Δ[11-F2]/Δt)/[11-F2]0, [11-F2]0 = 32.4 mM, TBAF = tetrabutylammonium fluoride. The red line is a smoothed-curve guide.](/image/article/2012/SC/c1sc00515d/c1sc00515d-f2.gif) | ||
| Fig. 2 Dependence of the rate of decay of cis-F2Au(CH2SiMe3)(IPr) (11-F2) on F− concentration, as determined by the method of initial rates. kobs = (Δ[11-F2]/Δt)/[11-F2]0, [11-F2]0 = 32.4 mM, TBAF = tetrabutylammonium fluoride. The red line is a smoothed-curve guide. | ||
A proposed mechanistic scheme accounting for the various observed reaction pathways is shown in Scheme 1. Consistent with the inverse rate dependence on fluoride concentration, we propose initial fluoride dissociation from an alkylgold(III) difluoride complex to reveal an activated, 3-coordinate complex of the type [(IPr)Au(F)(R)]+. Such a complex with R = Me has been characterized crystallographically in dimeric form.12 This key intermediate can then mediate R–F reductive elimination via a classical 3-centered transition state, consistent with the observed retention of stereochemistry observed for 6, followed by trapping of the [(IPr)Au]+ fragment with the dissociated fluoride. The coordinatively unsaturated nature of a [(IPr)Au(F)(R)]+ species would also allow for β-H elimination either by a traditional Au-mediated mechanism18 or by deprotonation of the β position by F−. We further propose that such a 3-coordinate, cationic species places a significant degree of positive charge character in the AuIII–R bond, resulting in the alkyl migrations and other carbocation-like rearrangements described herein in lieu of direct C–F reductive elimination. The large and negative ΔS‡ for the decay of 11-F2 is unusual for a dissociative process and could be indicative of a highly ordered transition state necessary for tandem methyl migration/reductive elimination. It is also possible that solvent reorganization is necessary for solvation of F− and contributes to this entropic term.19
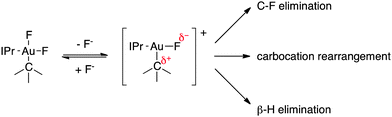 | ||
| Scheme 1 Proposed mechanistic scenario. | ||
Iodination of Au–C(sp3) bonds
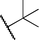
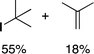
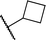
It has been established previously11 that CH3I reductive elimination from trans-I2Au(Me)(IPr) similarly proceeds through transient [(IPr)Au(Me)(I)]+. We thus chose to examine the degree of carbocation-like character possessed by such cationic alkylgold(III) iodide intermediates relative to their alkylgold(III) fluoride analogues.Product mixtures derived from halogenation of selected (IPr)AuR complexes with I2 are shown in Table 4. First it is worth reiterating that though C–F reductive elimination was not observed even at elevated temperatures for R = Me, clean C–I reductive elimination was observed at room temperature.11,12 While β-H elimination and cyclobutyl ring contraction/opening did occur to some extent for the bulkier iodide complexes, certain carbocation-like rearrangement such as methyl migration and cyclopropyl ring-opening which occurred readily for the fluoro complexes did not occur to any detectable extent for the iodo complexes. For example, the neo-pentyl and trimethylsilylmethyl derivatives produced the primary rather than tertiary iodoalkane products exclusively (Table 4). It follows that the carbocationic character of alkylgold(III) complexes is accentuated by fluoride ligands. Lastly, we note that, like C–F reductive elimination, C–I reductive elimination from gold(III) complexes has been shown previously to proceed with stereoretention.14 For no case other than (IPr)AuMe was the alkylgold(III) diiodide intermediate observable by NMR spectroscopy prior to iodoalkane extrusion.11
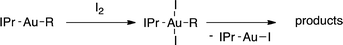
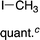
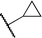
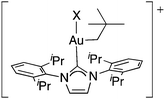
DFT analysis of relevant alkylgold(III) intermediates
In order to examine the nature of the cationic [(IPr)Au(F)(R)]+ intermediates, we initiated a preliminary DFT study. The optimized structure of cis-F2Au(CH2tBu)(IPr) (10-F2) exhibited the expected square planar geometry with a Au–CH2 distance of 2.110 Å and Au–F distances of 2.014 and 2.112 Å (cis and trans to the alkyl group, respectively). Minimization of [(IPr)Au(CH2tBu)(F)]+ (10-F+) from various starting geometries converged to an optimized T-shaped geometry, with the vacant coordination site occurring trans to the alkyl ligand. The Au–CH2 distance in 10-F+ was elongated to 2.180 Å despite dissociation of the trans ligand and onset of positive molecular charge. The calculated LUMO (LUMO = lowest unoccupied molecular orbital) of 10-F+, shown in Fig. 3, was σ*(Au–alkyl) in character and exhibited a through-space antibonding overlap between the σ*(Au–alkyl) contribution and that from an adjacent σ(C–CH3) bond, suggesting an electronic mechanism for methyl migration. Mülliken population analysis indicated that the LUMO of 10-F+ had larger %C contribution (43%) and smaller %Au contribution (20%) than the LUMO of 10-F2 (26%C, 31%Au).![Calculated LUMO (BPV86/LANL2DZ/6-311G++**, implicit CH2Cl2 solvation, 0.04 isocontour) of [(IPr)Au(CH2tBu)(F)]+ (10-F+).](/image/article/2012/SC/c1sc00515d/c1sc00515d-f3.gif) | ||
| Fig. 3 Calculated LUMO (BPV86/LANL2DZ/6-311G++**, implicit CH2Cl2 solvation, 0.04 isocontour) of [(IPr)Au(CH2tBu)(F)]+ (10-F+). | ||
In order to understand the special carbocation-like behavior of [(IPr)Au(F)(R)]+ intermediates, we analyzed the entire halide series [(IPr)Au(X)(CH2tBu)]+ (10-X+, X = F, Cl, Br, and I) by analogous DFT methods. The entire series of complexes was predicted to have roughly similar Au–Calkyl distances, electrostatic charges on CH2, and LUMO compositions (Table S4†). A striking relationship was observed, however, between the identity of the halide ligand X and the natural charge on Au (Table 5). According to natural population analysis, the electrostatic charge on Au ranged from 0.48e in 10-I+ to 0.86e in 10-F+. Because the natural charge on the alkyl CH2 unit remained roughly constant (0.14e for 10-I+ to 0.11e for 10-F+) across the series, this translated to a particularly polarized Au–Calkyl bond for 10-F+, as measured by the difference in charge between Au and CH2 (Table 5). In other words, the fluoro ligand imparts a build-up of positive charge on Au, thereby partially ionizing the Au–Calkyl bond and making (IPr)AuF a particularly effective leaving group. These factors are likely contributors to the unusual carbocation-like behaviour observed in Au-mediated C(sp3)–F reductive elimination reactions.
Conclusions
Alkylgold(III) fluoride complexes are proposed as the active electrophiles in many Au(I)/Au(III) catalytic cycles involving nucleophilic coupling partners.20 For such reactions it has been proposed12,14,21 that:1) C–C coupling is enabled by the relatively long lifetime of alkylgold(III) fluoride intermediates towards C–F reductive elimination
2) C–C coupling occurs by direct attack of nucleophiles on the alkyl group rather than by transmetalation to Au followed by reductive elimination
3) the presence of fluoride ligands enhances the reactivity of alkylgold(III) intermediates towards cross-coupling.
The results communicated herein are fully consistent with these assertions. Gold(III)-mediated C(sp3)–F reductive elimination clearly is less facile than, for example, gold(III)-mediated C(sp3)–I reductive elimination, based both on the differing reactivity of F2Au(Me)(IPr) and I2Au(Me)(IPr) as well as the longer lifetimes of F2Au(R)(IPr) intermediates relative to the corresponding I2Au(R)(IPr) intermediates. The predominantly Calkyl-centric nature of the LUMO in complexes like 10-F+ provides an electronic basis for direct attack of nucleophiles onto Calkyl rather than Au. Apparently one role of fluoride in such cross-coupling reactions, in addition to forming strong B–F or Si–F bonds, is to accentuate carbocation-like character by having a highly ionic interaction with AuIII relative to other, more covalent AuIII–X bonds (such as X = I). This discrepancy is evident both in the differing electronic structures of 10-F+ and 10-I+ as well as in their distinct reductive elimination reactivity patterns (Tables 1–4). We therefore suspect that AuIII–F bonds will continue to play an important role in Au(I)/Au(III) catalysis development, and attempts to incorporate C–F reductive elimination into such cycles are currently underway.
Acknowledgements
This work was supported by NIHGMS (RO1 GM073932). N.P.M. was supported by a NIH Kirchstein-NRSA postdoctoral fellowship. The UC Berkeley Molecular Graphics and Computational Facility is supported by the NSF (CHE-0840505). Prof. Robert Bergman provided helpful suggestions.Notes and references
- K. Müller, C. Faeh and F. Diederich, Science, 2007, 317, 1881 CrossRef.
- T. Furuya, A. S. Hamlet and T. Ritter, Nature, 2011, 473, 470 CrossRef CAS.
- D. F. McMillen and D. M. Golden, Annu. Rev. Phys. Chem., 1982, 33, 493 CrossRef CAS.
- Selected examples: (a) V. V. Grushin, Acc. Chem. Res., 2010, 43, 160 CrossRef CAS; (b) N. D. Ball and M. S. Sanford, J. Am. Chem. Soc., 2009, 131, 3796 CrossRef CAS; (c) T. Furuya, D. Benitez, E. Tkatchouk, A. Strom, P. Tang, W. A. Goddard III and T. Ritter, J. Am. Chem. Soc., 2010, 132, 3793 CrossRef CAS; (d) D. A. Watson, M. Su, G. Teverovskiy, Y. Zhang, J. Garcia-Fortanet, T. Kinzel and S. L. Buchwald, Science, 2009, 325, 1661 CrossRef CAS.
- Selected examples: (a) J. A. Akana, K. X. Bhattacharyya, P. Müller and J. P. Sadighi, J. Am. Chem. Soc., 2007, 129, 7736 CrossRef CAS; (b) M. H. Katcher and A. G. Doyle, J. Am. Chem. Soc., 2010, 132, 17402 CrossRef CAS; (c) C. Hollingworth, A. Hazari, M. N. Hopkinson, M. Tredwell, E. Benedetto, M. Huiban, A. D. Gee, J. M. Brown and V. Gouverneur, Angew. Chem., Int. Ed., 2011, 50, 2613 CrossRef CAS.
- For example: (a) T. Furuya and T. Ritter, Org. Lett., 2009, 11, 2860 CrossRef CAS; (b) K. L. Hull, W. Q. Anani and M. S. Sanford, J. Am. Chem. Soc., 2006, 128, 7134 CrossRef CAS; (c) X. Wang, T.-S. Mei and J.-Q. Yu, J. Am. Chem. Soc., 2009, 131, 7520 CrossRef CAS.
- (a) S.-B. Zhao, J. J. Becker and M. R. Gagné, Organometallics, 2011, 30, 3926 CrossRef CAS; (b) A. W. Kaspi, I. Goldberg and A. Vigalok, J. Am. Chem. Soc., 2010, 132, 10626 CrossRef CAS.
- Selected examples: (a) C. M. Anderson, M. Crespo, G. Ferguson, A. J. Lough and R. J. Puddephatt, Organometallics, 1992, 11, 1177 CrossRef CAS; (b) R. P. Hughes, R. B. Laritchev, L. N. Zakharov and A. L. Rheingold, Organometallics, 2005, 24, 4845 CrossRef CAS.
- (a) J. Choi, D. Y. Wang, S. Kundu, Y. Choliy, T. J. Emge, K. Krogh-Jespersen and A. S. Goldman, Science, 2011, 332, 1545 CrossRef CAS. See also: (b) J. Terao, H. Todo, H. Watabe, A. Ikumi, Y. Shinohara and N. Kambe, Pure Appl. Chem., 2008, 80, 941 CrossRef CAS and references therein.
- C(sp3)–F reductive elimination can be hypothesized to occur for certain existing catalytic cycles. For example: T. Wu, G. Yin and G. Liu, J. Am. Chem. Soc., 2009, 131, 16354 CrossRef CAS.
- V. J. Scott, J. A. Labinger and J. E. Bercaw, Organometallics, 2010, 29, 4090 CrossRef CAS.
- N. P. Mankad and F. D. Toste, J. Am. Chem. Soc., 2010, 132, 12859 CrossRef CAS.
- J. F. Hartwig, Inorg. Chem., 2007, 46, 1936 CrossRef CAS.
- E. Tkatchouk, N. P. Mankad, D. Benitez, W. A. Goddard, III and F. D. Toste, J. Am. Chem. Soc., 2011, 133, 14293 CrossRef CAS.
- C–C homocoupling rather than C–F coupling occurs when similar reactions are carried out with R3PAuR' complexes: (a) A. S. K. Hashmi, T. D. Ramamurthi and F. Rominger, J. Organomet. Chem., 2009, 694, 592 CrossRef CAS; (b) ref. 12 .
- A. A. Zuzek, S. C. Reynolds, D. S. Glueck, J. A. Golen and A. L. Rheingold, Organometallics, 2011, 30, 1812 CrossRef CAS.
- D. S. Laitar, P. Müller, T. G. Gray and J. P. Sadighi, Organometallics, 2005, 24, 4503 CrossRef CAS.
- A. Tamaki, S. A. Magennis and J. K. Kochi, J. Am. Chem. Soc., 1974, 96, 6140 CrossRef CAS.
- Selected examples of reductive elimination with large and negative ΔS‡: (a) D. L. Packett and W. C. Trogler, Inorg. Chem., 1988, 27, 1768 CrossRef CAS; (b) M. P. Brown, R. J. Puddephatt and C. E. E. Upton, J. Chem. Soc., Dalton Trans., 1974, 2457 RSC.
- Recent reviews: (a) K. M. Engle, T.-S. Mei, X. Wang and J.-Q. Yu, Angew. Chem., Int. Ed., 2011, 50, 1478 CrossRef CAS; (b) M. N. Hopkinson, A. D. Gee and V. Gouverneur, Chem.–Eur. J., 2011, 17, 8248 CrossRef CAS.
- (a) W. E. Brenzovich Jr., D. Benitez, A. D. Lackner, H. P. Shunatona, E. Tkatchouk, W. A. Goddard III and F. D. Toste, Angew. Chem., Int. Ed., 2010, 49, 5519 CrossRef; (b) W. E. Brenzovich Jr., J.-F. Brazeau and F. D. Toste, Org. Lett., 2010, 12, 4728 CrossRef; (c) A. D. Melhado, W. E. Brenzovich Jr., A. D. Lackner and F. D. Toste, J. Am. Chem. Soc., 2010, 132, 8885 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Synthetic, spectroscopic, kinetic, crystallographic, and computational details. CCDC reference numbers 837541. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c1sc00515d |
| This journal is © The Royal Society of Chemistry 2012 |