Effects of pore modification on the templating of guest molecules in a 2D honeycomb network
Minna T.
Räisänen†
a,
Anna G.
Slater (née Phillips)
b,
Neil R.
Champness
*b and
Manfred
Buck
*a
aEaStCHEM School of Chemistry, University of St Andrews, North Haugh, St Andrews, KY16 9ST, United Kingdom. E-mail: mb45@st-andrews.ac.uk; Fax: +44 1334 463808; Tel: +44 1334 463800
bSchool of Chemistry, University of Nottingham, University Park, Nottingham, NG7 2RD, United Kingdom. E-mail: Neil.Champness@nottingham.ac.uk; Fax: +44 1159 513563; Tel: +44 1159 513505
First published on 4th October 2011
Abstract
1,7-Diadamantanethioperylene-3,4:9,10-tetracarboxylic diimide, (Ad-S)2–PTCDI, adsorbed on Au(111) from solution was investigated by scanning tunneling microscopy (STM) and X-ray photoelectron spectroscopy (XPS). (Ad-S)2–PTCDI forms a well-ordered monolayer whose structure is described by a (2√63 × √19)R19.1° chiral unit cell containing four molecules. Codeposition of (Ad-S)2–PTCDI with 1,3,5-triazine-2,4,6-triamine (melamine) yields a honeycomb network whose (7√3 × 7√3)R30° unit cell is identical to the unsubstituted PTCDI/melamine analogue. The effect of the adamantyl thioether moieties on the adsorption of guest molecules is investigated using adamantane thiol and C60. While the thioether units do not affect the packing of adamantane thiol molecules a pronounced influence is seen in the case of fullerene. Pore modification involving different combinations of enantiomers of (Ad-S)2–PTCDI give rise to distinctly different arrangements of C60 molecules. The diversity of patterns is further increased by the presence of unsubstituted PTCDI molecules.
1. Introduction
Two-dimensional supramolecular networks, prepared on different substrates both by vapour deposition in an ultrahigh vacuum (UHV) environment and from solution, can be exploited as templates to direct the adsorption of guest entities.1–8 As long as periodicity, pore area, geometry and chemical functionality can be varied independently these systems provide an unprecedented precision and flexibility in the control over arrangement of guest entities. However, since supramolecular networks rely on relatively weak non-covalent interactions and are usually determined by a balance of different interactions, the possibility of a change in the overall structure upon modification of a building block is inherent to these systems, thus making systematic tuning of structures and properties challenging. While this is particularly pronounced for one component systems, as highlighted by derivatives of perylene-3,4:9,10-tetracarboxydiimide (PTCDI),9 bicomponent systems might be more tolerant provided that the synthon involves sufficiently strong directional interactions such as the triple hydrogen bonding between melamine and PTCDI.9As another step towards the controlled design of supramolecular structures we focus here on the aspect of pore modification by investigating how variations of the inner structure of a pore affects the incorporation of guest molecules. A bimolecular PTCDI/melamine network was chosen which has been studied previously10–17 and proven to be sufficiently robust to act as a flexible template in solution based processing.15 Extending previous studies of bromine and propylthio (PrS) substituted PTCDI derivatives9,18,19 the effect of the sterically bulky adamantyl thioether appendage is investigated here. Compared to the smaller Br and the flat lying PrS moiety, the three dimensional structure of the adamantyl groups is expected to exert a significantly different influence on the arrangement of guest molecules. The adamantyl unit is attached to the PTCDI core on opposite sides in the so-called bay regions (Fig. 1). This substitution breaks the mirror symmetries of the PTCDI molecule and as a consequence (Ad-S)2–PTCDI becomes chiral when adsorbed on a surface. If (Ad-S)2–PTCDI behaved like the unsubstituted molecule codeposition with melamine9,15,16,18 it would yield a honeycomb network whose pores are modified by the adamantyl moieties, as schematically shown in Fig. 1. Since the adamantyl moieties are well separated the pore area available for guest molecules will be critically dependent on their size.
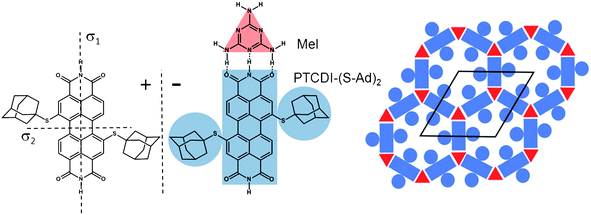 | ||
| Fig. 1 Mirror images (±) of the structure of 1,7-di(adamantanethio)perylene-3,4:9,10-tetracarboxylic diimide, (Ad-S)2–PTCDI, and the motif of the hydrogen bonding between the PTCDI derivative and melamine (Mel). The bimolecular network with the adamantyl-functionalised pores is shown on the right together with the unit cell. | ||
For this reason we have chosen two guest molecules which differ substantially in size. One is C60 which is also of general interest due to its physical properties.20,21 However, due to the high mobility of fullerenes highly ordered surface structures are difficult to achieve at room temperature. Thus in the context of host–guest surface chemistry7,9–14,22–30 the porous networks offer a valuable strategy for precisely controlling the arrangement, intermolecular distances, and interactions of fullerene molecules.20,31,32
The other guest molecule is adamantane thiol (AdSH). With a van der Waals radius of approximately 3.5 Å33 it is significantly smaller than the 10 Å sized C6034,35 and it can be expected that the effect of the pore modification is substantially different for the two molecules.
2. Experimental
All starting materials were used as received unless otherwise stated. Perylene-3,4:9,10-tetracarboxylic acid (Aldrich) was brominated according to a literature procedure17 and converted to N,N′-bis(2-ethylhexyl)dibromoperylene tetracarboxylicdiimide with 2-ethylhexylamine in butanol/H2O.18Column chromatography was performed on silica gel (Merck silica gel 60, 0.2–0.5 mm, 50–130 mesh). 1H NMR and 13C NMR spectra were obtained on a Bruker 300 MHz spectrometer. Microanalyses were performed by Stephen Boyer, London Metropolitan University. MS spectra (MALDI-TOF-MS) were determined on a Voyager-DE-STR mass spectrometer.Synthesis
Sample preparation and characterisation
Au/mica substrates (300 nm gold; Georg Albert PVD) were flame-annealed before sample preparation. (Ad-S)2–PTCDI SAMs were prepared by immersing Au(111) substrate into a hot (348–373 K) DMF solution of (Ad-S)2–PTCDI (210 μM or 1 mM) for 1 min after which the sample was rinsed with DMF and dried under a flow of N2. The (Ad-S)2–PTCDI/melamine network was prepared by modifying the previously reported procedure used for preparation of the unsubstituted PTCDI–melamine network.15–17 A saturated DMF solution of melamine and a 1 mM DMF solution of (Ad-S)2–PTCDI were mixed in DMF to result in 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :11 and 1:80 dilutions, respectively. The mixture was then heated in an oven at 348 K for 30 min before the Au substrate was immersed into it for 1 min at the elevated temperature. After immersion the sample was dipped into DMF at room temperature and blown dry with N2.
:11 and 1:80 dilutions, respectively. The mixture was then heated in an oven at 348 K for 30 min before the Au substrate was immersed into it for 1 min at the elevated temperature. After immersion the sample was dipped into DMF at room temperature and blown dry with N2.
For obtaining a hybrid structure with thiol SAM, (Ad-S)2–PTCDI/melamine modified Au substrate was immersed into 400 μM 1,3,5-trichlorobenzene solution of adamantane thiol for 90 s after which the sample was rinsed with EtOH and blown dry with N2. (Ad-S)2–PTCDI/melamine and PTCDI–melamine networks were exposed to C60 at room temperature by immersion of the network modified Au(111) substrates into a 1,3,5-trichlorobenzene solution of C60 for 20 s after which they were blown dry with N2.
STM images were recorded under ambient conditions with a PicoScan (Molecular Imaging) microscope and tips prepared by mechanically cutting a Pt/Ir (80:20) wire (0.25 mm diameter, Advent Research Materials Ltd.). The STM data were collected in constant current mode.
XPS spectra were recorded with Thermo VG Scientific Sigma Probe spectrometer at 9.6 × 10−9 torr. A monochromatic Al KαX-ray source was applied for all measurements with an analytical spot size of 0.5 mm2, a 37° takeoff angle, 40 ms dwell time, and pass energies of 40 eV (S 2p, Au 4f, C 1s, O 1s and N 1s). Samples were referenced against the internal Au 4f7/2 line at 84.0 eV. Data evaluation was carried using CasaXPS (Casa software Ltd).
3. Results and discussion
(Ad-S)2–PTCDI was prepared following the synthetic route shown in Scheme 1 in good overall yield. Introduction of the adamantylthioether unit to the perylene core was found to be most effective using a Pd(PPh3)4 catalysed coupling reaction between AdSH and Br2-PTCDI. Attempts to prepare (Ad-S)2–PTCDI in a method analogous to that used to prepare (PrS)2–PTCDI, via reaction of PrSH with Br2-PTCDI in the presence of base,9 were unsuccessful.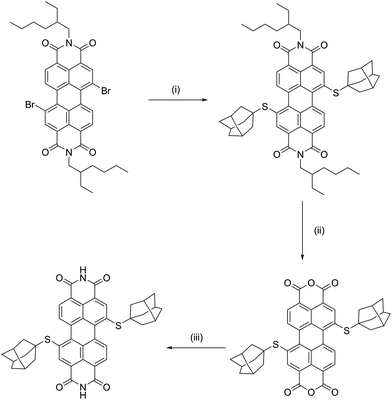 | ||
| Scheme 1 Synthetic pathway used to prepare (Ad-S)2–PTCDI. (i) AdSH, tBuONa, Pd(PPh3)4, toluene, 80–90 °C, 24h,; (ii) KOH, iPrOH, RT, 24h; (iii) NH4OH, THF, 50 °C, 24h. | ||
Monolayers of (Ad-S)2–PTCDI
Adsorption of (Ad-S)2–PTCDI at temperatures ranging between 348–373 K results in well ordered monolayers as illustrated by the STM image shown in Fig. 2a. The Fourier transform, Fig. 2a, reveals domains which are not in line with the symmetry axes of the substrate but which are aligned along directions differing by about 40°. In the magnified images (Fig. 2b) of the two domains labelled by the dashed and solid lines in Fig. 2a two clearly separated alternating groups of four protrusions each (highlighted by red and blue circles) are discernible which yield mirror domains. The distances between the protrusions of a group are too small for them to be identified with PTCDI moieties, i.e., the protrusions represent adamantyl units whereas the PTCDI moieties are not seen. The adamantyl units of a group are assigned to different (Ad-S)2–PTCDI molecules which means that the PTCDI units lie between protrusions of the two different groups.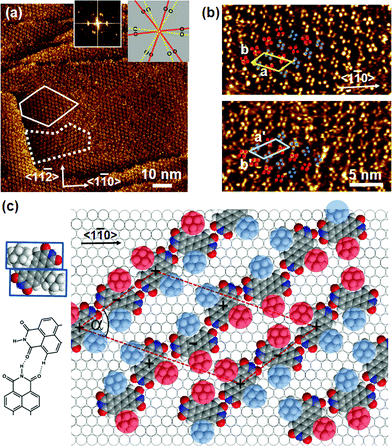 | ||
| Fig. 2 a) Large-scale STM image of (Ad-S)2–PTCDI on Au(111). The dotted and dashed lines frame two mirror domains which are also reflected in the Fourier transform (insets). The relationship between domains and diffraction spots is illustrated by the cartoon. It = 174 pA, Vbias = 180 mV b) Magnified images of the two domains marked in (a). Highlighted by red and blue circles are two groups of protrusions identified as the adamantyl moieties. c) Model of the chiral (Ad-S)2–PTCDI monolayer structure which is described by an oblique (2√63 × √19)R19.1° unit cell (a = 45.9 Å, b = 25.2 Å, α = 55.7°). The projected area of the space filling structure marked by two rectangles is shown on the left (for details see text). | ||
A model describing all experimental observations is depicted in Fig. 2c. This structure is described by the matrices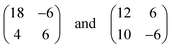 corresponding to oblique (2√63 × 2√19) unit cells containing four molecules and whose lattice vectors a (45.9 Å) and b (25.2 Å) are rotated by ∓19.1° and ±36.6° with respect to the <1
corresponding to oblique (2√63 × 2√19) unit cells containing four molecules and whose lattice vectors a (45.9 Å) and b (25.2 Å) are rotated by ∓19.1° and ±36.6° with respect to the <1![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) 0> direction. We note that while these dimensions are based on the average of several STM images, deviations by about ±5% from these dimensions are within the error margins of the experimental measurements. The monolayer structure is described by zig-zag rows of (Ad-S)2–PTCDI molecules with the PTCDI units interacting via the hydrogen bonding motif depicted in Fig. 2c. The unit cell is chiral but, similar to other cases such as heptahelicene,36 contains a racemic mixture of both enantiomers. While there is some uncertainty about the exact positioning of the adamantyl units relative to the PTCDI core due to the unknown angles associated with the C–S–C groups for the molecule adsorbed on Au, placement of the adamantyl units on the mirror axis σ2 (for definition see Fig. 1) can be excluded as this would be in conflict with the steric requirements of the PTCDI core and the observed distances between the adamantyl units.
0> direction. We note that while these dimensions are based on the average of several STM images, deviations by about ±5% from these dimensions are within the error margins of the experimental measurements. The monolayer structure is described by zig-zag rows of (Ad-S)2–PTCDI molecules with the PTCDI units interacting via the hydrogen bonding motif depicted in Fig. 2c. The unit cell is chiral but, similar to other cases such as heptahelicene,36 contains a racemic mixture of both enantiomers. While there is some uncertainty about the exact positioning of the adamantyl units relative to the PTCDI core due to the unknown angles associated with the C–S–C groups for the molecule adsorbed on Au, placement of the adamantyl units on the mirror axis σ2 (for definition see Fig. 1) can be excluded as this would be in conflict with the steric requirements of the PTCDI core and the observed distances between the adamantyl units.
Comparing the size per (Ad-S)2–PTCDI molecule of about 160 Å2, using a simple geometric approximation, as illustrated in Fig. 2c, with the area per molecule of about 240 Å2 calculated from the unit cell size shows that the coverage is well below a dense packing. The formation of this rather open structure which we consistently observed irrespective of the preparation conditions seems surprising considering that the substantial adsorption energy of perylene derivatives which is expected to be in the range of 2 eV37 should favour a maximisation of coverage, even though the value for (Ad-S)2–PTCDI might deviate to some extent due to the non-flat geometry of the free molecule and additional interactions of the sulfur with the gold substrate. The more open structure suggests a balance of contributions which, in addition to the adsorption energy comprises directional H-bonding interaction and interactions between the adamantyl units.
Compared to analogous compounds where, instead of adamantyl thioether groups, the PTCDI unit is substituted by bromine,9 PrS9,19 or benzoic acid18 the structure observed here is different. In contrast to the propyl thioether analogue where the intermolecular C–H⋯O interaction of the alkane chain with the carbonyl group of PTCDI yields a porous hexagonal structure, this type of interaction does not seem to be of decisive influence in the case of (Ad-S)2–PTCDI, perhaps precluded by the rigidity of the adamantyl moiety. It is also noted that the C–H⋯O interaction of propyl thioether results in an enantiomerically pure structure, within given islands, whereas (Ad-S)2–PTCDI seems to consist of a racemic mixture. It is also different from the hexagonal pattern of the benzoic acid derivative18 and the one dimensional structure of the bromine substituted PTCDI.9 The fact that all four substituted PTCDI molecules form different structures clearly demonstrates that the intermolecular hydrogen bonding between the imide units is not sufficiently strong to allow for a systematic variation of a basic pattern such as the hexagonal structure. The subtle balance between different contributions to the free energy of the system is further highlighted by the change in structure from PTCDI to the dianhydride analogue. For the PrS substituted perylenetetracarboxylic dianhydride (PTCDA) a dense molecular packing is found38 in agreement with the weaker interaction and, thus, small directional forces between anhydride compared to imide groups.
Coadsorption of (Ad-S)2–PTCDI and melamine
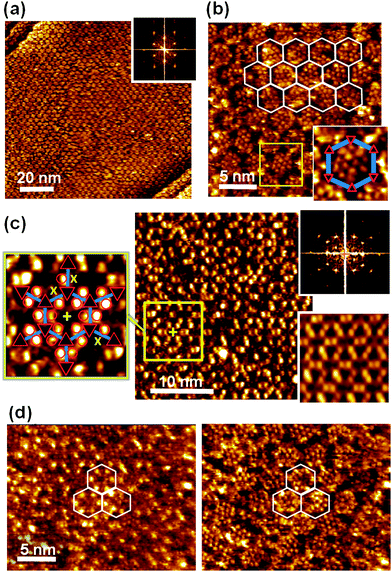
Provided the triple hydrogen bond between PTCDI and melamine is dominating the intermolecular forces, a honeycomb network structure should form whose pore geometry is modified by the adamantyl unit as illustrated in Fig. 1. Following our previously described solution-based method15–17(Ad-S)2–PTCDI and melamine are co-deposited from solution. While it is not immediately obvious from the large scale real space image shown in Fig. 3a, the well defined spots seen in the Fourier transform clearly demonstrates that a highly crystalline structure is formed. The apparent lack of order in the real space structure becomes even more obvious in the high magnification image of Fig. 3b, in particular when compared with the unsubstituted PTCDI/melamine honeycomb structure shown in the inset of Fig. 3b. The discrepancy between real space image and the Fourier transform becomes clear when analysing the structure in more detail. The upper part of Fig. 3c is a cutout from Fig. 3b marked by the rectangle. With the aid of the Fourier filtered image shown in the bottom part of Fig. 3c the network structure can be easily identified with dark regions corresponding to the melamine vortices of the network. Superimposing this network structure onto the unfiltered STM image reveals that the protrusions marked by the red circles exhibit a pattern. In the majority of cases the protrusions are paired and can be straightforwardly assigned to the adamantyl moieties of PTCDI compound. The hexagonal honeycomb structure is now evident and demonstrates that, in contrast to the pure (Ad-S)2–PTCDI layer hydrogen bonding now plays the decisive role in structure formation.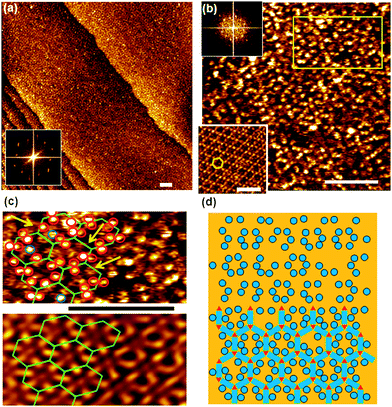 | ||
| Fig. 3 STM images of (Ad-S)2–PTCDI and melamine co-deposited on Au(111) from DMF solution. Iset = 15 pA, Vbias = 680 mV. All scale bars 10 nm. a,b) Large-scale (a) and high resolution images (b). Fourier transforms are shown as insets. For comparison a PTCDI-melamine network is shown in the lower left corner of b). c) Enlarged section of surface marked by the rectangle in b), showing data as acquired (top) and Fourier filtered (bottom). d) Model of the bimolecular network formed by (Ad-S)2–PTCDI and melamine with some adamantyl units missing. The apparent lack of order is illustrated in the top half where the PTCDI and melamine units have been omitted. | ||
The difficulty in identifying a regular structure in the real space image can be explained by two factors. Firstly a dynamic effect causes variations in the apparent height of the protrusions. Taking into account that the adamantyl units are well separated from each other, some residual conformational flexibility can be expected to result in a dynamic behaviour when the adamantyl units experiences the field of the scanning tip. The second reason is static in nature and arises from defects in the network. Marked by the arrows in Fig. 3c, some of the PTCDI sites show either only one protrusion or none at all. Furthermore, some protrusions as highlighted by the blue circles appear to be detached from the PTCDI units. This suggests that under the conditions of network preparation the thioether link is subject to some degradation, i.e., the adamantyl moiety can be detached from the perylene core but remains on the surface as adamantyl-thiolate. This explains the presence of both dislocated protrusions and PTCDI network units with one or even two adamantyl units missing. While the latter might also be explained by the dynamic imaging effect, the fact that it is not changing between scans is a strong indication of defects. The definite proof of the detachment of the thioadamantyl units is provided below when discussing the adsorption of C60 into the network pores.
Within the precision of our STM measurements, the dimensions and orientation of the adamantyl modified network is found to be identical to the structure of unsubstituted PTCDI, i.e., it has a periodicity of 35 Å and a (7√3 × 7√3)R30° unit cell indicated in Fig. 1. A close look at the exact position of the adamantyl units in Fig. 3c shows a narrow distribution in position. Even though there are cases where, within the resolution limit, the adamantyl moieties seem to be positioned symmetrically on the σ2 axis of the PTCDI core and in this case STM is unable to differentiate between different enantiomers. Mostly two adamantyl units attached to the same PTCDI core are on opposite sides of the σ2 mirror axis with both possible configurations occurring consistent with the presence of (+) and (−) enantiomers. This means that unless a pore consists of an enantiomerically pure arrangement of (Ad-S)2–PTCDI molecules there is a substantial variation in the distance between adjacent adamantyl units of a pore depending on whether the (Ad-S)2–PTCDI molecules of the same or opposite chirality are next to each other. This affects the adsorption of guest molecules as will be shown below. The experimental observations for the empty (Ad-S)2–PTCDI/melamine network are summarised in the cartoon of Fig. 3d. As experimentally observed, some of the adamantyl units are omitted from the figure and it can be seen that, in the absence of the honeycomb lattice, it is difficult to recognise the regular arrangement formed by the structure.
The formation of analogous bimolecular honeycomb networks by Br, and PrS substituted analogues as well as (Ad-S)2–PTCDI in combination with melamine, contrasts with the large variation in structures observed for monomolecular layers of the various substituted PTCDI compounds. The predictability in forming the bimolecular honeycomb networks evidences that the triple hydrogen bond formed between PTCDI and melamine is the dominating structural element in directing the bimolecular structure. Indeed, the imide–melamine interaction is sufficiently robust to allow for systematic variation of the network.
Even though the (Ad-S)2–PTCDI containing network results in a pronounced modification of the pore geometry, the presence of unsubstituted and monosubstituted PTCDI molecules (Ad-S)0,1–PTCDI) which are statistically distributed in the network at a level of about 20% limits the degree of control over the pore geometry. While the exact origin of the imperfections remains unclear at present, one explanation for the presence of defective building blocks are impurities present in the (Ad-S)2–PTCDI compound. Even though the chemical analysis of the bulk compound does not show a level of contamination above detection limit, a massive enrichment on the surface during the formation of the bimolecular network could occur if the adsorption energies and/or the adsorption kinetics of the respective compounds are very different.39 However, since the number of missing adamantyl protrusions in the STM images are very different for the monomolecular (Ad-S)2–PTCDI layer and the bimolecular network, a drastically different adsorption behaviour would be required for both systems. The other obvious explanation for the presence of imperfections is thermal degradation. The problem here is that preparation of the monomolecular (Ad-S)2–PTCDI layer under conditions of the network does not increase the defects. Therefore, the difference between the two types of layers is explained by a pronounced dependence of the thermal stability of (Ad-S)2–PTCDI under the experimental conditions used. While on the one hand studies of dialkylsulfides on gold excluded a cleavage of the C–S bond,39,40 a study of n-dioctadecyl sulfide found41 that cleavage is possible but strongly dependent on the preparation parameters such as temperature and, interestingly, also the choice of solvent. The occurrence of isolated circular protrusions which are marked by the blue circles in Fig. 3c could be taken as an indication that C–S bond cleavage indeed occurs, producing separate adamantyl thiolate species during the network preparation. Considering the similarity in the preparation conditions used for both bimolecular and monomolecular arrays it is reasonable to conclude that the melamine acts as a cleavage promoting base.
(Ad-S)2–PTCDI/melamine network as template
To study the effect of pore modification by the adamantyl moieties, two molecules differing in size, namely adamantyl thiol and C60, were chosen as guest molecules. Both molecules have been shown to adsorb into pores14,15 and their rigid structure facilitates imaging at molecular resolution.Adamantane thiol adsorption
To avoid confusion between the adamantyl moieties attached to PTCDI and adamantane thiol guest molecules we refer to them in the following as AdN and AdG, respectively.The large scale image shown in Fig. 4a, together with the Fourier transform, indicates that analogous to the PTCDI-melamine network15 the adamantyl modified network is sufficiently stable to act as template for thiol adsorption from solution. This is fully confirmed by the high resolution image (Fig. 4b) where the AdG molecules are discernible inside the pores. To guide the eye the network pattern is highlighted by the hexagons in the upper part of the image. The network is reflected by a hexagonal mesh of depressions which arise from the melamine molecules as highlighted by the enlarged image of a pore shown in the inset. Unlike the unsubstituted PTCDI/melamine network,15 the (Ad-S)2–PTCDI analogue does not appear as a continuous hexagonal mesh of grooves. The presence of the AdN units affects the measured height above the PTCDI units, most likely due to the somewhat shorter distance between two adamantyl units of a (Ad-S)2–PTCDI molecule compared to two AdG molecules adsorbed on either side of an unsubstituted PTCDI molecule. Otherwise there is no obvious difference between the present network and the analogous network formed by unsubstituted PTCDI. In both cases 12–14 densely packed adamantyl units are observed with an intermolecular distance of around 7 Å which agrees with the value of 6.9 Å given for uniform adamantane thiol SAMs.33 This might not be too surprising considering the identical nature of pore modifier and guest molecule. Furthermore, the distance between two adjacent AdN moieties in a pore is sufficient for an AdG molecule to fit in between.
 | ||
| Fig. 4 STM images of (Ad-S)2–PTCDI/Melamine network with adamantane thiol, AdG, filled pores. a) Large scale image with Fourier transform shown as inset. Ub = −340 mV and It = 36 pA. b) Molecularly resolved image with network frame indicated. Ub = −340 mV and It = 174 pA. Inset shows magnified image of pore highlighted by the square. c) AdSH, AdG, filled network imaged under different tunneling conditions. Fourier transform and Fourier filtered image are shown on the right. Enlarged section of the network with structure overlaid depicted on the right. “x” mark adamantyl units missing from the (Ad-S)2–PTCDI moieties. Ub = 180 mV and It = 92 pA. d) Illustration of contrast change upon change of tunneling current. Same area scanned at Ub = −340 mV and It = 45 pA (left) and 174 pA (right), respectively. | ||
Somewhat unexpected is the lack of height contrast between AdN and AdG units in the image shown in Fig. 4b. Despite their similarity, AdN is attached to PTCDI and interacts with the substrate via a thioether group whereas AdG is adsorbed as a thiolate. Therefore, one could expect that differences in the bonding to the substrate is also reflected in the tunneling resistance. Indeed as Fig. 4c illustrates, AdN and AdG can appear very differently. A different set of tunneling parameters gives rise to a pronounced contrast between the two types of adamantyl moieties. The change from negative to positive bias highlights now AdN whereas the AdG molecules appear dark and cannot be resolved. In agreement with the results for the empty network, some of the PTCDI moieties are missing an AdN unit as demonstrated by the enlarged image of the area marked by the yellow square. Whereas most PTCDI units have two AdN units (marked by red circles), some defects marked by “x” are discernible. It is interesting to note that, compared to the empty network, imaging of the filled network under conditions where the AdN moieties are highlighted gives a better resolved image of the adamantyl moieties. They seem to be better defined with regard to both variations in apparent height and position. This is also reflected by the Fourier filtered image where in the pore filled network the AdN units appear as separate protrusions, in contrast to the corresponding blurred image of the empty network (Fig. 3c). This can be rationalised by a reduced conformational flexibility of the AdN units due to the presence of densely packed AdG thiolates.
That the contrast variation is indeed originating from the tunneling is evidenced by Fig. 4d where images of the same area but acquired under different tunneling conditions are shown. The change from highlighted AdN units (left) when imaged at lower currents to the same tunneling contrast of the AdN and AdG moieties at higher currents demonstrates the possibility to discriminate between the two differently attached adamantyl units. However, while the tunneling parameters do change the relative heights of the AdN and AdG additional factors can also affect the contrast. Differences as shown in Fig. 4 can even occur within a single scan, i.e. with tunneling parameters remaining unchanged, thus, evidencing that the state of the STM tip is also crucial for the imaging contrast. Since the tip state is crucial it also is difficult to provide exact parameters for a respective imaging mode irrespective of general trends like the current dependent contrast.
Fullerene adsorption
While the size of the adamantyl unit allows for a seemless fit of AdG molecules into the AdN modified pore of the (Ad-S)2–PTCDI/melamine network, this is expected to be very different for the significantly larger C60. To illustrate the effect of pore modification by the adamantyl moieties, we show C60 adsorbed into an unmodified PTCDI/melamine network for comparison. As demonstrated in Fig. 5a, adsorption of the fullerenes from solution yields an arrangement of mostly seven molecules per pore, identical to experiments where the C60 was deposited in a UHV environment.14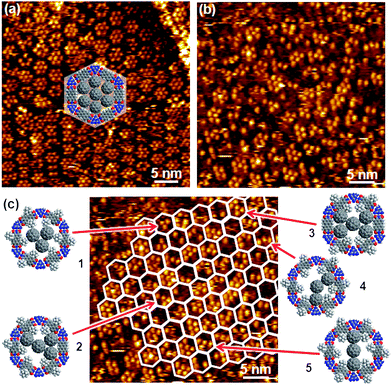 | ||
| Fig. 5 STM images of C60 deposited into networks of PTCDI/melamine (a) and (Ad-S)2–PTCDI/melamine (b) on Au(111). Imaging parameters: I = 28 pA, Vbias = 680 mV (a) and I = 28 pA, Vbias = 340 mV (b). c) same image as in b) with the honeycomb pattern of the network indicated as a guide to the eye. The models exemplify different arrangements of C60 molecules determined by the combination of (Ad-S)2–PTCDI enantiomers and missing adamantyl units. For details see text. | ||
The analogous experiment carried out with the (Ad-S)2–PTCDI/melamine network yields a very different picture. In contrast to the uniform clusters seen in Fig. 5a, there is a large variation with regard to both the number of C60 molecules which ranges between 2 and 7 and geometry of the clusters as seen from Fig. 5b. Similar to the case shown in Fig. 3, the lack of uniform regular features makes it difficult to even identify the hexagonal network. However, the templating effect of the network becomes obvious if the network pattern is overlaid as shown in Fig. 3c. Irrespective of their size and shape, all clusters are strictly confined to the hexagonal pores of the network.
There are two reasons for the diversity of clusters one of which is the defects in the network by missing AdN units as already discussed above (Fig. 3c,d). The occurrence of five or more C60 molecules in a pore can only be explained by this type of network imperfection. We note at this point that the number of missing AdN moieties seems to be significantly higher compared to the bare empty network. Recalling that the pure (Ad-S)2–PTCDI layer does not show any indication of missing adamantyl units, the number of defects seem to increase with increasing network modification. At this point we can only speculate as to the reason for the enhanced proportion of defects but as the thioether link is clearly prone to cleavage then additional forces exerted by the adsorption of C60 would further increase the defect density.
The second reason for the occurrence of different C60 clusters is ascribed to the arrangement of the (Ad-S)2–PTCDI molecules. As indicated above two enantiomers of the PTCDI can be differentiated, unless the adamantyl units are located on the σ2 mirror axis (see Fig. 1). Whether or not a fullerene can fit between two adjacent adamantyl units depends critically on the combination of enantiomers within a given pore. For two neighbouring (Ad-S)2–PTCDI molecules of the same chirality there is insufficient space for a C60 to fit between the adamantyl units, whereas in the case of neighbouring adamantyl groups of opposing chirality there is sufficient space. Assuming that the (+) and (−) enantiomers are incorporated with equal probability, i.e. this is not affected by the chirality of the neighbouring (Ad-S)2–PTCDI molecules, a number of adamantyl patterns becomes possible among which a regular pore structure consisting of only one enantiomer is the least likely of all configurations. For an enantiomerically pure pore geometry at most three C60 molecules could be accommodated in the centre of the pore, as illustrated by model 1 in Fig. 5c. Indeed a configuration matching this geometry is infrequently observed. Compared to other C60 molecules which are located at the edge of the pore, the group of three is centred and, in accordance with the model, the corners of the triangular arrangement of the fullerenes point towards the melamine molecules of the hydrogen-bonded network. Apart from this case of an enantiomerically pure pore geometry, where the AdN units are arranged in a regular fashion, the combination of (+) and (−) enantiomers yields different spacings, thus giving rise to different fullerene patterns. Structures 4 and 5 are examples where C60 molecules are located between two adamantyl units right at the melamine vortices of the network. The number of possible C60 arrangements is further increased by missing AdN moieties. Any adjacent fullerenes located along a pore edge require a missing adamantyl unit beside neighbouring (Ad-S)2–PTCDI molecules of opposite enantiomeric arrangement. These latter cases are the underlying structures 2 and 3 shown in Fig. 5c.
On the one hand, the variety of C60 configurations compiled in Fig. 5c clearly demonstrate the opportunities for controlling the arrangement of guest molecules but also reveal the precision required for a full control over molecular geometries. The present investigation complements previous studies where PTCDI modified with two PrS or two bromo groups served as building blocks in the bimolecular honeycomb structure formed on Ag/Si(111)-√3 × √3R30°.9 Contrasting the influence of the adamantyl moieties, the bromo groups proved to be too small to affect the adsorption of C60 and similarly to the unsubstituted PTCDI seven molecules were accommodated in the pores. The PrS groups, on the other hand, had a partial blocking effect on the C60 adsorption although in an irregular manner.9 In the case of more rigid groups, such as benzoic acid moieties,18 a higher degree of control over molecular adsorption can be achieved. A similar approach for shaping pore structures has been explored with metal–organic coordination networks (MOCNs) based on Fe-1,4-dicarboxylic benzoic acid and Fe-1,2,4-tricarboxylic benzoic acid on Cu(100).7 In these cases the cavities of both MOCNs accommodated one C60 molecule. Thus the additional –COOH side group of the 1,2,4-tricarboxylic benzoic acid molecules penetrating into the cavities did not block fullerene adsorption on the surface but instead resulted in an increased host–guest interaction.
XPS measurements
For the chemical analysis of the supramolecular network and the adsorption of thiols into the pores, the sulfur signal is of particular interest as the binding energies differ for thiolates and thioethers.42 The S2p spectra of the different systems are compiled in Fig. 6a. Spectrum 1 shows a uniform adamantane thiol SAM with a peak around 162 eV and a shoulder at 163.3 eV, assigned to the 2p3/2 and 2p1/2 doublet at binding energies characteristic of thiolates. For the pure (Ad-S)2–PTCDI monolayer (spectrum 2) the peak appears at 163.3 eV as expected for a thioether species. The PTCDI/melamine network with the pores filled by adamantane thiol yields a spectrum (4) which is identical to the uniform adamantane thiol SAM (1) apart from the lower intensity due to the reduction in area of the SAM by the hydrogen-bonded network. Analogously, the combination of (Ad-S)2–PTCDI with melamine to the network (5) yields a position of the sulfur peak identical to that of the uniform (Ad-S)2–PTCDI layer. Again the network structure reduces the density and, thus, intensity of the signal. Finally, the network-SAM hybrid structure (3) where the adamantane thiolate molecules filling the network pores and the thioether species of the network coexist yields a superposition of both signals as reflected by a position of the sulfur peak in between the thiolate at 162 eV and the thioether at 163.3 eV. For completeness the signal of a PTCDI-melamine network is shown (6) to illustrate the signal/noise ratio.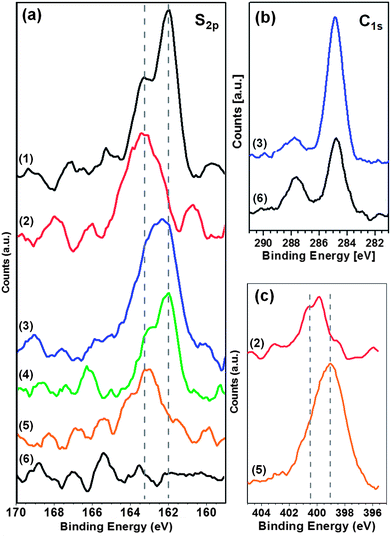 | ||
| Fig. 6 XP spectra of PTCDI/melamine networks and an adamantanethiol SAM on Au(111). S2p (a), C1s (b) and N1s (c) regions are shown for pure AdSH-SAM (1), pure (Ad-S)2–PTCDI monolayer (2), (Ad-S)2–PTCDI/Mel network with AdSH, AdG, filled pores (3), PTCDI/Mel-network filled with AdSH, AdG, filled pores (4), empty (Ad-S)2–PTCDI/Mel network (5), empty PTCDI/Mel network (6). | ||
As far as the other elements are concerned there are, as expected, no significant differences. The C1s spectrum consists, in all cases, of a peak around 284.5 eV and a second peak of lower intensity around 287.5 eV which originate from the aliphatic/aromatic carbon and carbonyl species, respectively. The AdSH filled (Ad-S)2–PTCDI/melamine network and the PTCDI/melamine network are shown as illustrative examples with the biggest difference between systems (Fig. 6b). Comparing both spectra, the peak at 284.5 eV is higher for the pore filled network due to the additional AdSH molecules. Conversely, the carbonyl peak is weaker than in the PTCDI/melamine case due to the attenuation of the photoelectrons by the AdSH layer. We note that the attenuation is amplified due to the geometry of the experiment as the photoelectrons emitted an angle of 53° from the surface normal were measured.
The N1s spectra are also very similar apart from the difference between layers consisting of (Ad-S)2–PTCDI molecules only and of networks combining (Ad-S)2–PTCDI and melamine (Fig. 6c). The spectrum of the former is characterised by a single nitrogen peak at ∼400 eV from the imide nitrogen whereas in the network spectrum the signal is dominated by the melamine nitrogen which peaks at 399 eV. In accordance with the higher nitrogen content of melamine the signal is higher compared to pure (Ad-S)2–PTCDI despite the more open structure of the network.
Conclusions
We have demonstrated that it is possible to introduce a bulky 3D substituent as pore modifier of the PTCDI-melamine surface-based hydrogen bonded network. The influence of the large adamantyl thioether moieties on the adsorption of the host molecules is sensitively dependent on the size of the host molecules. While adamantane thiol fits seamlessly into the modified pores, the situation is pronouncedly different for fullerenes due to the surface-based chirality of the (Ad-S)2–PTCDI molecule. The statistical combination of the enantiomers gives rise to different templating pore geometries, thus yielding different arrangements of C60 molecules. Partial cleavage of the adamantyl thioether substituents upon preparation of the bimolecular (Ad-S)2–PTCDI/melamine array further increases the number of possible fullerene arrangements. Even though at present the different patterns of fullerene guest molecules are realised in a statistical fashion, the system presented here demonstrates the opportunities for the arrangement of guest entities through modification of pore geometries. We continue to pursue our understanding of the formation of surface-based hydrogen-bonded networks in order to accomplish full control over the pore design.Acknowledgements
This work was supported by the UK Engineering Physical Sciences Research Council (EPSRC). NRC gratefully acknowledges the receipt of a Royal Society Leverhulme Trust Senior Research Fellowship. MTR acknowledges the financial support of the Emil Aaltonen Foundation and the Academy of Finland.Notes and references
- F. Cicoira, C. Santato and F. Rosei, Top. Curr. Chem., 2008, 285, 203–267 CrossRef CAS.
- J. V. Barth, Surf. Sci., 2009, 603, 1533–1541 CrossRef CAS.
- J. A. A. W. Elemans, S. B. Lei and S. De Feyter, Angew. Chem., Int. Ed., 2009, 48, 7298–7332 CrossRef CAS.
- S. Furukawa and S. De Feyter, Top. Curr. Chem., 2009, 287, 87–133 CrossRef CAS.
- T. Kudernac, S. B. Lei, J. A. A. W. Elemans and S. De Feyter, Chem. Soc. Rev., 2009, 38, 402–421 RSC.
- N. Lin, S. Stepanow, M. Ruben and J. V. Barth, Top. Curr. Chem., 2009, 287, 1–44 CrossRef CAS.
- S. Stepanow, M. Lingenfelder, A. Dmitriev, H. Spillmann, E. Delvigne, N. Lin, X. B. Deng, C. Z. Cai, J. V. Barth and K. Kern, Nat. Mater., 2004, 3, 229–233 CrossRef CAS.
- A. G. Slater, P. H. Beton and N. R. Champness, Chem. Sci., 2011, 2, 1440–1448 RSC.
- L. M. A. Perdigao, A. Saywell, G. N. Fontes, P. A. Staniec, G. Goretzki, A. G. Phillips, N. R. Champness and P. H. Beton, Chem.–Eur. J., 2008, 14, 7600–7607 CrossRef CAS.
- L. M. A. Perdigao, E. W. Perkins, J. Ma, P. A. Staniec, B. L. Rogers, N. R. Champness and P. H. Beton, J. Phys. Chem. B, 2006, 110, 12539–12542 CrossRef CAS.
- F. Silly, A. Q. Shaw, K. Porfyrakis, G. A. D. Briggs and M. R. Castell, Appl. Phys. Lett., 2007, 91 CAS.
- A. Saywell, G. Magnano, C. J. Satterley, L. M. A. Perdigao, N. R. Champness, P. H. Beton and J. N. O'Shea, J. Phys. Chem. C, 2008, 112, 7706–7709 CAS.
- J. A. Theobald, N. S. Oxtoby, N. R. Champness, P. H. Beton and T. J. S. Dennis, Langmuir, 2005, 21, 2038–2041 CrossRef CAS.
- J. A. Theobald, N. S. Oxtoby, M. A. Phillips, N. R. Champness and P. H. Beton, Nature, 2003, 424, 1029–1031 CrossRef CAS.
- R. Madueno, M. T. Raisanen, C. Silien and M. Buck, Nature, 2008, 454, 618–621 CrossRef CAS.
- C. Silien, M. T. Raisanen and M. Buck, Angew. Chem., Int. Ed., 2009, 48, 3349–3352 CrossRef CAS.
- C. Silien, M. T. Raisanen and M. Buck, Small, 2010, 6, 391–394 CrossRef CAS.
- A. G. Phillips, L. M. A. Perdigao, P. H. Beton and N. R. Champness, Chem. Commun., 2010, 46, 2775–2777 RSC.
- A. J. Pollard, E. W. Perkins, N. A. Smith, A. Saywell, G. Goretzki, A. G. Phillips, S. P. Argent, H. Sachdev, F. Muller, S. Hufner, S. Gsell, M. Fischer, M. Schreck, J. Osterwalder, T. Greber, S. Berner, N. R. Champness and P. H. Beton, Angew. Chem., Int. Ed., 2010, 49, 1794–1799 CrossRef CAS.
- D. Bonifazi, O. Enger and F. Diederich, Chem. Soc. Rev., 2007, 36, 390–414 RSC.
- A. A. R. Watt, M. R. Sambrook, K. Porfyrakis, B. W. Lovett, H. El Mkami, G. M. Smith and G. A. D. Briggs, Chem. Commun., 2006, 1944–1946 RSC.
- S. J. H. Griessl, M. Lackinger, F. Jamitzky, T. Markert, M. Hietschold and W. M. Heckl, J. Phys. Chem. B, 2004, 108, 11556–11560 CrossRef CAS.
- G. B. Pan, J. M. Liu, H. M. Zhang, L. J. Wan, Q. Y. Zheng and C. L. Bai, Angew. Chem., Int. Ed., 2003, 42, 2747–2751 CrossRef CAS.
- P. A. Staniec, L. M. A. Perdigao, A. Saywell, N. R. Champness and P. H. Beton, ChemPhysChem, 2007, 8, 2177–2181 CrossRef CAS.
- M. Stohr, M. Wahl, H. Spillmann, L. H. Gade and T. A. Jung, Small, 2007, 3, 1336–1340 CrossRef CAS.
- S. Yoshimoto, Y. Honda, O. Ito and K. Itaya, J. Am. Chem. Soc., 2008, 130, 1085–1092 CrossRef CAS.
- D. Bonifazi, H. Spillmann, A. Kiebele, M. de Wild, P. Seiler, F. Y. Cheng, H. J. Guntherodt, T. Jung and F. Diederich, Angew. Chem., Int. Ed., 2004, 43, 4759–4763 CrossRef CAS.
- E. Mena-Osteritz and P. Bauerle, Adv. Mater., 2006, 18, 447–451 CrossRef CAS.
- H. Spillmann, A. Kiebele, M. Stohr, T. A. Jung, D. Bonifazi, F. Y. Cheng and F. Diederich, Adv. Mater., 2006, 18, 275–279 CrossRef CAS.
- A. Kiebele, D. Bonifazi, F. Y. Cheng, M. Stohr, F. Diederich, T. Jung and H. Spillmann, ChemPhysChem, 2006, 7, 1462–1470 CrossRef CAS.
- M. Li, K. Deng, S. B. Lei, Y. L. Yang, T. S. Wang, Y. T. Shen, C. R. Wang, Q. D. Zeng and C. Wang, Angew. Chem., Int. Ed., 2008, 47, 6717–6721 CrossRef CAS.
- L. Sanchez, R. Otero, J. M. Gallego, R. Miranda and N. Martin, Chem. Rev., 2009, 109, 2081–2091 CrossRef CAS.
- A. A. Dameron, T. J. Mullen, R. W. Hengstebeck, H. M. Saavedra and P. S. Weiss, J. Phys. Chem. C, 2007, 111, 6747–6752 CAS.
- A. Marchenko and J. Cousty, Surf. Sci., 2002, 513, 233–237 CrossRef CAS.
- J. A. Gardener, G. A. D. Briggs and M. R. Castell, Phys. Rev. B: Condens. Matter Mater. Phys., 2009, 80, 235434 CrossRef.
- R. Fasel, M. Parschau and K.-H. Ernst, Nature, 2006, 439, 449–452 CrossRef CAS.
- J. Ziroff, P. Gold, A. Bendounan, F. Forster and F. Reinert, Surf. Sci., 2009, 603, 354–358 CrossRef CAS.
- J. C. Russell, M. O. Blunt, G. Goretzki, A. G. Phillips, N. R. Champness and P. H. Beton, Langmuir, 2010, 26, 3972–3974 CrossRef CAS.
- C. Jung, O. Dannenberger, Y. Xu, M. Buck and M. Grunze, Langmuir, 1998, 14, 1103–1107 CrossRef CAS.
- C. J. Zhong, R. C. Brush, J. Anderegg and M. D. Porter, Langmuir, 1999, 15, 518–525 CrossRef CAS.
- H. Takiguchi, K. Sato, T. Ishida, K. Abe, K. Yase and K. Tamada, Langmuir, 2000, 16, 1703–1710 CrossRef CAS.
- T. Weidner, N. Ballav, U. Siemeling, D. Troegel, T. Walter, R. Tacke, D. G. Castner and M. Zharnikov, J. Phys. Chem. C, 2009, 113, 19609–19617 CAS.
Footnote |
| † Present address: Laboratory of Inorganic Chemistry, Department of Chemistry, University of Helsinki, P. O. Box 55, FI-00014 University of Helsinki, Finland. |
| This journal is © The Royal Society of Chemistry 2012 |