DOI:
10.1039/C1SC00621E
(Minireview)
Chem. Sci., 2012,
3, 53-57
Cooperative Lewis acid/N-heterocyclic carbene catalysis
Received
31st August 2011
, Accepted 27th September 2011
First published on 12th October 2011
Abstract
Lewis acid activation with N-heterocyclic carbene (NHC) catalysis has presented new opportunities for enantioselective reaction development. Recent findings illustrate that Lewis acids can play an important role in homoenolate annulations by: enhancement of the reactivity, reversal of the diastereo- or regioselectivity, and activation of previously inactive electrophiles. Additionally, the incorporation of a Lewis acid into Brønsted base-catalyzed conjugate addition allowed for an increase in yields.
Introduction
The ability to facilitate synthetic transformations without stoichiometric reagents is an ongoing study in organic synthesis. N-Heterocyclic carbene (NHC) catalysis has emerged as a powerful method in this regard, allowing for access to catalytically generated acyl anions, homoenolates, enolates, and α-acylvinyl anion equivalents.1 These reactive intermediates have been added to a wide range of electrophiles, assembling complex and biologically active structural motifs.1 Even with the aformentioned advances, NHC-driven processes can be limited with respect to the stereoselectivity, regioselectivity of the bond formation, or the type of electrophile. We sought to improve these carbene catalyzed processes and expand the repertoire of the electrophile class by exploring a cooperative catalytic approach.2 Specifically, we envisaged the integration of Lewis acids (LA) with NHC catalysis as a way to provide two simultaneous modes of activation (i.e., cooperative catalysis). However, this potentially promising strategy is fundamentally inauspicious since NHCs are known to be strong ligands for many transition metals. Reported herein are recent developments in this novel NHC/LA cooperatively catalytic approach which defy conventional wisdom regarding the potential incompatibilty of Lewis acids and bases. This minireview focuses on advances in cooperative NHC/LA combinations; a comprehensive account of related exciting discoveries involving sequential NHC catalysis with either transition metals3 or in combination with enamine catalysis4 is not possible given the limits of the present format, but are similarly exciting in terms of new directions.
Improving the reactivity
The γ-lactam is an important structural motif that is prevalent in many biologically active natural products.5 Despite its significance, there are limited reported syntheses of N–H γ-lactams.6 We have been investigating formal cycloadditions by combining NHC-generated homoenolate equivalents with simple electrophiles in order to access cyclic scaffolds. We envisioned that these γ-lactam derivatives could be constructed by a formal [3 + 2] cycloaddition of NHC-generated homoenolate equivalents and N-acyl hydrazones (1) (Scheme 1).7 Initial attempts employing only NHC-catalysis resulted in product formation with low to moderate yield and selectivity, but with incomplete consumption of the starting material. In order to enhance the reactivity/electrophilicity of these N-acyl hydrazones, we explored the use of LA activation in combination with NHC-catalysis. After an extensive survey of potentially compatible Lewis acids, we discovered that employing Mg(Ot-Bu)2 in combination with an NHC resulted in an increase in yield and selectivity. After optimization of additional reaction parameters, this homoenolate annulation was facilitated using low loadings of both the NHC and the LA (5 mol% of each), furnishing the desired lactam (3) in good to excellent yield with excellent diastereo- and enantioselectivity.
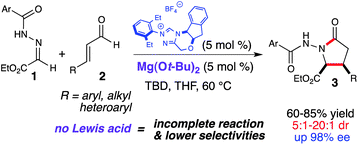 |
| | Scheme 1
Homoenolate annulation with N-acyl hydrazones | |
The proposed cooperative catalytic reaction pathway for this reaction is depicted in Scheme 2. The deprotonated azolium (NHC) adds to the α,β-unsaturated aldehyde (2), which undergoes a 1,2-proton transfer to generate the homoenolate equivalent (I). The homoenolate then undergoes addition to the hydrazone, which is activated by Mg(II) alkoxide (II). Following C–C bond formation, enol III undergoes tautomerization and dissociation of Mg(Ot-Bu)2. Intramolecular acylation of IV regenerates the NHC and furnishes γ-lactam 3. Interestingly, preliminary initial rate data indicates that the reaction is overall inverse first order with respect to Mg(II) concentration.
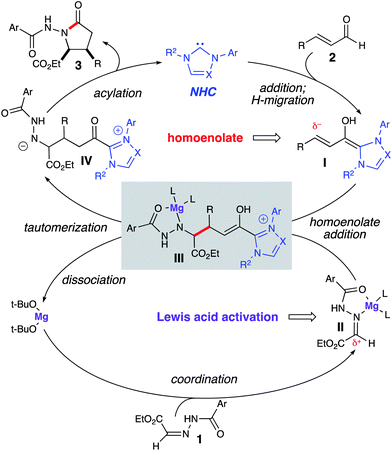 |
| | Scheme 2 Proposed pathway for homoenolate addition to hydrazones | |
Reversal of facial selectivity
In 2006, Nair reported the racemic synthesis of trans 1,3,4-trisubstituted cyclopentenes by homoenolate annulation of chalcones.8 A subsequent report by Bode allowed access to optically active cis cyclopentenes using a chiral NHC catalyst.9 The aformentioned process was limited mainly to (E)-ethyl 4-oxo-2-butenoates as the coupling electrophile. When attempting to employ chalcones in the asymmetric variant the transcyclopentene was observed (9![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 dr), but with only moderate enantioselectivity (55% ee). However, excellent enantioselectivity was achieved for the cis diastereomer (99% ee). We viewed this reaction as a good opportunity to explore a NHC/LA cooperative catalytic approach and hypothesized that a LA would allow for activation of the conjugate acceptor and provide increased cis diastereoselectivity through preorganization of the reactants (Scheme 3).10 Our investigation was initiated by examining different Lewis acids. Metals such as zinc or scandium triflate resulted in complete inhibition of this reaction. Mg(Ot-Bu)2, which proved to be effective with homoenolate annulation of N-acyl hydrazones (vide supra), afforded predominately the trans diastereomer (2:1). However, when employing titanium(IV) iso-propoxide (20 mol%), the desired cis cyclopentene (6) (20:1 dr) was isolated in good yield with excellent enantioselectivity (97% ee). Further investigation showed that performing the reaction with substoichiometric amounts of 2-propanol (20 mol%) allowed for higher yields and shorter reaction times. The proposed reaction pathway for this annulation is outlined in Scheme 4. Coordination of titanium(IV) to the enal (4) and 1,2-addition of the deprotonated azolium promotes the formation of the extended Breslow intermediate (I). Subsequent coordination of the chalcone allows for efficient substrate organization and activation of the enone towards the conjugate addition (II). Following C–C bond formation,11,12bis-enolate III undergoes protonation, tautomerization, and an intramolecular aldol forming the cyclopentane ring (IV). An ensuing acylation and decarboxylation cascade affords the cis cyclopentene (6). We hypothesize that 2-propanol increases the reaction rate by adding to the titanium in intermediate IV, thereby allowing the formation of the tertiary alkoxide for the acylation step and catalyst regeneration. Recent DFT calculations by Domingo and coworkers12 has shown that titanium a) lowers the Gibbs free energy for this NHC-catalyzed annulation reaction, and b) makes the cis stereoselective C–C bond-formation (II → III) more favorable than the trans.
:1 dr), but with only moderate enantioselectivity (55% ee). However, excellent enantioselectivity was achieved for the cis diastereomer (99% ee). We viewed this reaction as a good opportunity to explore a NHC/LA cooperative catalytic approach and hypothesized that a LA would allow for activation of the conjugate acceptor and provide increased cis diastereoselectivity through preorganization of the reactants (Scheme 3).10 Our investigation was initiated by examining different Lewis acids. Metals such as zinc or scandium triflate resulted in complete inhibition of this reaction. Mg(Ot-Bu)2, which proved to be effective with homoenolate annulation of N-acyl hydrazones (vide supra), afforded predominately the trans diastereomer (2:1). However, when employing titanium(IV) iso-propoxide (20 mol%), the desired cis cyclopentene (6) (20:1 dr) was isolated in good yield with excellent enantioselectivity (97% ee). Further investigation showed that performing the reaction with substoichiometric amounts of 2-propanol (20 mol%) allowed for higher yields and shorter reaction times. The proposed reaction pathway for this annulation is outlined in Scheme 4. Coordination of titanium(IV) to the enal (4) and 1,2-addition of the deprotonated azolium promotes the formation of the extended Breslow intermediate (I). Subsequent coordination of the chalcone allows for efficient substrate organization and activation of the enone towards the conjugate addition (II). Following C–C bond formation,11,12bis-enolate III undergoes protonation, tautomerization, and an intramolecular aldol forming the cyclopentane ring (IV). An ensuing acylation and decarboxylation cascade affords the cis cyclopentene (6). We hypothesize that 2-propanol increases the reaction rate by adding to the titanium in intermediate IV, thereby allowing the formation of the tertiary alkoxide for the acylation step and catalyst regeneration. Recent DFT calculations by Domingo and coworkers12 has shown that titanium a) lowers the Gibbs free energy for this NHC-catalyzed annulation reaction, and b) makes the cis stereoselective C–C bond-formation (II → III) more favorable than the trans.
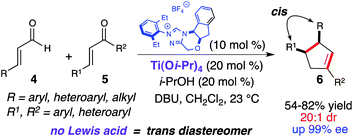 |
| | Scheme 3
Homoenolate annulation with chalcones | |
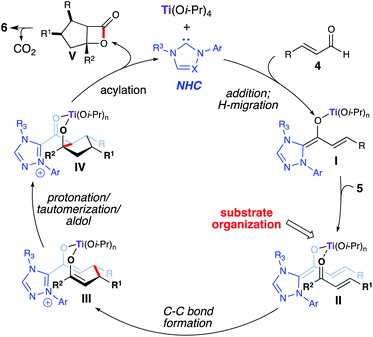 |
| | Scheme 4 Proposed pathway for homoenolate annulation with chalcones | |
Enal
dimerization
Achiral NHC/chiral Lewis acid cooperative catalysis
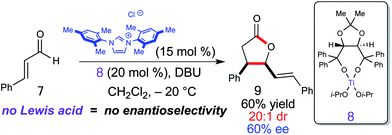
With successful integration of Lewis acids into NHC-catalysis with lactam and cyclopentene formations above, we turned our attention toward controlling chirality by employing chiral Lewis acids and an achiral carbene catalyst. If this approach provided enantiomerically enriched products, this would allow for modulation of enantioselectivity by either chiral Lewis acid, chiral NHC, or both! This aspect of cooperative carbene catalysis is especially attractive, since in many cases the ability of a single catalyst to induce high levels of enantioselectivity can be challenging. The synthesis of γ-butyrolactones using NHC-catalysis has been the subject of many publications over the last several years.13 We chose to investigate an NHC/chiral Lewis acid combination with chiral titanium alkoxides since they are prevalent in the literature and easily prepared.14 When combining cinnamaldehyde (7) with (R,R)-Ti-TADDOL (8) and 15 mol% of IMes-derived carbene at −20 °C only the cis γ-butyrolactone (9) was observed with a 60% ee (Scheme 5).10 Although the enantioinduction is moderate, this proof of concept combining achiral carbenes with chiral Lewis acids offers new opportunities for access to enantioenriched materials through NHC-catalyzed processes.
Reversal of regioselectivity
In 2004, Glorius13a and Bode13b reported the synthesis of γ-butyrolactones by an NHC-catalyzed homoenolate annulations with aldehydes. This reaction can proceed without a second aldehyde or carbonyl compound, thereby leading to dimerization of the starting material. With IMes and DBU, the 1,2-addition product is favored over the 1,4-addition product (Scheme 6, eq. 1).15 Although this reaction is interesting, formation of these aforementioned dimer products (γ-lactone or β-lactone) was not done selectively and control of the regioselecitivity (1,2-additionversus1,4-addition) is not well understood. We anticipated that incorporating an oxophilic Lewis acid in this NHC-catalyzed dimerization might provide a more ordered transition state for the homoenolate addition, as well as activate the coupling enal toward the conjugate addition. Additionally, we hoped to develop a highly enantioselective variant of this dimerization which had not yet been reported (Scheme 6, eq. 2).16
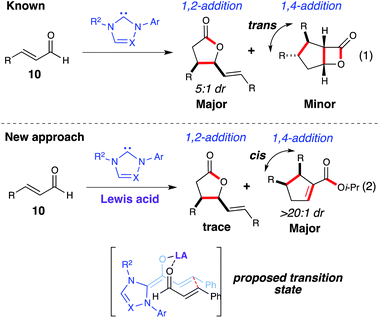 |
| | Scheme 6 NHC-catalyzed enal dimerization | |
Screening of several Lewis-acid alkoxides such as Mg(Ot-Bu)2, Ba(Oi-Pr)2, Zr(Oi-Pr)4, and Sr(Oi-Pr)2 resulted in either decomposition or recovery of the starting material after 48 h. However, the homoenolate 1,4-addition was favored when employing Ti(Oi-Pr)4 in the reaction, furnishing the respective cyclopentene (12) as a single diastereomer (>20:1 dr). This highly diastereoselective dimerization was rendered enantioselective by utilizing a novel tryptophan-derived triazolium (Scheme 7).17 This dimerization worked well with β-aryl substituted enals (11), providing the 2,3,4-trisubstituted cyclopentenes (12) in good yield (59–72%) with excellent diastereoselectivity (>20:1 dr) and enantioselectivity (84–90% ee). As with the homoenolate annulation with chalcones, we believe that the titanium plays a role in substrate preorganization and activation toward the conjugate addition over the 1,2-addition (γ-lactone) (Scheme 6).
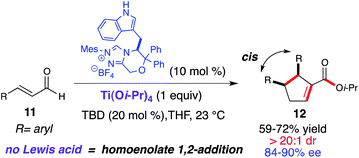 |
| | Scheme 7 NHC-catalyzed enantioselective enal dimerization | |
Activation of a previous inactive electrophile
Our strategy of activating conjugate acceptors with titanium in combination with NHC-catalysis prompted us to investigate other conjugate acceptors that were completely inactive to NHC-catalyzed homoenolate conjugate additions. If successful, this new approach would expand the utility of NHC/Lewis acid methodology and provide other synthetically useful products.
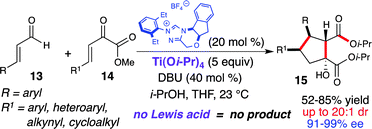
In 2009, Nair reported the NHC-catalyzed formation of racemic β-hydroxycyclopentanecarboxylates by homoenolate annulations to chalcones.18 This work described the simultaneous formation of four contiguous stereogenic centers, albeit with low stereocontrol for the initial bond formation. Based on this work and previous homoenolate additions, we envisioned that the highly reactive β,γ-unsaturated α-ketoesters (14) would also participate in similar reactivity to afford more decorated compounds with a complementary stereochemical outcome. Unfortunately, conjugate addition of the homoenolate intermediate to the β,γ-unsaturated α-ketoester was not observed when just using an NHC catalyst (Scheme 8).19 However, running this reaction with a stoichiometric amount of Ti(Oi-Pr)4 furnished a highly functionalized cyclopentanol (15) in 67% yield with moderate diastereoselectivity (7:1 dr) and excellent enantioselectivity (90% ee). Further investigation revealed that an increase in the titanium loading and the addition of 2-propanol resulted in an increase in yield (84%), diastereoselectivity (20:1 dr), and enantioselectivity (95% ee).
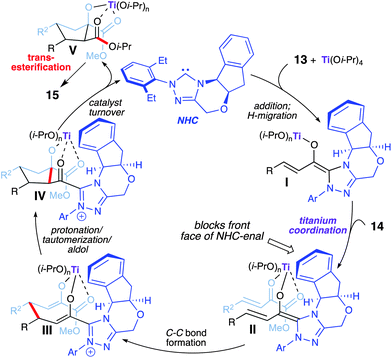
The proposed reaction pathway is shown in Scheme 9. The addition of the NHC to the titanium coordinated enal and a subsequent formal proton transfer promotes the formation of extended Breslow intermediate I. The coordination of the β,γ-unsaturated α-ketoester (14) then occurs from the back face of the carbene-aldehyde-titanium(IV) intermediate thereby allowing for high enantio-induction during the initial C–C bond formation. The fully assembled intermediate II (complex of NHC + Ti + enal + ketoester) undergoes C–C bond formation,11,12 yielding bis-enolate III. An organized protonation, tautomerization, and intramolecular aldol sequence provides the cyclopentane ring (IV). Acylation/catalyst turnover with 2-propanol gives mixed esterV. Finally, transesterification to the diisopropyl ester furnishes cyclopentanol 15. The various titanium–oxygen interactions/ligations within intermediate IV prevents intramolecular acylation, which explains why formation of the β-lactone or the corresponding cyclopentene was not observed.
Lewis acid activation in Brønsted catalysis
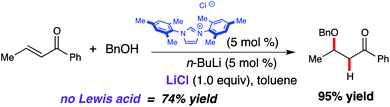
The integration of Lewis acidic metals into NHC-catalyzed reactions is not limited to homoenolate processes. While investigating the conjugate addition of alcohols to enones catalyzed by free carbenes (i.e., fully deprotonated species), it was observed that lithium chloride (LiCl) enhanced the overall yield of the process (Scheme 10).20 While catalytic amounts of LiCl are generated via n-BuLi deprotonation of IMes, sequestering the lithium cation by adding 12-crown-4 resulted in a decreased yield (74% vs. 88%). However, addition of 1.0 equivalent of LiCl resulted in nearly quantitative yield. Although the mechanism of this process is still under investigation, the participation of lithium as a Lewis acid may activate the enone toward the conjugate addition of the alcohol.
Conclusions
The combination of N-hetereocyclic carbene catalysis and Lewis acid activation has open new directions in the area of asymmetric organocatalysis. With a proper, albeit somewhat empirical, choice of Lewis acid, the equilibrium between free species and NHC-LA complex seemingly favors disassociation, subsequent activation, and productive reactivity. Magnesium(II) tert-butoxide was used to enhance the electrophilicity of N-acyl hydrazones, allowing for a more efficient synthesis of γ-lactams. To date, titanium(IV) iso-propoxide has been incorporated into four carbene catalyzed processes. Using titanium cooperative catalysis, we have been able to 1) reverse the facial selectivity in the homoenolate annulation of chalcones, 2) reverse regioselectivity in enal dimerization, 3) introduce moderate enantioselectivity with chiral Ti(IV) Lewis acids and an achiral carbene in the synthesis of γ-butyrolactones, and 4) activate an electrophile (β,γ-unsaturated α-ketoesters) which had previously been unreactive in NHC-generated homoenolate processes. Lastly, lithium cations in NHC reactions have been observed to enhance the yield in Brønsted base-catalyzed conjugate addition of alcohols to enones. Overall, these initial reports have demonstrated the compatibility of mild Lewis acids with strongly nucleophilic N-heterocyclic carbenes. Further investigations are clearly necessary in order to fully understand these systems and provide a comprehensive picture of this catalysis strategy. The unlikely combination of these seemingly incompatible catalysts has provided innovative means for which to modulate the selectivity and reactivity of NHC-catalyzed transformations. Lewis acid integration with carbene catalysis, along with complimentary and equally exciting modes of catalysis (e.g., transition metal and enamine organocatalysis), will undoubtedly provide new avenues for reaction discovery.
Acknowledgements
We thank the National Institute of General Medical Science (R01GM73072) for support of this work. D.T.C. thanks Northwestern University for a GAANN Fellowship (2008–2009) and the ACS Division of Organic Chemistry Graduate for a fellowship sponsored by Organic Syntheses/Organic Reactions (2011–2012).
References
-
(a) D. Enders and T. Balensiefer, Acc. Chem. Res., 2004, 37, 534 CrossRef CAS;
(b) D. Enders, O. Niemeier and A. Henseler, Chem. Rev., 2007, 107, 5606 CrossRef CAS;
(c) V. Nair, S. Vellalath and B. P. Babu, Chem. Soc. Rev., 2008, 37, 2691 RSC;
(d) E. M. Phillips, A. Chan and K. A. Scheidt, Aldrichim Acta, 2009, 43, 55 Search PubMed;
(e) A. T. Biju, N. Kuhl and F. Glorius, Acc. Chem. Res., 2011 DOI:10.1021/ar2000716. For a recent report on the importance of ortho, ortho-disubstituted triazoliums see:
(f) J. Mahatthananchai and J. W. Bode, Chem. Sci., 2011 10.1039/C1SC00397F.
- For reviews on NHC catalysis combined with transition metals, enamine catalysis, and Lewis acids, see:
(a) K. Hirano, I. Piel and F. Glorius, Chem. Lett., 2011, 40, 786 CrossRef CAS;
(b) N. T. Patil, Angew. Chem., Int. Ed., 2011, 50, 1759 CrossRef CAS. For a recent example of Brønsted acid/NHC cooperative catalysis see:
(c) X. Zhao, D. A. DiRocco and T. Rovis, J. Am. Chem. Soc., 2011, 133, 12466 CrossRef CAS.
-
(a) H. Zhou, E. J. Campbell and S. T. Nguyen, Org. Lett., 2001, 3, 2229 CrossRef CAS;
(b) J. Wu, X. Sun, S. Ye and W. Sun, Tetrahedron Lett., 2006, 47, 4813 CrossRef CAS;
(c) T. Nemoto, T. Fukuda and Y. Hamada, Tetrahedron Lett., 2006, 47, 4365 CrossRef CAS;
(d) R. Lebeuf, K. Hirano and F. Glorius, Org. Lett., 2008, 10, 4243 CrossRef CAS;
(e) J. M. He, S. B. Tang, S. C. Tang, J. Liu, Y. Q. Sun, X. F. Pan and X. G. She, Tetrahedron Lett., 2009, 50, 430 CrossRef CAS;
(f) Z. Y. Chen, X. X. Yu and J. Wu, Chem. Commun., 2010, 46, 6356 RSC;
(g) R. S. Reddy, J. N. Rosa, L. F. Veiros, S. Caddick and P. M. P. Gois, Org. Biomol. Chem., 2011, 9, 3126 RSC.
-
(a) S. P. Lathrop and T. Rovis, J. Am. Chem. Soc., 2009, 131, 13628 CrossRef CAS;
(b) D. Enders, A. Grossmann, H. Huang and G. Raabe, Eur. J. Org. Chem., 2011, 4298 CrossRef CAS;
(c) C. B. Jacobsen, K. L. Jensen, J. Udmark and K. A. Jørgensen, Org. Lett., 2011, 13, 4790 CrossRef CAS;
(d) K. E. Ozboya and T. Rovis, Chem. Sci., 2011, 2, 1835 RSC.
- D. Q. Tan, K. S. Martin, J. C. Fettinger and J. T. Shaw, Proc. Natl. Acad. Sci. U. S. A., 2011, 108, 6781 CrossRef CAS.
-
(a) R. B. Lettan, C. V. Galliford, C. C. Woodward and K. A. Scheidt, J. Am. Chem. Soc., 2009, 131, 8805 CrossRef CAS. For γ-lactams by NHC catalysis see:
(b) P.-C. Chiang and J. W. Bode, TCI MAIL, 2011, 149, 2 Search PubMed. For an additional example ref. 2c.
- D. E. A. Raup, B. Cardinal-David, D. Holte and K. A. Scheidt, Nat. Chem., 2010, 2, 766 CrossRef CAS.
- V. Nair, S. Vellalath, M. Poonoth and E. Suresh, J. Am. Chem. Soc., 2006, 128, 8736 CrossRef CAS.
-
(a) P. C. Chiang, J. Kaeobamrung and J. W. Bode, J. Am. Chem. Soc., 2007, 129, 3520 CrossRef CAS;
(b) P. C. Chiang, M. Rommel and J. W. Bode, J. Am. Chem. Soc., 2009, 131, 8714 CrossRef CAS.
- B. Cardinal-David, D. E. A. Raup and K. A. Scheidt, J. Am. Chem. Soc., 2010, 132, 5345 CrossRef CAS.
- L. R. Domingo, R. J. Zaragoza and M. Arno, Org. Biomol. Chem., 2010, 8, 4884 CAS.
- L. R. Domingo, R. J. Zaragoza and M. Arno, Org. Biomol. Chem., 2011, 9, 6616 CAS.
-
(a) C. Burstein and F. Glorius, Angew. Chem., Int. Ed., 2004, 43, 6205 CrossRef CAS;
(b) S. S. Sohn, E. L. Rosen and J. W. Bode, J. Am. Chem. Soc., 2004, 126, 14370 CrossRef CAS;
(c) W. Ye, G. L. Cai, Z. Y. Zhuang, X. S. Jia and H. B. Zhai, Org. Lett., 2005, 7, 3769 CrossRef CAS;
(d) V. Nair, S. Vellalath, M. Poonoth, R. Mohan and E. Suresh, Org. Lett., 2006, 8, 507 CrossRef CAS;
(e) V. Nair, S. Vellalath, M. Poonoth, E. Suresh and S. Viji, Synthesis, 2007, 3195 CrossRef CAS;
(f) K. Hirano, I. Piel and F. Glorius, Adv. Synth. Catal., 2008, 350, 984 CrossRef CAS;
(g) Y. Ishida, Y. Matsuoka and K. Saigo, Tetrahedron Lett., 2008, 49, 2985 CrossRef;
(h) Y. Ishida, Y. Matsuoka, D. Sasaki and K. Saigo, Chem.–Eur. J., 2008, 14, 9215 CrossRef;
(i) Y. Li, Z.-A. Zhao, H. He and S.-L. You, Adv. Synth. Catal., 2008, 350, 1885 CrossRef CAS;
(j) L.-H. Sun, L.-T. Shen and S. Ye, Chem. Commun., 2011, 47, 10136 RSC.
-
(a) R. O. Duthaler and A. Hafner, Chem. Rev., 1992, 92, 807 CrossRef CAS;
(b) D. Seebach, A. K. Beck and A. Heckel, Angew. Chem., Int. Ed., 2001, 40, 92 CrossRef CAS.
- M. Wadamoto, E. M. Phillips, T. E. Reynolds and K. A. Scheidt, J. Am. Chem. Soc., 2007, 129, 10098 CrossRef CAS.
- D. T. Cohen, B. Cardinal-David, J. M. Roberts, A. A. Sarjeant and K. A. Scheidt, Org. Lett., 2011, 13, 1068 CrossRef CAS.
- B. E. Maki, A. Chan and K. A. Scheidt, Synthesis, 2008, 1306 CAS.
-
(a) V. Nair, B. P. Babu, S. Vellalath, V. Varghese, A. E. Raveendran and E. Suresh, Org. Lett., 2009, 11, 2507 CrossRef CAS;
(b) J. W. Bode and J. Kaeobamrung, Org. Lett., 2009, 11, 677 CrossRef.
- D. T. Cohen, B. Cardinal-David and K. A. Scheidt, Angew. Chem., Int. Ed., 2011, 50, 1678 CrossRef CAS.
- E. M. Phillips, M. Riedrich and K. A. Scheidt, J. Am. Chem. Soc., 2010, 132, 13179 CrossRef CAS.
|
| This journal is © The Royal Society of Chemistry 2012 |
Click here to see how this site uses Cookies. View our privacy policy here.