Transducing methyltransferase activity into electrical signals in a carbon nanotube–DNA device†
Hanfei
Wang
a,
Natalie B.
Muren
b,
David
Ordinario
c,
Alon A.
Gorodetsky
c,
Jacqueline K.
Barton
*b and
Colin
Nuckolls
*a
aDepartment of Chemistry, Columbia University, New York, NY 10027, USA. E-mail: cn37@columbia.edu; Fax: (+1) 212-932-1289
bDivision of Chemistry and Chemical Engineering, California Institute of Technology, Pasadena, CA 91125, USA. E-mail: jkbarton@caltech.edu; Fax: (+1) 626-577-4976
cDepartment of Chemical Engineering & Materials Science, University of California, Irvine, CA 92697, USA
First published on 20th October 2011
Abstract
This study creates a device where the DNA is electronically integrated to serve as both the biological target and electrical transducer in a CNT–DNA–CNT device. We detect DNA binding and methylation by the methyltransferase M.SssI at the single molecule level. We demonstrate sequence-specific, reversible binding of M.SssI and protein-catalyzed methylation that alters the protein-binding affinity of the device. This device, which relies on the exquisite electrical sensitivity of DNA, represents a unique route for the specific, single molecule detection of enzymatic activity.
Introduction
Here we describe a molecular-scale electrical device that can sense an individual reaction between a DNA duplex and a methyltransferase enzyme. The device consists of a DNA duplex that is chemically wired between carbon nanotube (CNT) leads and can be operated as a field effect transistor (Fig. 1). In general, CNTs and nanowires incorporated in field-effect transistors offer valuable detection platforms because their conductivity is modulated by binding of analytes.1–5 One drawback of these devices is that the conductivity changes are not specific and depend upon the characteristics of the analyte and its mode of interaction with the CNT.6,7 Interference associated with non-specific binding of analytes presents a challenge to detection, particularly in single molecule measurements. We and others have previously shown that single wall CNT devices provide a general means to measure the electrical properties of single molecules incorporated into an oxidatively cut gap in the CNT.8–16 These measurements can be carried out in aqueous solution, making them useful for detection of biomolecules. In one such experiment, a DNA duplex wired into the gap in the CNT was shown to be sensitive to perturbations in base stacking,16 consistent with observations made using ensemble electrochemical studies.17–20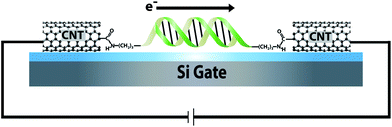 | ||
| Fig. 1 Electronic integration of DNA into a CNT device. Oxidative cutting of the CNT creates a gap with carboxylic acid point contacts on the cut ends. Amine-modified DNA is then covalently attached to these point contacts by peptide coupling and forms an electronic bridge across the gap. The resulting DNA-mediated path for current flow through the device is sensitive to perturbations that disrupt charge transport. | ||
In this study, we probe the specific reaction of a methlytransferase. The methyltransferase M.SssI binds the sequence 5′-CG-3′ and, in the presence of the cofactor S-adenosyl methionine (SAM), methylates the cytosine base.21Methyltransferases such as M.SssI, and the closely related M.HhaI, perform this reaction on DNA through a base flipping mechanism that is clearly visualized from the crystal structure of M.HhaI bound to DNA with its cytosine base completely rotated out of the π-stacked core of the DNA duplex.22,23 In electrochemical studies this base flipping action interrupts charge transport through DNA.24 In this study, we show that CNT–DNA–CNT devices that contain a conductive, bridging segment of DNA duplex can also detect binding and base flipping by M.SssI by disruption of DNA-mediated charge transport.
Results and discussion
Device preparation
To fabricate the devices used in this study we utilized our previously published procedure.11–16,25,26Chemical vapor deposition is used to grow sparse single walled CNTs on a doped silicon wafer containing 300 nm of native oxide on its surface. Electron beam lithography then opens a nanoscale window in PMMA over a section of the nanotube. The exposed portion of the tube can then be cut with an oxidative plasma etch, leaving carboxylic acids on the termini of the tubes. The nanotubes were treated with a 2-(N-morpholino)ethansulfonic acid (MES) buffer solution containing the amide activating/coupling agents EDCI and Sulfo-NHS, activating the carboxylic acids for amide coupling with 3′- and 5′-amine-modified DNA, and in so doing, covalently bridging the gap in the CNT with a single DNA duplex (Fig. 1). We perform electrical characterization of the devices in ambient conditions with the silicon wafer serving as a global back gate.Sensing methyltransferase activity
We record measurements as a function of the methyltransferase binding and reactivity to evaluate the current flow through the device. The scheme for a typical M.SssI binding experiment on a DNA-bridged device is given in Fig. 2, and the corresponding conductance-voltage characteristics are given in Fig. 3. For these initial experiments the DNA sequence has more than one 5′-CG-3′ binding site for M.SssI. However, given the footprint of M.SssI which spans at least 6 base pairs on each side of the binding site,27 the 15-mer DNA segment would only accommodate binding by a single M.SssI protein at a central site. We observe an average conductance of 1.4 ± 0.8 μS for the 3 devices tested (Fig. 3, left column). Upon incubation of these devices with 6 nM M.SssI, as well as 300 μM of the requisite SAM cofactor, we observed a 91 ± 15% reduction in the conductance. Notably, the M.SssI concentration utilized for our experiments is just below the binding affinity of this enzyme (Kd = 11 nM).28 The crystal structure of M.HhaI bound to methylated DNA shows that base flipping occurs even when the protein is bound to this substrate,23 although the binding affinity is reduced.29–31 In these single-molecule devices, it is apparent that the protein remains bound with the base flipped out, even after the methylation reaction is complete. Washing these devices with buffer is necessary to dissociate the enzyme and recover the conductance to close to the initial value.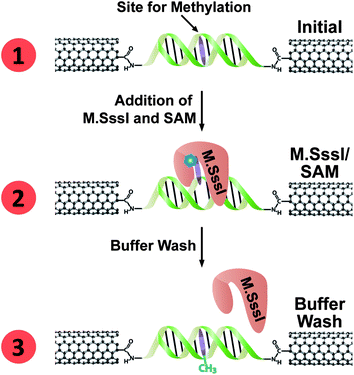 | ||
| Fig. 2 Electrical detection of M.SssI binding at a DNA-bridged CNT device. (1) A DNA segment containing the M.SssI binding site (with target base to be methylated shown in purple) forms a conductive bridge between the two ends of a gap cut in a CNT device. (2) Upon addition of M.SssI and SAM cofactor (represented by the blue hexagon), the methyltransferase binds the DNA at its recognition site, and flips the base to be methylated out of the DNA π-stack, thereby cutting off charge transport through the DNA. M.SssI remains bound with the base flipped even after the methylation reaction is complete. (3) Upon rinsing, M.SssI dissociates from the DNA; the methylated base re-inserts into the DNA π-stack and restores charge transport through the device. | ||
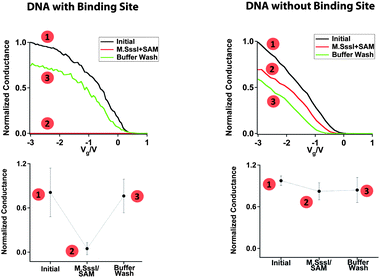 | ||
| Fig. 3 Sequence specific M.SssI binding and disruption of charge transport through the DNA molecules. For each, the numbered points correspond to the numbered steps illustrated in Fig. 2. Left column: M.SssI binding at a DNA-bridged device containing the M.SssI recognition site. The sequence was H2N-5′-CGGCCCGGCCGCGCG-3-NH2. Right column: Lack of M.SssI binding at a DNA-bridged device that does not contain the M.SssI recognition site. The sequence was H2N-5′-ATTAAATTAATATAT-3′-NH2. Typical normalized conductance curves (top) and average relative conductance values (bottom) are shown for the respective DNA-bridged devices in (1) buffer before addition of M.SssI, (2) buffer with M.SssI and SAM, and (3) buffer after M.SssI has been rinsed away. The buffer conditions were 50 mM NaCl, 10 mM Tris-HCl, 10 mM MgCl2, 1 mM Dithiothreitol, pH 7.9. The conductance of each device was normalized with respect to the highest conductance value. | ||
To test for sequence-specific binding, we bridge the nanotube gap with a sequence that lacks the M.SssI binding site (Fig. 3, right column). Before treatment with the methyltransferase, the devices have an average conductance 1.5 ± 0.4 μS. Even after extended incubation (>1 h) of these devices with M.SssI and the SAM cofactor, we did not see any dramatic changes in the characteristics. Clearly, the correct sequence is necessary for the M.SssI binding and subsequent base flipping that shuts off current flow through the device.
The cofactor SAM is required for the methyltransferase activity, because it is the source of the electrophilic methyl group and is necessary for the specific binding of the enzyme to the substrate. We tested the conductance of the devices in the absence and presence of SAM. To simplify the analysis, we utilized devices that only have one centrally located 5′-CG-3′ binding site for M.SssI. When a DNA-bridged device was incubated with M.SssI in the absence of the SAM cofactor, we observed a small attenuation in its conductance. This may be due to the introduction of more scattering sites in the channel of the device upon protein binding.12 However, when the same device was subsequently incubated with M.SssI in the presence of the SAM cofactor, we observed a >90% attenuation of its conductance. In turn, the conductance of the device could be recovered to its original value with a buffer wash. This is shown in Fig. S1.†
We next investigate how methylation of the M.SssI binding site influences the measurement of subsequent activity by M.SssI. To address this question, we again utilize devices with a single M.SssI binding site and first expose these devices to M.SssI and SAM in order to methylate the site. Similar to the experiments described above, we observe a large decrease in the current flow through the device that recovers to close to its original conductivity when the protein is washed away (Fig. 4, steps 1–4). If we then re-subject this now methylated device to M.SssI and SAM, we see no change in the conductance (Fig. 4, Steps 5–7). This result reflects the reduced affinity of M.SssI for methylated DNA. Initially, the 6 nM concentration of M.SssI is high enough for binding and base flipping to occur at the non-methylated substrate. With the protein washed away, the added methyl group itself does not inhibit DNA charge transport as it does not perturb the base stack. When the device is re-subjected to 6 nM M.SssI, however, this concentration is no longer high enough to initiate binding to the now methylated DNA. These changes in the binding potential of the DNA are sensitively translated here from the single molecule level as an electrical output; chemical modification of the DNA wire by the protein alters subsequent protein binding and the resulting electrical response.
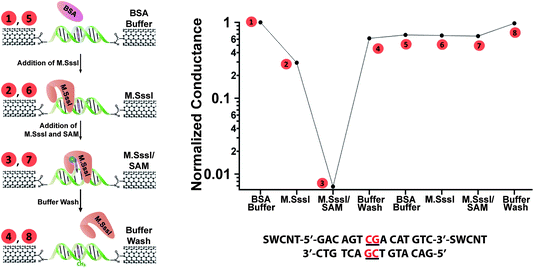 | ||
| Fig. 4 M.SssI-catalyzed DNA methylation alters the protein-binding affinity of the device. A device containing a single M.SssI binding site was taken through the illustrated steps (left) and the corresponding relative conductances (top right) were measured in (1) buffer containing 10 nM BSA before addition of M.SssI, (2) buffer and M.SssI without SAM, (3) buffer and M.SssI with SAM (represented by the blue hexagon), and (4) buffer after M.SssI has been rinsed away. Note that current attenuation is only observed for step (3). After methylation, the device was taken through the same sequence of steps (5)–(8) with no attenuation observed. The sequence was H2N-5′-GACAGTCGACATGTC -3′-NH2, with the single 5′-CG-3′ binding site located in the middle. The buffer conditions were 50 mM NaCl, 10 mM Tris-HCl, 10 mM MgCl2, 1 mM Dithiothreitol, pH 7.9. | ||
Taken together, these experiments show that the DNA segment that has been covalently wired into these devices is simultaneously a sensitive recognition element and transducer of methyltransferase binding. The base flipping by M.SssI at its specific recognition sequence disrupts charge transport through the DNA base pair π-stack and causes nearly complete attenuation of the device conductance. These devices can be used to sensitively distinguish DNA that lacks the 5′-CG-3′ binding site, as M.SssI does not bind and base flip this DNA as well as DNA that contains the binding site. This device represents a unique and promising route for specific, single molecule measurements of binding and activity by methyltransferases and other DNA-binding proteins.
Conclusion
Thus, we demonstrate, for the first time, DNA binding and methylation by the methyltransferase M.SssI at the single molecule level using an electrical device. These studies take the next step in complexity up from our earlier studies in which we used these devices to detect DNA cutting by a restriction enzyme.16 Unlike these previous studies in which detection was achieved by irreversible destruction of the device, here we detect the sequence-specific binding and activity of M.SssI in a way that leaves the device intact, yet reports on the enzymatic reaction. We observe the result of the chemical modification to DNA carried out by M.SssI, which alters the protein binding affinity of the device but not its conductivity. Thus, this work generalizes our CNT platform for measurements of binding and activity by other proteins whose activity disrupts DNA-mediated charge transport such as transcription factors and enzymes that perturb the DNA base pair π-stack upon binding and reaction.Acknowledgements
We are grateful to the National Institute of Health (GM61077 to JKB), the Office of Naval Research (N00014-09-1-1117 to JKB and CN), the National Science Foundation NIRT (ECCS-07-07748 to CN), and the National Science Foundation (CHE0936923 to AAG) for their financial support.Notes and references
- G. Zheng, F. Patolsky, Y. Cui, W. U. Wang and C. M. Lieber, Nat. Biotechnol., 2005, 23, 1294 CrossRef CAS.
- R. J. Chen, S. Bangsaruntip, K. A. Drouvalakis, N. W. S. Kam, M. Shim, Y. Li, W. Kim, P. J. Utz and H. Dai, Proc. Natl. Acad. Sci. U. S. A., 2003, 100, 4984 CrossRef CAS.
- F. Patolsky, G. Zheng and C. M. Lieber, Anal. Chem., 2006, 78, 4260 CrossRef CAS.
- S. Sorgenfrei, C.-Y. Chiu, R. L. Gonzalez Jr, Y.-J. Yu, P. Kim, C. Nuckolls and K. L. Shepard, Nat. Nanotechnol, 2011, 11, 1093 Search PubMed.
- D. Kauffman and A. Star, Chem. Soc. Rev., 2008, 37, 1197 RSC.
- K. Balasubramanian and M. Burghard, Anal. Bioanal. Chem., 2006, 385, 452 CrossRef CAS.
- C. B. Jacobs, M. J. Peairs and B. J. Venton, Anal. Chim. Acta, 2010, 662, 105 CrossRef CAS.
- S. Liu, X. Zhang, W. Luo, Z. Wang, X. Guo, M. L. Steigerwald and X. Fang, Angew. Chem., Int. Ed., 2011, 50, 2496 CrossRef CAS.
- S. Liu, G. H. Clever, Y. Takezawa, M. Kaneko, K. Tanaka, X. Guo and M. Shionoya, Angew. Chem., Int. Ed., 2011, 50, 8886 CrossRef CAS.
- S. Roy, H. Vedala, A. D. Roy, D. Kim, M. Doud, K. Mathee, H. Shin, N. Shimamoto, V. Prasad and W. Choi, Nano Lett., 2008, 8, 26 CrossRef CAS.
- X. Guo, J. P. Small, J. E. Klare, Y. Wang, M. S. Purewal, I. W. Tam, B. H. Hong, R. Caldwell, L. Huang, S. O'Brien, J. Yan, R. Breslow, S. J. Wind, J. Hone, P. Kim and C. Nuckolls, Science, 2006, 311, 356 CrossRef CAS.
- A. C. Whalley, M. L. Steigerwald, X. Guo and C. Nuckolls, J. Am. Chem. Soc., 2007, 129, 12590 CrossRef CAS.
- X. Guo, A. Whalley, J. E. Klare, L. Huang, S. O'Brien, M. Steigerwald and C. Nuckolls, Nano Lett., 2007, 7, 1119 CrossRef CAS.
- A. Feldman, M. Steigerwald, X. Guo and C. Nuckolls, Acc. Chem. Res., 2008, 41, 1731 CrossRef CAS.
- X. Guo and C. Nuckolls, J. Mater. Chem., 2009, 19, 5470 RSC.
- X. Guo, A. A. Gorodetsky, J. Hone, J. K. Barton and C. Nuckolls, Nat. Nanotechnol., 2008, 3, 163 CrossRef CAS.
- S. O. Kelley, R. E. Holmlin, E. D. A. Stemp and J. K. Barton, J. Am. Chem. Soc., 1997, 119, 9861 CrossRef CAS.
- S. O. Kelley, E. M. Boon, J. K. Barton, N. M. Jackson and M. G. Hill, Nucleic Acids Res., 1999, 27, 4830 CrossRef CAS.
- S. O. Kelley and J. K. Barton, Science, 1999, 283, 375 CrossRef CAS.
- J. C. Genereux and J. K. Barton, Chem. Rev., 2010, 110, 1642 CrossRef CAS.
- P. Renbaum, D. Abrahamove, A. Fainsod, G. Wilson, S. Rottem and A. Razin, Nucleic Acids Res., 1990, 18, 1145 CrossRef CAS.
- S. Klimasauskas, S. Kumar, R. J. Roberts and X. Cheng, Cell, 1994, 76, 357 CrossRef CAS.
- M. O'Gara, S. Klimasauskas, R. Roberts and X. Cheng, J. Mol. Biol., 1996, 261, 634 CrossRef CAS.
- E. M. Boon, J. E. Salas and J. K. Barton, Nat. Biotechnol., 2002, 20, 282 CrossRef CAS.
- X. Guo, S. Xiao, M. Myers, Q. Miao, M. Steigerwald and C. Nuckolls, Proc. Natl. Acad. Sci. U. S. A., 2009, 106, 691 CrossRef CAS.
- X. Guo, M. Myers, S. Xiao, M. Lefenfeld, R. Steiner, G. Tulevski, J. Tang, J. Baumert, F. Leibfarth, J. Yardley, M. Steigerwald, P. Kim and C. Nuckolls, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 11452 CrossRef CAS.
- A. Razin and P. Renbaum, J. Mol. Biol., 1995, 248, 19 CrossRef.
- M. V. Darii, O. V. Kirsanova, V. L. Drutsa, S. N. Kochetkov and E. S. Gromova, Mol. Biol., 2007, 41, 110 CrossRef CAS.
- R. J. Roberts and S. Klimasauskas, Nucleic Acids Res., 1995, 23, 1388 CrossRef.
- S. Klimasauskas, E. Weinhold, S. Serva, E. Merkiene and G. Vilkaitis, J. Biol. Chem., 2001, 276, 20924 CrossRef.
- A. K. Dubey and R. J. Roberts, Nucleic Acids Res., 1992, 20, 3167 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c1sc00772f |
| This journal is © The Royal Society of Chemistry 2012 |