Integration of rapid DNA hybridization and capillary zone electrophoresis using bidirectional isotachophoresis†
Supreet S.
Bahga‡
,
Crystal M.
Han‡
and
Juan G.
Santiago
*
Department of Mechanical Engineering, Stanford University, 440 Escondido Mall, Bldg. 530, Room 225, Stanford, CA 94305, USA. E-mail: juan.santiago@stanford.edu; Fax: +650 723 7657; Tel: +650 723 5689
First published on 15th October 2012
Abstract
We present a method for rapid, sequence-specific detection of multiple DNA fragments by integrating isotachophoresis (ITP) based DNA hybridization and capillary zone electrophoresis (CZE) using bidirectional ITP. Our method leverages the high preconcentration ability of ITP to accelerate slow, second-order DNA hybridization kinetics, and the high resolving power of CZE to separate and identify reaction products. We demonstrate the speed and sensitivity of our assay by detecting 5 pM, 39 nt ssDNA target within 3 min, using a molecular beacon probe. We also demonstrate the feasibility of our assay for multiplexed detection of multiple-length ssDNA targets by simultaneously detecting 39 and 90 nt ssDNA targets.
DNA hybridization is essential to a wide range of diagnostics and biological sample processing steps. For example, DNA hybridization is deployed in methods for genetic profiling1 and many nucleic acid based pathogen detection assays.2 However, slow, second-order hybridization kinetics at low concentrations of DNA samples often results in long analysis times,3 limiting assay speed and applicability.
We here present a novel combination of two recently demonstrated methods for DNA analysis. The first is the use of isotachophoresis (ITP) to achieve rapid, sequence-specific DNA hybridization.4 The second is bidirectional ITP which uses a strong counter-migrating pH gradient across a cationic ITP interface to disrupt ITP preconcentration and trigger capillary zone electrophoresis separation (CZE).5,6 This combination is unique and relevant as it integrates the rapid hybridization enabled by ITP with high resolution separation of reaction products by CZE. ITP can rapidly mix and preconcentrate single-stranded DNA (ssDNA) fragments within a small (order 10 pL) reaction zone, thus accelerating the rate of hybridization. The method was first proposed by Goet et al.,7 and its experimental demonstration was later presented by Persat and Santiago8 who applied it to the profiling of micro-RNA. Bercovici et al.9 applied ITP based hybridization to the extraction and detection of 16S ribosomal RNA (rRNA) of E. coli in human urine samples. Recently, Bercovici et al.4 presented a detailed study of the physicochemical processes which govern DNA hybridization under ITP focusing. The latter study used molecular beacons10 (MBs) to quantify reaction rates, and demonstrated 14![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000-fold reduction in hybridization time.
000-fold reduction in hybridization time.
MBs provide a sequence-specific fluorescence signal increase upon hybridization,10 but also limit sensitivity and dynamic range of assay due to the background signal from unreacted MBs.11 We here present a method that directly addresses the issue of a relatively high background signal in ITP based hybridization assays by effecting CZE separation to separate unreacted labelled probes from target–probe hybrids. Further, CZE separation of DNA reaction products provides a simple way to extend the functionality of ITP based hybridization assays for multiplexed, sequence-specific detection of nucleic acids. We trigger CZE separation using bidirectional ITP which uses the interaction of anionic and cationic ITP fronts to effect strong changes in the physicochemical environment of the focused analyte zone (see Bahga et al.5,6). Here, the interaction of these fronts disrupts ITP focusing of analytes (products of DNA hybridization reaction) and initiates CZE. Analysis of DNA hybridization reaction products using CZE was originally demonstrated by Chen et al.12 as a method for removal of the background signal from the products of hybridization reaction between a synthetic oligonucleotide and a fluorescently labelled probe. Later, Bianchi et al.13 applied hybridization and CZE on polymerase chain reaction (PCR) products to detect HIV-1 genomic sequences. However, these and other previous studies performed a slow, off-line DNA hybridization followed by manual transfer and loading of the hybridization products into an electrophoresis setup.
Our combination integrates rapid ITP-hybridization and CZE separation in a single, integrated process where no manual steps are required after initiating ITP. We demonstrate the speed and sensitivity of our assay by detecting 5 pM ssDNA target within 3 min, using a molecular beacon probe. We also discuss the applicability of our technique for multiplexed detection of variable length ssDNA targets.
In ITP, analytes focus between zones of high effective mobility leading electrolyte (LE) ions and low effective mobility trailing electrolyte (TE) ions. ITP is characterized by self-sharpening and electromigrating interfaces (or ion-concentration shock waves14) between adjacent zones, which prevent focused analytes from diffusing over time. Our assay uses bidirectional ITP5,15 in a single channel to interact anionic and cationic ITP shock waves, and leverages this interaction to trigger CZE. As reviewed by Bahga et al.,5 this approach differs significantly from the traditional applications of bidirectional ITP such as the work of Thormann et al.16 and Hirokawa et al.,17,18 who used it for simultaneous isotachophoretic analysis of anions and cations. In contrast to our assay, anionic and cationic ITP shock waves in the experiments of Thormann et al.16 and Hirokawa et al.17,18 propagate away from each other, and so do not interact. Our experiments require two oppositely charged pairs of LE and TE ions. Here we term these ions LE+, TE+, LE−, and TE−. LE and TE denote the leading and trailing electrolyte ions, respectively, and + and − correspond to cations and anions, respectively.
Fig. 1a–c show schematics of our technique and protocol. We fill the separation channel with a mixture of LE− and LE+ ions. We fill the right (anodic) reservoir with the TE+/LE− mixture, and similarly the left (cathodic) reservoir with a mixture of LE+, TE−, ssDNA target, and molecular beacon probe. When an electric field is applied along the channel (Fig. 1b), the target and the probe preconcentrate, mix, and react at the interface of LE− and TE− zones while propagating rightwards. Simultaneously, a cationic ITP shock forms between LE+ and TE+ zones near the right reservoir, and propagates leftwards. Anionic ITP preconcentration of the target and molecular beacon probe results in rapid hybridization and corresponding increase in the fluorescence signal. Later, when the anionic and the cationic ITP shocks interact (Fig. 1c), LE+ is replaced with TE+ as the counter-ion for anionic ITP. This counter-ion exchange decreases the local pH of anionic ITP zones to a value at which effective mobility of LE− becomes significantly lower than the mobilities of all DNA fragments. Thus, shock wave interaction causes the target, probe, and target–probe hybrid to migrate into the LE zone, triggering electrophoretic separation of the products of hybridization and unreacted species (see Bahga and Santiago6 for a review of methods to integrate ITP and CZE). The electropherogram signal shows peaks corresponding to the target–probe hybrid and the unreacted probe (the unreacted target has no attached fluorophore). The separation of products removes the background signal associated with unreacted MB probes, and enables simultaneous identification of multiple length targets.
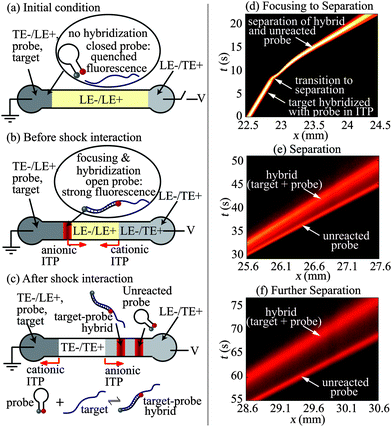 | ||
| Fig. 1 Schematic (a–c) and experimental visualization (d–f) of preconcentration and hybridization of nucleic acids followed by electrophoretic separation using bidirectional ITP. (a) The channel is initially filled with LE+/LE− mixture. The left (cathodic) reservoir is filled with a mixture of TE−, LE+, ssDNA target and probe (molecular beacon). The right (anodic) reservoir is filled with the TE+/LE− mixture. (b) When voltage is applied, the target and the probe focus at the anionic ITP interface and propagate rightwards. Simultaneously, a cationic ITP interface forms near the right reservoir and propagates leftwards. Anionic ITP preconcentration dramatically accelerates hybridization between the target and probe. (c) At a later time, the cationic ITP interface interacts with focused hybridization products, and triggers CZE separation. The resulting electropherogram shows peaks corresponding to the fluorescent unreacted probe and the target–probe hybrid. (d–f) Spatiotemporal plots showing the intensity of fluorescent probe in channel versus the distance along the channel axis, x, and time, t. (d) Initially (t < 10 s), ssDNA target and MB probe mix, focus, and hybridize in anionic ITP. At x = 23 mm and t = 10 s, a counter migrating cationic ITP interface interacts with the focused analyte zones, initiating CZE separation. (e and f) Resolved peaks of fluorescent hybrid and unreacted probe in the CZE mode. | ||
Here, we chose the following electrolyte chemistry: 150 mM sodium bicarbonate for the LE+/LE− mixture; 10 mM sodium hydroxide and 34 mM Hepes for the LE+/TE− mixture; and 100 mM pyridine and 50 mM hydrochloric acid for the TE+/LE− mixture (cationic TE). Note that the cationic TE mixture is titrated with hydrochloric acid instead of carbonic acid. However this does not affect our bidirectional ITP experiments as chloride ion remains in the anodic reservoir and does not enter the separation channel. To the LE+/LE− mixture, we added 1% w/w of hydroxyl ethyl cellulose (HEC) to serve as a sieving matrix for DNA separations. We also added 1% w/w polyvinylpyrrolidone to all electrolyte solutions to suppress electroosmotic flow.19 As explained in the ESI†, this electrolyte chemistry enables coupling of ITP and CZE via shock interaction in bidirectional ITP. Briefly, interaction of ITP shocks replaces sodium (LE+, pKa,+1 = 13.7) with pyridine (TE+, pKa,+1 = 5.2) as the counter-ion for anionic ITP, and very quickly decreases the pH of anionic ITP zones from approximately 8 to 5.5. At this lower pH, the mobility of LE− (carbonic acid) drops significantly below the mobility of nucleic acids resulting in disruption of ITP and initiation of CZE.
We performed bidirectional ITP experiments involving hybridization between the MB probe and two synthetic ssDNA targets (39 and 90 nt) to demonstrate rapid DNA hybridization and separation. The 5′ and 3′ terminals of the MB probe were labelled with Cy5 and Black Hole Quencher 2 (BHQ2), respectively: 5′-/Cy5/CCG AGC [CAT CGT TTA CGG CGT GGA CTA CCA GGG] GCT CGG/BHQ2/-3′. Here, 27 bases within the brackets indicate a probe sequence complementary to a part of the 16S rRNA of E. coli bacteria.9 The 39 and 90 nt ssDNA targets have a common 27 nt long portion of sequence which is perfectly complementary to the bracketed probe sequence. See ESI† for the complete sequences of the 39 and 90 nt ssDNA targets.
We first performed bidirectional ITP experiments to visualize and demonstrate ITP hybridization followed by CZE separation of the reaction products. Details of experimental setup and protocol are provided in the ESI†. For these visualizations, we used relatively high concentrations of a 90 nt ssDNA target (20 nM) and a 39 nt MB probe (500 nM) so as to capture full field images using a CCD camera. Fig. 1d–f show representative spatiotemporal plots of the experimentally measured fluorescence intensity in the channel (scalar) versus distance along the channel (abscissa) and time (ordinate). Fig. 1d shows that, prior to the interaction of anionic and cationic ITP shocks (t < 10 s), the ssDNA target and MB probe mix, focus, and hybridize in a narrow anionic ITP zone. The focused analyte zone in anionic ITP shows a strong signal and migrates rightwards at a constant velocity (i.e., a positive slope for t < 10 s in Fig. 1d). The strong fluorescence signal during ITP is a result of preconcentration as well as hybridization-induced increase in the fluorescence signal from the MB probe. At about t = 10 s, the focused ssDNA target and MB probe interact with a counter-migrating cationic ITP front, and within less than 1 s previously focused analytes begin to separate in CZE mode. Initiation of separation is indicated by the sudden change in the migration speed (inverse of the slope in t vs. x plot) of the fluorescent zone around t = 10 s. Fig. 1e and f show separation of the unreacted probe and the target–probe hybrid at later times. As is usual with CZE, the analyte zones diffuse as they gradually separate over time. Note that in Fig. 1e and f separated peaks corresponding to the unreacted MB probe with quenched fluorescence and the target–probe hybrid yield a comparable fluorescence signal. This is a consequence of the excess amount of MB probe with incomplete fluorescence quenching; inefficiencies in fluorescence quenching of MBs typically result only in an order of 50-fold reduction in fluorescent intensity.10
Next, we performed experiments to demonstrate rapid and high-sensitivity sequence-specific detection of a single 39 nt ssDNA target using our assay. We fixed the 39 nt MB probe concentration at 200 pM, and varied the target concentration from 5 to 30 pM. The total assay time for each experiment was less than 3 min, including 120 s of hybridization time (40 s in unidirectional ITP) prior to CZE separation. From the model of Bercovici et al.,4 we estimate that ITP here increased the hybridization rate by a factor of about 5000. That is, the approximately 40% completion of the DNA hybridization in our 120 s hybridization would otherwise require 7 days to achieve the same degree of completion using off-line hybridization. The inset of Fig. 2 shows a representative electropherogram (raw experimental data) for the case of 30 pM ssDNA target. The left and the right peaks correspond to regions containing the unreacted probe and the target–probe hybrid, respectively. Excess, unreacted MB probe and inefficiencies in its fluorescence quenching yield peak fluorescent intensity comparable to that of the target–probe hybrid peak. Quantifying fluorescence from the target–probe hybrid in such a case is extremely difficult without removal of the background signal from the unreacted probe. The electropherogram shown in the inset of Fig. 2 also highlights the high resolving power of this integrated ITP-CZE separation; here we separate and resolve single-stranded 39 nt probe from its double-stranded hybrid of the same length within about 50 s of separation time.
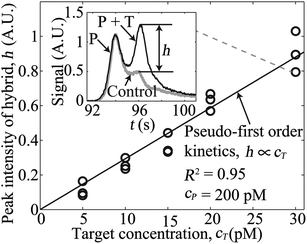 | ||
| Fig. 2 Variation of peak intensity of the target–probe hybrid for five initial concentrations of a 39 nt ssDNA target. We varied the target concentration from 5 to 30 pM, while keeping the probe concentration fixed at 200 pM. The inset plot shows representative raw, electropherogram signals observed after hybridization and separation. The grey curve is a negative control (no target), and the black curve is for the case of 30 pM initial target concentration. Here, time t = 0 corresponds to the onset of bidirectional ITP following 40 s of unidirectional ITP. The signal (black curve) shows two peaks corresponding to the unreacted probe (P) and the target–probe hybrid (P + T). The contribution to signal from the target–probe hybrid is denoted by the signal component h as shown. The main plot shows how observed signal, h, of the target–probe hybrid zone increases linearly (R2 = 0.95 for linear fit) with the initial target concentration. | ||
We subtracted the signal of a control experiment with a MB probe and no target (grey curve in the inset of Fig. 2) from the measured electropherograms to extract the contribution to the signal of the target–probe hybrid peak, h (see details in ESI†). In the main plot of Fig. 2, we show variations of the magnitude of target–probe hybrid peak h for five initial concentrations of the target. The target–probe peak signal component h is linearly proportional to the initial target concentration, and a linear fit to the data (passing through the origin) yields R2 value of 0.95. The linear increase in peak height in the current case is expected because hybridization reactions obey pseudo-first-order kinetics in the presence of excess amount of probe (see details in ESI†). Here, we demonstrated 5 pM detection sensitivity of our assay within 3 min, which is 20-fold improvement over previous ITP-hybridization assays with no background signal removal.4,8,9
Our sensitivity and assay time are on par with 3 pM sensitivity and 10 min run time demonstrated by Garcia-Schwarz and Santiago.11 The latter study used ITP for rapid nucleic acid hybridization and combined this with a functionalized gel to remove the background fluorescence signal associated with the unreacted fluorescent probe. However, we note that the method of Garcia-Schwarz and Santiago requires more experimental preparation to functionalize the on-chip gel, and does not discriminate between nucleic acid fragments with varying lengths (it instead discriminates based on sequence). Further, our method is significantly more sensitive and faster than existing electrophoretic separation based DNA hybridization assays such as Southern blotting20 (order 10–100 μM sensitivity and 24 h assay time) and its capillary electrophoresis based alternatives (10–100 nM sensitivity and 1.5 h assay time).12,13 We hypothesize that further improvements in detection sensitivity to sub-picomolar limits are likely possible with our method by using linear, labeled-DNA probes which have higher hybridization rates than MBs. Such higher hybridization rates would come at the cost of lower stringency and higher background signal compared with MBs. The latter limitation of linear DNA probes can be addressed by using longer channels for higher CZE separation resolution and thereby improved background signal removal.
One limitation of our assay is the limited choices of electrolyte systems that can ensure coupling of ITP and CZE via shock interaction in bidirectional ITP. Detailed guidelines to select electrolytes for coupling ITP and CZE using bidirectional ITP can be found in Bahga et al.5 along with several practical examples. We note that, besides bidirectional ITP, several other coupled ITP-CE methods exist,6 which can potentially be applied to this problem.
Lastly, we have performed experiments to demonstrate the feasibility of our assay for multiplexed detection of multiple-length ssDNA targets by simultaneously detecting 39 and 90 nt targets. We refer the readers to Fig. S5 of the ESI† for the measured electropherograms. Briefly, we observed three well-resolved peaks corresponding to the unreacted probe and two target–probe hybrids for 39 and 90 nt targets. We identified the target–probe hybrid peaks by noting the relative change in peak intensities when the concentrations of 39 and 90 nt targets were individually varied in the initial sample mixture.
In summary, we have demonstrated integration of ITP-based DNA hybridization and CZE separation of reaction products using bidirectional ITP, and applied it to sequence-specific detection of two ssDNA targets with a fluorescently labeled DNA probe. Our method combines high preconcentration ability of ITP to accelerate slow DNA hybridization kinetics, and high resolving power of CZE to separate and identify reaction products. CZE separation of the products of DNA hybridization reaction removes the background signal associated with unreacted probes. This allows quantification of fluorescence from the target–probe hybrid even when the signal from unbound MB well exceeds the hybridization signal. CZE separation of reaction products also provides a way of extending the functionality of ITP-based hybridization to detect multiple-length DNA targets. Potential extensions of our method include multiplexed detection of a larger number of DNA targets sharing a common portion of their sequences, and application to the identification of sequence variations in restriction fragments. Detection of such DNA targets has applications in mutation studies21 and diagnosis of genetic diseases.22 However, we note that care must be taken while designing MBs so as to reduce the possibility of non-specific hybridization. As demonstrated by Tyagi and Kramer23 MBs with optimally designed stem-loop structure can discriminate single nucleotide mismatch with reasonable accuracy.
Acknowledgements
S.S.B. is supported by a Mayfield Stanford Graduate Fellowship and a Kodak Fellowship. C.M.H. is supported by a Robert and Katherine Eustis Stanford Graduate Fellowship. We gratefully acknowledge support from Defence Advanced Research Projects Agency (DARPA) under contract number HR0011-12-C-0080, program manager Daniel J. Wattendorf.Notes and references
- J. Wang, Nucleic Acids Res., 2000, 28, 3011–3016 CrossRef CAS.
- O. Lazcka, F. J. Del Campo and F. X. Munoz, Biosens. Bioelectron., 2007, 22, 1205–1217 CrossRef CAS.
- R. Wieder and J. G. Wetmur, Biopolymers, 1981, 20, 1537–1547 CrossRef CAS.
- M. Bercovici, C. M. Han, J. C. Liao and J. G. Santiago, Proc. Natl. Acad. Sci. U. S. A., 2012, 109, 11127–11132 CrossRef CAS.
- S. S. Bahga, R. D. Chambers and J. G. Santiago, Anal. Chem., 2011, 83, 6154–6162 CrossRef CAS.
- S. S. Bahga and J. G. Santiago, Analyst, submitted Search PubMed.
- G. Goet, T. Baier and S. Hardt, Lab Chip, 2009, 9, 3586–3593 RSC.
- A. Persat and J. G. Santiago, Anal. Chem., 2011, 83, 2310–2316 CrossRef CAS.
- M. Bercovici, G. V. Kaigala, K. E. Mach, C. M. Han, J. C. Liao and J. G. Santiago, Anal. Chem., 2012, 83, 4110–4117 CrossRef.
- S. Tyagi and F. R. Kramer, Nat. Biotechnol., 1996, 14, 303–308 CrossRef CAS.
- G. Garcia-Schwarz and J. G. Santiago, Anal. Chem., 2012, 84, 6366–6369 CrossRef CAS.
- J. W. Chen, A. S. Cohen and B. L. Karger, J. Chromatogr., 1991, 559, 295–305 CrossRef CAS.
- N. Bianchi, C. Mischiati, G. Feriotto and R. Gambari, Nucleic Acids Res., 1993, 21, 3595–3596 CrossRef CAS.
- M. Y. Zhukov, Zh. Vychisl. Mat. Mat. Fiz., 1984, 24, 549–565 CAS.
- S. S. Bahga and J. G. Santiago, Electrophoresis, 2012, 33, 1048–1059 CrossRef CAS.
- W. Thormann, D. Arn and E. Schumacher, Electrophoresis, 1985, 6, 10–18 CrossRef CAS.
- T. Hirokawa, K. Watanabe, Y. Yokota and Y. Kiso, J. Chromatogr., A, 1993, 633, 251–259 CrossRef CAS.
- T. Hirokawa, J. Chromatogr., A, 1994, 686, 158–163 CrossRef CAS.
- D. Milanova, R. D. Chambers, S. S. Bahga and J. G. Santiago, Electrophoresis, 2011, 32, 3286–3294 CrossRef CAS.
- E. M. Southern, J. Mol. Biol., 1975, 98, 503–517 CrossRef CAS.
- D. Botstein, R. L. White, M. Skolnick and R. W. Davis, Am. J. Hum. Genet., 1980, 32, 314–331 CAS.
- C. Stolle, et al. , Hum. Mutat., 1998, 12, 417–423 CrossRef CAS.
- S. Tyagi and F. R. Kramer, Nat. Biotechnol., 1996, 14, 303–308 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available: Choice of electrolyte chemistry for bidirectional ITP; experimental setup; channel geometry and experimental protocol; signal analysis of hybridization data from point detector; modelling of the concentration of target–probe hybrids in bidirectional ITP; demonstration of multiplexed detection; and ssDNA target sequence information. See DOI: 10.1039/c2an36249j |
| ‡ S.S.B. and C.M.H. contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2013 |