 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Antibody orientation on biosensor surfaces: a minireview
Anke K.
Trilling
ab,
Jules
Beekwilder
a and
Han
Zuilhof
*bc
aPlant Research International, Wageningen, The Netherlands
bLaboratory of Organic Chemistry, Wageningen University, Wageningen, The Netherlands. E-mail: Han.Zuilhof@wur.nl
cDepartment of Chemical and Materials Engineering, King Abdulaziz University, Jeddah, Saudi Arabia
First published on 22nd January 2013
Abstract
Detection elements play a key role in analyte recognition in biosensors. Therefore, detection elements with high analyte specificity and binding strength are required. While antibodies (Abs) have been increasingly used as detection elements in biosensors, a key challenge remains – the immobilization on the biosensor surface. This minireview highlights recent approaches to immobilize and study Abs on surfaces. We first introduce Ab species used as detection elements, and discuss techniques recently used to elucidate Ab orientation by determination of layer thickness or surface topology. Then, several immobilization methods will be presented: non-covalent and covalent surface attachment, yielding oriented or random coupled Abs. Finally, protein modification methods applicable for oriented Ab immobilization are reviewed with an eye to future application.
 Anke K. Trilling | Anke K. Trilling received her MSc in Biotechnology at Ecole supérieure de biotechnologie Strasbourg (F) in 2008. Currently, she is a Ph.D. candidate under supervision of Dr J. Beekwilder and Prof. H. Zuilhof, developing new strategies to self-assemble llama antibodies uniformly on tailor-made surfaces. |
 Jules Beekwilder | Jules Beekwilder received an MSc in Chemical Biology at Utrecht University (1989), and did his Ph.D. at Leiden University (1996). He did his Postdoctoral research at Leiden University (1996) and CPRO-DLO in Wageningen (1997–2000). From 2000 he has been a permanent staff researcher at Plant Research International in Wageningen (NL). One of his research lines is the use of recombinant antibodies for biosensing. |
 Han Zuilhof | Han Zuilhof is the Chair of Organic Chemistry at Wageningen University. His interests focus on surface-bound (bio-)organic chemistry and bionanotechnology. He obtained both an MSc in Chemistry and an MA in Philosophy, before deciding that Chemistry was the more social of the two disciplines. After obtaining a Ph.D. in Chemistry (Leiden University, 1994; highest honors), he did postdoctoral work at the University of Rochester, NY, and at Columbia University. Subsequently he joined the faculty at Wageningen University, and has been a Professor of Organic Chemistry since 2007. He is also a Distinguished Adjunct Professor of Chemical Engineering at the King Abdulaziz University in Jeddah, Saudi Arabia, is the founder of Surfix, and serves on the Editorial Advisory Board of Langmuir and Applied Surface Science. |
1 Introduction
In the last decade, a wide variety of different biosensors emerged. Sensor specificity relies strongly on the properties of the immobilized detection element, which has stimulated the use of antibodies (Abs) or fragments thereof. In 1971,1 Abs were used for the first time in an enzyme-linked immunosorbent assay (ELISA) to quantitatively detect analytes. Nowadays, antigen–Ab interactions can be detected by a variety of techniques, including quartz crystal microbalance (QCM), surface plasmon resonance (SPR) and electrochemical impedance spectroscopy (EIS).Abs with better affinities and higher stabilities have been selected to improve biosensor performance. Further sensor optimization was directed towards surface preparation of biosensors aiming to promote specific binding and suppress non-specific binding. For this purpose site-specific coupling and immobilization of proteins are of great interest.2–4
Here we review recently applied Ab immobilization strategies. In the field of proteins, Abs represent a small class of glycoproteins with a well-defined structure. Since Abs possess only one binding site, it can be highly advantageous to orient these molecules to improve biosensor performance, with improvement factors as high as 200 being reported upon orientation.5 Similar, albeit smaller, effects have also been reported by several other groups.6–14 The first section of this minireview presents different types of Abs applied as detection elements. Next, methods are reviewed that are used to elucidate the orientation of immobilized Abs. Then, various recently applied non-covalent and covalent methods for Ab immobilization are summarized, including oriented and random immobilization of both native and engineered Ab species. Finally, we explore to what extent protein modification techniques have already been implemented for Abs, and where new opportunities open up to bring the advantages of protein orientation closer to application in future biosensors.
2 Abs as detection elements
There are several types of Ab structures that are currently being focused on in biosensing (Fig. 1). Immunoglobulin Gs (IgGs, ∼150 kDa, 143 × 77 × 40 Å3)15 consist of two light and two heavy chains, linked by disulfide bonds to form the characteristic Y-shape. The chains are divided into constant (C) and variable (V) regions. On the second heavy chain constant region (CH2), IgGs have carbohydrate moieties (Fig. 1).16 The variable domain bears three hypervariable regions, known as complementarity-determining regions (CDRs), which are responsible for the specific Ab–antigen interaction. The diversity in this area allows the endless supply of Abs with different specificity and binding strength (affinity).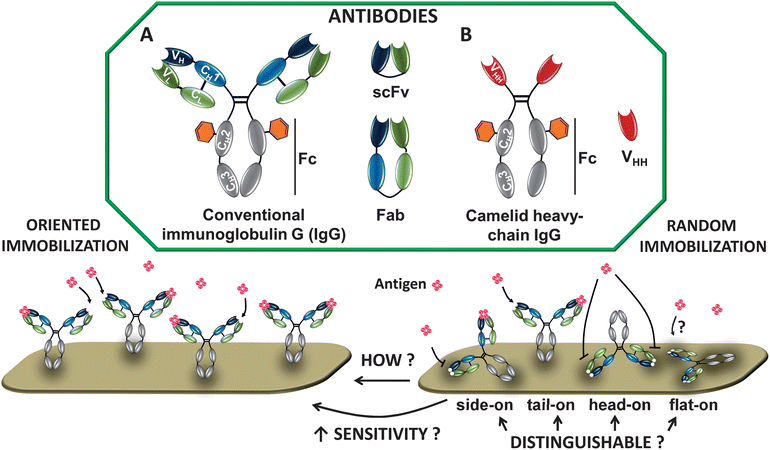 | ||
| Fig. 1 Schematic depiction of natural Abs. (A) Immunoglobulin G (IgG) consists of 2 heavy chains (gray/blue) and 2 light chains (green). Antigens bind to the variable regions VL and VH. The CH2 domain bears a carbohydrate moiety (orange hexagon); scFv = single-chain variable fragment composed of VL and VH; Fab = antigen-binding Ig fragment. (B) Camelid heavy-chain Ab contains only 2 heavy chains, which are composed of one variable (VHH; antigen-binding unit) and two constant (CH2 and CH3) domains. Abs can be immobilized in an oriented or random fashion (see example for IgG). | ||
Molecular engineering enabled the minimization of conventional Abs into smaller and more stable Ab-derived fragments. Examples include Fabs (antigen binding fragment), or the even smaller single-chain variable fragments (scFv, ∼27 kDa), both of which still retain antigen-binding specificity. Further size reduction into monomeric single domain Abs (sdAbs), the VH or VL, resulted in loss of affinity towards the antigen,17 making time-consuming selection and affinity maturation necessary.
Apart from IgGs, two other Ab classes are becoming important. First, camelid Abs (found in dromedary, llama, alpaca and camel) (Fig. 1).18 These Abs lack the light chain and the first constant domain of the heavy chain (CH1), leaving one single domain for effective antigen binding, known as VHH or nanobody. In VHH, the C-terminus is situated at the opposite site of the antigen binding region and represents an optimal target for functionalization. Second, another novel immunoglobulin with one variable domain (VNAR), called novel antigen receptor (IgNAR), was discovered in cartilaginous fish, such as sharks.19
VHH, VNAR and sdAbs possess superior stability, and are highly soluble, small (∼15 kDa) monomeric binding domains that are very useful as detection elements in biosensors. Their value for biotechnological applications has been recently reviewed.20–22 Antigen-specific Ab fragments can be selected from large libraries by a variety of methods, reviewed in the literature.23,24
3 Techniques to study antibody orientation
Providing information of Ab orientation on the surface is of fundamental interest. Immobilized IgG can adopt four exemplary molecular orientations: side-on (one Fc and one Fab attached to the surface), tail-on (Fc attached to the surface), head-on (both Fabs attached to the surface) or flat-on (all three fragments attached to the surface) (Fig. 1). For the highest analyte binding, Abs should display free antigen-binding regions after immobilization. Controlling the orientation will therefore lead to better analyte binding resulting in improved biosensor sensitivity.Many techniques have been used to elucidate the presence and binding function of immobilized Abs. Fourier transform infrared reflection (FTIR) spectroscopy is used to characterize the presence of specific chemical groups, and various fluorescence microscopies help to visualize efficient binding of analyte to Ab-functionalized surfaces.25,26 In fact, spectroscopic techniques are predominately used to roughly confirm an effective Ab orientation, with only a relatively small set of studies that investigate the orientation of Abs by comparing Abs immobilized in different directions. For example, SPR can be used to calculate the Ab coverage, and the relationship between the adsorbed amount and molecular orientation on the surface has been used earlier to distinguish between tail/head-on and side/flat-on orientation.27 But minimal direct information about Ab orientation can be deduced with such approaches. Here we present a selection of the techniques recently applied to characterize Ab orientation in a more direct manner by measuring the layer thickness or by scanning of the surface (Fig. 2). It should be kept in mind that this list is not comprehensive, that it focuses due to space considerations on a few representative recent cases, and that each of these techniques still provides only limited information; to get the full picture, several techniques are ideally combined.
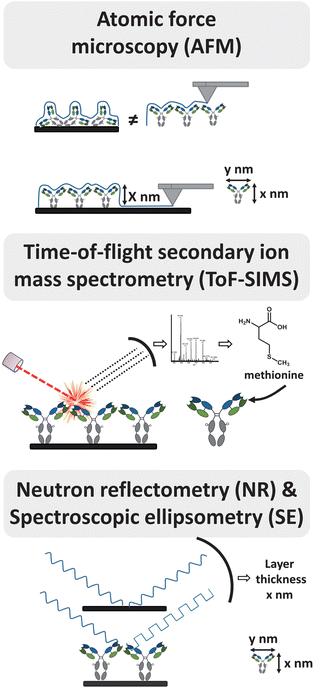 | ||
| Fig. 2 Selection of techniques to study antibody orientation. For detailed information see text. | ||
3.1 Atomic force microscopy (AFM)
In atomic force microscopy (AFM), surfaces are scanned by a nano-scale tip, immobilized at the end of a cantilever, yielding resolutions below a nanometer. 3-D structures of soft biomaterials such as proteins can be visualized by AFM, resulting in a topographical surface map.28 From this information, local properties of the surface such as the degree of coverage, the thickness of the layer, and the shape of the proteins can be deduced (Fig. 2).AFM is often used to deduce Ab orientation by determining the dimensions of the Abs. Sarkar and co-workers29 employed noncontact mode AFM to measure the total thickness of a surface coating before and after immobilization of IgG onto a protein A-coated surface. The measured change of height after IgG incubation corresponded roughly to the long axis of IgG, suggesting tail-on position of IgG.30 Chen et al.27 used AFM to measure the height of the Ab layer immobilized on a calixarene monolayer to distinguish between tail-on and side-on orientation. AFM has also been used to investigate Ab orientation by scanning 5 nm Au nanoparticles on surfaces as size standard,31 and to measure the width of the IgG arms.32
Likewise, AFM can be used to elucidate the surface topology. Kim et al.33 compared the surface roughness of Abs captured by cysteine-functionalized protein G (Cys-protein G) to that of a surface that captured Abs by native protein G. The more uniform height image obtained for Cys-protein G-modified surfaces was interpreted as more uniformly oriented Abs. AFM was also used to study the time-dependent conformational change of Fabs immobilized on gold in the absence and presence of stabilizing polyethylene glycol (PEG) layers.34 Directly immobilized Fab fragments showed a fast decrease in height accompanied by a decrease of the antigen-binding ability. In contrast, Fabs stabilized by the PEG layer displayed a diminished height decrease and better antigen binding abilities, suggesting a slowed down change of conformation or/and orientation by the co-immobilized PEG-layer.
3.2 Time-of-flight secondary ion mass spectrometry (ToF-SIMS)
High-resolution time-of-flight secondary ion mass spectrometry (ToF-SIMS) emerged as a powerful method to obtain evidence about the structure of the surface by providing biophysical information about the molecular structure. This technique combines a high chemical specificity with a good surface sensitivity (sampling depth 1–3 nm) by ion bombardment of a surface with a pulsed primary ion beam.35 The resulting positively or negatively charged ions are analyzed by a time-of-flight mass analyzer, yielding a fingerprint of the proteins (Fig. 2). As the sampling depth is very shallow, these data can be used to interpret the orientation of immobilized proteins, and the more recently developed milder bombardments with Ar clusters may improve this further.ToF-SIMS was used by Baio et al.7 to show intensity differences of secondary ions originating from asymmetrically located amino acids in the protein. Distinct orientations of a variable fragment (HuLys Fv) were achieved on two different substrates – each reacting with a specific, tailor-made moiety at one of the termini. Comparison of the intensity ratios of specific secondary ions suggested that HuLys Fv was indeed oriented in different ways on the two different substrates, although it is difficult to specify any of these orientations based on the data. In another study8 ToF-SIMS was used to characterize the orientation of randomly biotinylated and site-specifically biotinylated Abs (IgGs, F(ab')2 and Fabs) on streptavidin surfaces. ToF-SIMS results could not be linked to specific locations near the analyte binding site of site-specific biotinylated Abs, but unique peaks were observed in oriented Abs that were absent in randomly immobilized controls. This indicated that site-specific and randomly biotinylated Abs were assembled in distinct orientations. The data analysis can be greatly facilitated by principal component analysis (PCA), as shown in an analogous study by the group of Lee.36 They showed that random biotinylated IgGs at high concentrations yield the same ToF-SIMS spectra as site-specific biotinylated IgGs. This suggests that with high concentrations orientation was achieved for both biotinylated Abs. At the same time Liu et al.37 showed that head-on orientation (by immobilizing protein A on the surface) can be distinguished from tail-on orientation (by immobilizing the antigen on the surface) of immobilized Abs by combining ToF-SIMS with PCA. Amino acids characteristic of the Fab and Fc fragments could be used to provide an image ‘map’ of Ab orientations across the patterned surfaces.
3.3 Dual polarization interferometry (DPI)
Dual Polarization Interferometry (DPI), an optical wave-guide-based analytical technique, can be used to obtain information on molecular dimensions (layer thickness), packing (layer refractive index, density) and stoichiometry (mass). The measured layer thickness can provide information on the arrangement of Abs on surfaces when combined with known dimensions of the molecule.Song et al.38 used dimensions of Abs as determined by X-ray crystallography in correlation with layer thickness and surface coverage (or mass) to distinguish between possible Ab orientations. The surface coverage of random immobilized Abs on a monolayer and oriented Abs on protein G surfaces was determined by DPI and compared to the reference values of ‘theoretical’ saturated surface coverages for random and oriented Abs. Tail-on orientation of IgG Abs on a protein G layer was further suggested by determining the layer thickness by DPI, which revealed a thickness of the Ab layer on protein G corresponding to the long axis of the Y-shaped Ab.
3.4 Neutron reflectometry (NR)
Neutron reflectometry (NR) is a neutron diffraction technique to determine the thickness and composition of molecular layers on surfaces with a sensitivity of 2–3 Å.39 The technique involves directing a beam of neutrons onto a flat surface, and measurement of the intensity of the reflected radiation as a function of angle or neutron wavelength. Comparison of layer thickness with the molecular dimensions of Abs allows differentiation between flat-on, side-on and head-on/tail-on orientations of the Abs (Fig. 2).Using NR Zhao et al.40 determined the orientation of Abs adsorbed on silicon wafers at low concentration. The observed thickness of adsorbed Abs corresponded to the short axial length of the Ab, suggesting a flat-on orientation. In contrast, Abs immobilized via the Fc region onto an engineered protein A-like (ZZctOmpA) surface adopt a tail-on orientation.15
3.5 Spectroscopic ellipsometry (SE)
Spectroscopic ellipsometry (SE) analyses the state of polarized light reflected from multilayer reflective samples. The layer thickness can be deduced by a model-based analysis based on how the light interacts with the surface (Fig. 2). This makes SE a valuable tool to investigate optical parameters and deposition kinetics of thin film structures.Bae et al.41 determined the thickness of protein layers consisting of Abs bound to thiolated protein G oriented on gold. The measured thickness suggested that Abs are immobilized in such a manner that the Fc domain is bound to the protein G layer, with the antigen binding domain facing away from the surface.
4 Immobilization strategies
Strategies for immobilization may result in specific or random orientation of the Abs. The orientation is dependent on the self-organizing capacity of the antibodies, which may be steered by specific reactive groups on the surface, on the antibody, or on both. Specific orientation of immobilized Abs is not easily achieved, since Abs usually carry several copies of reactive groups.It is essential to immobilize Abs on surfaces without changing their binding activity and specificity. Therefore immobilization strategies should be mild. These strategies can usually be made compatible with the surfaces of various materials, by functionalizing the surface with specific groups. Surfaces can be used either directly or functionalized with (mono)layers, either as two-dimensional surfaces or as three-dimensional matrices. Gold, glass, copper, silicon nitride and silicon surfaces or magnetic beads represent only a small selection of surfaces used for Ab immobilization.29,32,42–45 Below we will review several strategies of Ab immobilization. They can be distinguished by non-covalent or covalent coupling chemistry.
4.1 Non-covalent immobilization
Immobilization of untreated Abs can be mediated by an intermediate protein directly coupled to the surface, such as protein A and protein G. These proteins display five and two binding domains specific to the Fc portion of Abs, respectively. This results predominantly in tail-on orientation. Improvement of biosensor performance by orienting Abs with protein A or protein G has been shown in several studies when compared to their randomly immobilized counterparts.10,26,43,46 Further improvements have been achieved by orientation of protein A or G. Feng et al.47 optimized the approach by forming highly organized aggregates of IgG and protein A. Immobilized IgG–protein A aggregates yielded three-dimensional structures on the surface with IgGs exposing their analyte binding sites. Johnson and Mutharasan48 showed that the pH used for protein G adsorption on gold surfaces influences the protein G orientation and subsequent Ab binding. Often, exposed Cys residues are used for oriented immobilization of protein A or G. Thiolated protein G was used for immobilization onto a copper surface.42 Lee et al.49 prepared cysteine-functionalized protein G multimers to improve Ab immobilization on magnetic silica nanoparticles. Recombinant Cys-protein G trimers were engineered by repeated linking of protein G monomers via a flexible linker. The use of such Cys-protein G trimers improved Ab immobilization and enhanced the biosensor sensitivity by 10-fold compared to a Cys-protein G monomer setup. Brun et al.15 fused protein A domains genetically to a Cys-exposing variant of E. coli protein ompA, which was embedded in a PEG monolayer on a gold surface, and allowed oriented binding and presentation of antibodies. Ko et al.50 fused a gold-binding protein (GBP) to protein A, resulting in GBP–ProtA. Compared to native protein A, this fusion protein self-assembled at a higher density on gold surfaces and bound more IgG. Tajima et al.11 enzymatically conjugated protein A onto a substrate to achieve a “super-oriented IgG” bound to oriented protein A. The strict control of the IgG orientation resulted in an approximately 100-fold higher affinity than the partially oriented IgG, when protein A was physisorbed on the surface.Classically, non-covalent binding of Abs on surfaces is achieved by physical adsorption, avoiding an intermediate protein. Making use of ionic bonds, electrostatic and hydrophobic interactions and van der Waals forces results in non-covalent immobilization. Physical adsorption gives low control over the orientation of the Abs, even though immobilization of Abs using a pneumatic nebulizer is fast and reproducible.51 Zhao et al.52 studied how solution pH, salt concentration and surface chemistry affect Ab adsorption onto silica surfaces. The salt concentration and pH did influence the amount of adsorbed Ab and analyte binding, but did not influence the orientation of immobilized Abs. Abs predominantly adopted flat-on orientation on the investigated surfaces. Um et al.53 introduced tail-on orientation in bound Abs by the electrochemical immobilization onto poly-(2-cyano-ethylpyrrole)-coated gold electrodes. Induction by cyclic voltammetry favored electrostatic interactions between the cyano group on the surface and the hydroxyl group of the Ab present in the Fc region. Electrochemically immobilized Abs showed an improved analyte binding compared to physisorbed Abs, which was attributed to orientation effects. Abs adsorbed on hydroxyapatite nanoparticles mainly orient themselves in the tail-on position due to steric hindrance on the round surface.54 Harmsen14 showed improved orientation of adsorbed VHHs due to genetic fusion of peptide tags to the C-terminus, situated opposite to the analyte binding site. It was suggested that these tags trigger oriented binding on polystyrene surfaces by hydrophobic interactions with the surface. Nevertheless, physical interactions are generally weak and sensitive to changes in condition such as pH, temperature or salt concentration. Typically, biosensors using non-covalent binding may therefore suffer from poor analytical performance due to lower operational and storage stability. Specific directional interactions between the surface and part of the Ab therefore provide a step forward, and are an intermediate towards covalent and fully irreversible immobilization. As an example, a more stable immobilization resulting in orientation can be achieved when the thiol group is utilized for Ab immobilization on surfaces such as gold. Disulfide bonds are a common feature of intact Abs and thiol groups can be obtained under mild reduction conditions (Fig. 3). Balevicius et al.55 showed that oriented Ab fragments can bind 2.5 times more analyte than the intact Ab immobilized in a random fashion using amine groups. An elegant light-assisted approach for Ab immobilization was shown by Ventura et al.56 Disulfide bonds were broken upon absorption of UV light by nearby aromatic amino acids, yielding reactive thiol groups that are effective for oriented binding onto gold electrodes. Adsorption of thiol-exposing Fab fragments onto gold was also used to show that co-immobilization of densely packed polyethylene glycol layers improved the time-dependent analyte binding ability of immobilized Fab fragments.34 Albeit being site-specific, this method yields monovalent Abs, and too harsh reduction conditions might inactivate Ab fragments due to the unintentional reduction of internal disulfide bonds. Another immobilization method introducing orientation involves the fusion of a polyhistidine (His6) affinity-tag on the C- or N-terminus of the recombinant protein. The His6 tag shows a high affinity (K = 107 M−1) to Ni2+, Co2+ and Cu2+ surfaces. The tetradentate ligand nitriloacetate (NTA) forms hexagonal complexes with divalent metal ions, leaving two binding sites available for chelation with a histidine residue. The His6 tag on the C-terminus of a variable fragment (HuLys Fv) was used to control orientation onto a gold substrate.7 When compared to HuLys Fv immobilized onto maleimide-terminated monolayers via an N-terminal cysteine, binding via the C-terminal His6 tag to Ni-loaded NTA-terminated monolayers showed a 10-fold higher SPR signal upon analyte binding. However, because the binding affinity of the His6 tag is for several approaches still not high enough, these experiments might suffer from undesired protein dissociation.
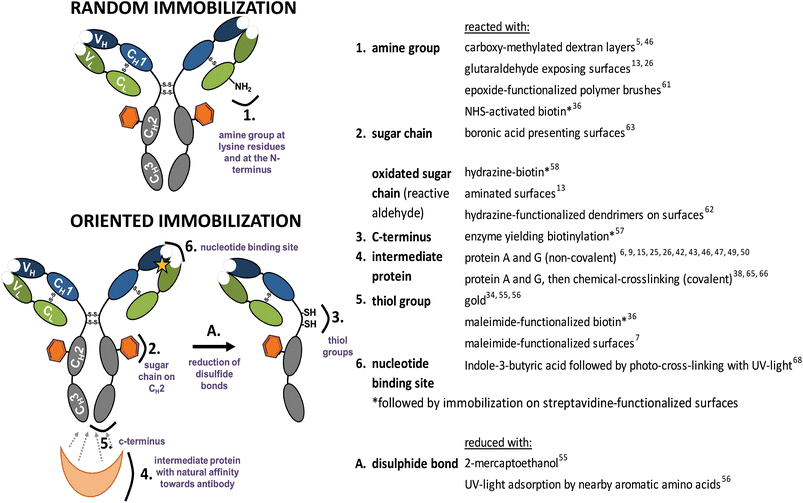 | ||
| Fig. 3 Functional groups used for random and oriented Ab immobilization onto surfaces. | ||
Stronger non-covalent protein immobilization can be achieved by the use of the streptavidin–biotin interaction, one of the strongest non-covalent interactions known in biology. Depending on the applied biotinylation method, Abs can be immobilized in a random or oriented fashion. Abs randomly biotinylated at the amine groups were compared to oriented Abs, site-specifically biotinylated at the hinge region.36 With increased immobilization concentration both biotinylated Abs adopted tail-on orientation, while site-specific biotinylated IgG became oriented at a slightly faster rate. Cho et al.8 compared the binding signal of random and site-specific biotinylated IgG and Fab on distinct streptavidin-coated surfaces. In a sandwich-type immunoassay, the group reported a 2 to 3 times higher binding signal for site-specifically biotinylated Ab species. Several other approaches to add a biotin moiety site-specifically to Abs have been employed lately. Biotinylation specifically at the C-terminus of the Ab was achieved using the enzyme carboxypeptidase Y.57 Kang and co-workers58 achieved site-specific biotinylation of Abs using a sugar moiety. Oxidation of sugar chains yielded aldehydes reactive towards hydrazine–biotin. Further, in vivo biotinylation of VHH was successfully explored by our group using the Avi-tag.5 This tag is recognized by the BirA enzyme, and biotinylation occurs at the lysine position of the tag. This in vivo biotinylation is somewhat time-consuming, but is also extremely effective as it improved the analyte binding by more than a 200-fold.
4.2 Covalent immobilization
Covalent immobilization does, in principle, provide the best entry point to combine longevity of the Ab-modified surface with a high sensitivity due to a specific orientation. Therefore, a lot of efforts are currently undertaken to investigate and improve this area, including both by now well-known reactions of naturally present moieties and the developments of tailor-made, i.e. man-made, modifications thereof.So far the chemistry deployed to immobilize Abs has been limited to classical protein chemistries. Amine (NH2) groups present in the lysine amino acid side-chain on the Ab surface can be used for covalent immobilization in a random fashion (Fig. 3). Widely used, e.g. for SPR studies, is the immobilization of Abs onto carboxy-methylated dextran layers on gold surfaces.5,46,55 Recently, it has been shown that this immobilization is not completely random. Prior to covalent conjugation, Abs need to undergo physisorption. Orientation during physisorption depends on the surface pKa, isoelectric point of the Ab and the pH of the used immobilization buffer, by optimizing the conditions one orientation can be favored.59,60 By attaching antibodies via the amine group to protein-repellent zwitterionic polymer brushes Nguyen et al.44 prepared surfaces recognizing the antigen while preventing nonspecific adsorption of other proteins. Epoxide-functionalized polymer brushes allow another possibility to immobilize Abs via the amine group.61 Similarly, immobilization via the amine group can be achieved by the use of glutaraldehyde-functionalized surfaces.13,26
Another covalent approach to orientation makes use of a unique carbohydrate moiety at the Fc part of Abs (Fig. 3). Specific oxidation on the carbohydrate vicinal hydroxyl group via the use of periodate sodium generates aldehydes. These aldehydes are reactive towards aminated surfaces13 or hydrazine-functionalized dendrimers on surfaces,62 resulting in oriented covalent Ab coupling. Ho et al.63 used a boronic acid-presenting surface to orient Abs via the carbohydrate moiety. Boronic acids form cyclic boronate esters with 1,2- and 1,3-diols present in carbohydrates of Abs, and thus provide an additional anchoring point, with chemistry that is largely orthogonal to other methods discussed in here.
Also the Fc region itself is used for oriented immobilization. Batalla et al.64 used heterofunctionally activated agarose matrices displaying metal chelate groups. These lead to oriented non-covalent attachment of the Ab via the histidine-rich Fc portion of the Ab. Then, reaction of exposed amine groups with matrix-bound glyoxyl groups was promoted by a change of pH. The formed reversible Schiff's base bonds can then be mildly reduced by e.g. NaB(CN)H3 to obtain irreversible oriented and covalently attached Abs. Another possibility to immobilize Abs via the Fc part makes use of intermediate proteins described before. To circumvent stability problems of the non-covalent interaction between protein A/protein G and the Fc region, chemical crosslinking was successfully applied using cyanamide,65 dimethyl pimelimidate (DMP)66 or the homobifunctional linker bis(sulfosuccinimidyl) suberate.38 Analogously, thiol groups were explored for covalent oriented coupling of Abs, via reduction of the disulfide group present in the hinge region of Abs (Fig. 3), and subsequent coupling to a maleimide-functionalized surface.67
Another possibility to covalently immobilize Abs in an oriented manner makes use of the conserved nucleotide binding site (NBS) present in the conserved region of the variable domain of all Ab isotypes. This region, which has not been removed so far from the antigen-binding site, is rich in specific aromatic amino acids, and displays an affinity for indole-3-butyric acid. Exposure to 254 nm light allows irreversible photo-attachment of Abs onto an indole-3-butyric acid-terminated surface, while leaving the antigen binding site unaffected.68
These examples show the success of covalently attached Abs, but also point to the potential of using and developing milder (non-denaturing), bio-orthogonal reactions that allow control over the Ab direction. Today, protein coupling can be achieved by a range of highly reliable chemistries. However, most of these have not been deployed for Abs since they rely on protein engineering and recombinant production. Currently, bioorthogonal chemistries such as Diels–Alder reaction or Staudinger ligation are widely applied for protein immobilization or functionalization, but functional groups are often introduced via the amine group of lysine side-chains or the thiol group of cysteine, making such approaches unsuitable for the site-specific immobilization of Abs. Here we will limit the discussion to a selection of methods that would lead to site-specific introduction of functional groups.
Stamos et al.69 immobilized various proteins site specifically via the Diels–Alder cycloaddition reaction70 between an o-imino-quinone group in the protein and an acryloyl linker on the surface. To this end, site-specific genetic incorporation of the 3-NH2Tyr amino acid into proteins was performed.
The Staudinger ligation reported by Saxon and Bertozzi involves the reaction between an azide and a phosphine-containing ester or thioester yielding a covalent amide bond.71 Introduction of a functional azide-group into the protein can be achieved by the use of the methionine analogue azidohomoalanine.72 Unfortunately, this approach is only site-specific if no more than one methionine is surface accessible. To guarantee specificity, engineering of the protein is often required. Another technology to truly site-specifically incorporate azides was developed by the group of Schultz.73 Functionally unique unnatural amino acids such as p-azido-L-phenylalanine can be incorporated into proteins such as green fluorescent protein by expressing orthogonal tRNAs and aminoacyl-tRNA synthetases.74 Using this approach azides can be incorporated into antibodies in response to amber nonsense codons at the preferred protein position. Another approach to introduce an azide specifically at the N- or C-terminus uses expressed protein ligation (EPL).75 EPL generates recombinant protein thioesters by thiolysis of intein fusion proteins reactive towards synthetic peptides bearing an N-terminal cysteine, which yields a native amide bond. EPL has been employed in combination with a synthetic azide-containing reagent to produce azide-functionalized RNase A (azido-RNase A).76 Azido-RNase A was then site-specifically immobilized onto phosphine-terminated surfaces by a Staudinger ligation.
Sulfonylazides react with terminal alkynes under the catalysis of Cu(I) to form N-acylsulfonamides. This ‘click sulfonamide reaction’ (CSR)77,78 is related to the Cu(I)-catalyzed [3 + 2] azide–alkyne cycloaddition (CuAAC). Both reactions displayed specificity during protein immobilization using alkyne-modified mCerry-Ypt7 protein (at the C-terminus by ELP) on sulfonylazide- and azide-functionalized surfaces, respectively.79 A potential disadvantage of CuAAC is the used copper(I) catalyst. In contrast, the strain-promoted alkyne–azide cycloaddition (SPAAC) with cyclooctynes requires no additional reagent, but reacts spontaneously with azides. Different cyclooctyne variants used for surface functionalization and/or bioconjugation have been reviewed.80–82 Taking advantage of these mild and bioorthogonal reactions will most likely open up new approaches for the site-specific immobilization of Abs on surfaces. For example, Witte et al.83 prepared anti-GFP VHH with azide or cyclooctyne moieties at the C-terminus using a combined shortage-click strategy to prepare VHH dimers. Such functionalized VHHs could also be covalently immobilized using SPAAC onto cyclooctyne- or azide-functionalized surfaces, respectively.
5 Outlook
The approaches for Ab immobilization as presented in this minireview show that a wide variety of immobilization methods are available for various surfaces and different Ab species. However, the variety of available methods also illustrates that a universal method is not yet available.The limited amount of studies in which oriented Ab immobilization was quantitatively compared to random Ab immobilization shows that orientation can significantly (up to two orders of magnitude) improve the analyte binding signal. Therefore, the ever increasing demands in sensitivity should lead future efforts to oriented immobilization of Abs. The currently most widely used method, adsorption onto protein-coated surfaces, is effective, but also limited given the heterogeneous nature of such surfaces. Further improvements in orientation and therefore likely in sensor sensitivity will require involvement of tailor-made Ab modifications and/or novel bio-orthogonal surface-bound or Ab-directed chemistries. Specifically the ongoing exploration in protein coupling techniques is expected to hold significant promises for the field of Ab orientation in the future.
Notes and references
- E. Engvall and P. Perlmann, Immunochemistry, 1971, 8, 871–874 CrossRef CAS.
- P. Jonkheijm, D. Weinrich, H. Schröder, C. M. Niemeyer and H. Waldmann, Angew. Chem., Int. Ed., 2008, 47, 9618–9647 CrossRef CAS.
- P.-C. Lin, D. Weinrich and H. Waldmann, Macromol. Chem. Phys., 2010, 211, 136–144 CrossRef CAS.
- K. Hernandez and R. Fernandez-Lafuente, Enzyme Microb. Technol., 2011, 48, 107–122 CrossRef CAS.
- A. K. Trilling, M. M. Harmsen, V. J. B. Ruigrok, H. Zuilhof and J. Beekwilder, Biosens. Bioelectron., 2013, 40, 219–226 CrossRef CAS.
- A. Kausaite-Minkstimiene, A. Ramanaviciene, J. Kirlyte and A. Ramanavicius, Anal. Chem., 2010, 82, 6401–6408 CrossRef CAS.
- J. E. Baio, F. Cheng, D. M. Ratner, P. S. Stayton and D. G. Castner, J. Biomed. Mater. Res., Part A, 2011, 97, 1–7 CrossRef.
- I.-H. Cho, J.-W. Park, T. G. Lee, H. Lee and S.-H. Paek, Analyst, 2011, 136, 1412–1419 RSC.
- M. Park, J. Jose and J.-C. Pyun, Sens. Actuators, B, 2011, 154, 82–88 CrossRef.
- G. Shen, C. Cai, K. Wang and J. Lu, Anal. Biochem., 2011, 409, 22–27 CrossRef CAS.
- N. Tajima, M. Takai and K. Ishihara, Anal. Chem., 2011, 83, 1969–1976 CrossRef CAS.
- G. Yoo, M. Park, E. H. Lee, J. Jose and J. C. Pyun, Anal. Chim. Acta, 2011, 707, 142–147 CrossRef CAS.
- Y. Yuan, M. Yin, J. Qian and C. Liu, Soft Matter, 2011, 7, 7207–7216 RSC.
- M. M. Harmsen and H. P. D. Fijten, J. Immunoassay Immunochem., 2012, 33, 234–251 CrossRef CAS.
- A. P. Le Brun, S. A. Holt, D. S. H. Shah, C. F. Majkrzak and J. H. Lakey, Biomaterials, 2011, 32, 3303–3311 CrossRef CAS.
- E. M. Yoo, K. R. Chintalacharuvu, M. L. Penichet and S. L. Morrison, J. Immunol. Methods, 2002, 261, 1–20 CrossRef CAS.
- C. A. Borrebaeck, A. C. Malmborg, C. Furebring, A. Michaelsson, S. Ward, L. Danielsson and M. Ohlin, Biotechnology, 1992, 10, 697–698 CrossRef CAS.
- T. A. C. Hamers-Casterman, S. Muyldermans, G. Robinson, C. Hammers, E. Bajyana Songa, N. Bendahman and R. Hammers, Nature, 1993, 363, 446–448 CrossRef.
- A. S. Greenberg, D. Avila, M. Hughes, A. Hughes, E. C. McKinney and M. F. Flajnik, Nature, 1995, 374, 168–173 CrossRef CAS.
- L. Huang, S. Muyldermans and D. Saerens, Expert Rev. Mol. Diagn., 2010, 10, 777–785 CrossRef.
- D. Saerens, L. Huang, K. Bonroy and S. Muyldermans, Sensors, 2008, 8, 4669–4686 CrossRef CAS.
- C. A. K. Borrebaeck and C. Wingren, Methods Mol. Biol., 2011, 785, 247–262 CAS.
- P. J. Conroy, S. Hearty, P. Leonard and R. J. O'Kennedy, Semin. Cell Dev. Biol., 2009, 20, 10–26 CrossRef CAS.
- X. Zeng, Z. Shen and R. Mernaugh, Anal. Bioanal. Chem., 2012, 402, 3027–3038 CrossRef CAS.
- T. Q. Huy, N. T. H. Hanh, N. T. Thuy, P. V. Chung, P. T. Nga and M. A. Tuan, Talanta, 2011, 86, 271–277 CrossRef CAS.
- T. Q. Huy, N. T. H. Hanh, P. V. Chung, D. D. Anh, P. T. Nga and M. A. Tuan, Appl. Surf. Sci., 2011, 257, 7090–7095 CrossRef CAS.
- H. Chen, J. Huang, J. Lee, S. Hwang and K. Koh, Sens. Actuators, B, 2010, 147, 548–553 CrossRef.
- P. J. M. Murphy, M. Shannon and J. Goertz, J. Visualized Exp., 2011, 53, 1–5 Search PubMed.
- P. Dutta, S. Sawoo, N. Ray, O. Bouloussa and A. Sarkar, Bioconjugate Chem., 2011, 22, 1202–1209 CrossRef CAS.
- M. C. Coen, R. Lehmann, P. Gröning, M. Bielmann, C. Galli and L. Schlapbach, J. Colloid Interface Sci., 2001, 233, 180–189 CrossRef CAS.
- L. R. Farris and M. J. McDonald, Anal. Bioanal. Chem., 2011, 401, 1–9 CrossRef.
- S. Kumar, R. Ch, D. Rath and S. Panda, Mater. Sci. Eng., C, 2011, 31, 370–376 CrossRef CAS.
- E.-S. Kim, C.-K. Shim, J. W. Lee, J. W. Park and K. Y. Choi, Analyst, 2012, 137, 2421–2430 RSC.
- K. Yoshimoto, M. Nishio, H. Sugasawa and Y. Nagasaki, J. Am. Chem. Soc., 2010, 132, 7982–7989 CrossRef CAS.
- M. S. Wagner and D. G. Castner, Appl. Surf. Sci., 2004, 231, 366–376 CrossRef.
- J. W. Park, I. H. Cho, D. W. Moon, S. H. Paek and T. G. Lee, Surf. Interface Anal., 2011, 43, 285–289 CrossRef CAS.
- F. Liu, M. Dubey, H. Takahashi, D. G. Castner and D. W. Grainger, Anal. Chem., 2010, 82, 2947–2958 CrossRef CAS.
- H. Y. Song, X. Zhou, J. Hobley and X. Su, Langmuir, 2011, 28, 997–1004 CrossRef.
- J. R. Lu, E. M. Lee and R. K. Thomas, Acta Crystallogr., Sect. A: Found. Crystallogr., 1996, 52, 11–41 CrossRef.
- X. Zhao, F. Pan, B. Cowsill, J. R. Lu, L. Garcia-Gancedo, A. J. Flewitt, G. M. Ashley and J. Luo, Langmuir, 2011, 27, 7654–7662 CrossRef CAS.
- Y. M. Bae, B.-K. Oh, W. Lee, W. H. Lee and J.-W. Choi, Biosens. Bioelectron., 2005, 21, 103–110 CrossRef CAS.
- X. Liu, X. Wang, J. Zhang, H. Feng, X. Liu and D. Wong, Biosens. Bioelectron., 2012, 35, 56–62 CrossRef CAS.
- Y. Ryu, Z. Jin, M. Kang and H.-S. Kim, BioChip J., 2011, 5, 193–198 CrossRef CAS.
- A. T. Nguyen, J. Baggerman, J. M. J. Paulusse, H. Zuilhof and C. J. M. van Rijn, Langmuir, 2012, 28, 604–610 CrossRef CAS.
- Z. Yang, Y. Chevolot, T. Gehin, V. Dugas, N. Xanthopoulos, V. Laporte, T. Delair, Y. Ataman-Önal, G. Choquet-Kastylevsky, E. Souteyrand and E. Laurenceau, Langmuir, 2013 DOI:10.1021/la3041055.
- S. K. Vashist, C. K. Dixit, B. D. MacCraith and R. O'Kennedy, Analyst, 2011, 136, 4431–4436 RSC.
- B. Feng, S. Huang, F. Ge, Y. Luo, D. Jia and Y. Dai, Biosens. Bioelectron., 2011, 28, 91–96 CrossRef CAS.
- B. N. Johnson and R. Mutharasan, Langmuir, 2012, 28, 6928–6934 CrossRef CAS.
- J. H. Lee, H. K. Choi, S. Y. Lee, M. W. Lim and J. H. Chang, Biosens. Bioelectron., 2011, 28, 146–151 CrossRef CAS.
- S. Ko, C. J. Kim and D. Y. Kwon, NSTI-Nanotech, 2010, vol. 3, pp. 129–136 Search PubMed.
- J. Figueroa, S. Magaña, D. V. Lim and R. Schlaf, J. Immunol. Methods, 2011, 386, 1–9 CrossRef.
- X. Zhao, F. Pan, L. Garcia-Gancedo, A. J. Flewitt, G. M. Ashley, J. Luo and J. R. Lu, J. R. Soc., Interface, 2012, 9, 2457–2467 CrossRef CAS.
- H.-J. Um, M. Kim, S.-H. Lee, J. Min, H. Kim, Y.-W. Choi and Y.-H. Kim, Talanta, 2011, 84, 330–334 CrossRef CAS.
- M. Iafisco, E. Varoni, M. Di Foggia, S. Pietronave, M. Fini, N. Roveri, L. Rimondini and M. Prat, Colloids Surf., B, 2012, 90, 1–7 CrossRef CAS.
- Z. Balevicius, A. Ramanaviciene, I. Baleviciute, A. Makaraviciute, L. Mikoliunaite and A. Ramanavicius, Sens. Actuators, B, 2011, 160, 555–562 CrossRef CAS.
- B. D. Ventura, L. Schiavo, C. Altucci, R. Esposito and R. Velotta, Biomed. Opt. Express, 2011, 2, 3223–3231 CrossRef.
- E. J. Franco, H. Hofstetter and O. Hofstetter, J. Sep. Sci., 2006, 29, 1458–1469 CrossRef CAS.
- J. H. Kang, H. J. Choi, S. Y. Hwang, S. H. Han, J. Y. Jeon and E. K. Lee, J. Chromatogr., A, 2007, 1161, 9–14 CrossRef CAS.
- X. Yuan, D. Fabregat, K. Yoshimoto and Y. Nagasaki, Colloids Surf., B, 2012, 99, 45–52 CrossRef CAS.
- Z. Pei, H. Anderson, A. Myrskog, G. Dunér, B. Ingemarsson and T. Aastrup, Anal. Biochem., 2010, 398, 161–168 CrossRef CAS.
- Y. Liu, C. X. Guo, W. Hu, Z. Lu and C. M. Li, J. Colloid Interface Sci., 2011, 360, 593–599 CrossRef CAS.
- H. J. Han, R. M. Kannan, S. Wang, G. Mao, J. P. Kusanovic and R. Romero, Adv. Funct. Mater., 2010, 20, 409–421 CrossRef CAS.
- J.-a. A. Ho, W.-L. Hsu, W.-C. Liao, J.-K. Chiu, M.-L. Chen, H.-C. Chang and C.-C. Li, Biosens. Bioelectron., 2010, 26, 1021–1027 CrossRef CAS.
- P. Batalla, J. M. Bolívar, F. Lopez-Gallego and J. M. Guisan, J. Chromatogr., A, 2012, 1262, 56–63 CrossRef CAS.
- N. Bereli, G. Şener, H. Yavuz and A. Denizli, Mater. Sci. Eng., C, 2011, 31, 1078–1083 CrossRef CAS.
- G. Bergstroem and C.-F. Mandenius, Sens. Actuators, B, 2011, 158, 265–270 CrossRef CAS.
- M. M. Billah, C. S. Hodges, H. C. W. Hays and P. A. Millner, Bioelectrochemistry, 2010, 80, 49–54 CrossRef CAS.
- N. J. Alves, T. Kiziltepe and B. Bilgicer, Langmuir, 2012, 28, 9640–9648 CrossRef CAS.
- B. Stamos, L. Loredo, S. Chand, T. V. Phan, Y. Zhang, S. Mohapatra, K. Rajeshwar and R. Perera, Anal. Biochem., 2012, 424, 114–123 CrossRef CAS.
- U. M. Lindström, Chem. Rev., 2002, 102, 2751–2772 CrossRef.
- E. Saxon and C. R. Bertozzi, Science, 2000, 287, 2007–2010 CrossRef CAS.
- K. L. Kiick, E. Saxon, D. A. Tirrell and C. R. Bertozzi, Proc. Natl. Acad. Sci. U. S. A., 2002, 99, 19–24 CrossRef CAS.
- J. W. Chin, S. W. Santoro, A. B. Martin, D. S. King, L. Wang and P. G. Schultz, J. Am. Chem. Soc., 2002, 124, 9026–9027 CrossRef CAS.
- B. C. Bundy and J. R. Swartz, Bioconjugate Chem., 2010, 21, 255–263 CrossRef CAS.
- T. W. Muir, D. Sondhi and P. A. Cole, Proc. Natl. Acad. Sci. U. S. A., 1998, 95, 6705–6710 CrossRef CAS.
- J. Kalia, N. L. Abbott and R. T. Raines, Bioconjugate Chem., 2007, 18, 1064–1069 CrossRef CAS.
- I.-H. Cho, E.-H. Paek, H. Lee, J. Y. Kang, T. S. Kim and S.-H. Paek, Anal. Biochem., 2007, 365, 14–23 CrossRef CAS.
- M. P. Cassidy, J. Raushel and V. V. Fokin, Angew. Chem., Int. Ed., 2006, 45, 3154–3157 CrossRef CAS.
- T. Govindaraju, P. Jonkheijm, L. Gogolin, H. Schroeder, C. F. W. Becker, C. M. Niemeyer and H. Waldmann, Chem. Commun., 2008, 3723–3725 RSC.
- R. Manova, T. A. van Beek and H. Zuilhof, Angew. Chem., Int. Ed., 2011, 50, 5428–5430 CrossRef CAS.
- J. C. Jewett and C. R. Bertozzi, Chem. Soc. Rev., 2010, 39, 1272–1279 RSC.
- M. F. Debets, S. S. van Berkel, J. Dommerholt, A. J. Dirks, F. P. J. T. Rutjes and F. L. van Delft, Acc. Chem. Res., 2011, 44, 805–815 CrossRef CAS.
- M. D. Witte, J. J. Cragnolini, S. K. Dougan, N. C. Yoder, M. W. Popp and H. L. Ploegh, Proc. Natl. Acad. Sci. U. S. A., 2012, 109, 11993–11998 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2013 |