 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
FTIR microspectroscopy for rapid screening and monitoring of polyunsaturated fatty acid production in commercially valuable marine yeasts and protists†
Jitraporn Vongsvivut*a,
Philip Heraudb,
Adarsha Guptaa,
Munish Puria,
Don McNaughtonb and
Colin J. Barrowa
aCentre for Chemistry and Biotechnology (CCB), School of Life and Environmental Sciences, Deakin University, Pigdons Road, Waurn Ponds, Victoria 3217, Australia. E-mail: p.vongsvivut@deakin.edu.au; Fax: +61 3 5227 1040; Tel: +61 3 5227 2096
bCentre for Biospectroscopy, School of Chemistry, Monash University, Wellington Road, Clayton, Victoria 3800, Australia
First published on 31st July 2013
Abstract
The increase in polyunsaturated fatty acid (PUFA) consumption has prompted research into alternative resources other than fish oil. In this study, a new approach based on focal-plane-array Fourier transform infrared (FPA-FTIR) microspectroscopy and multivariate data analysis was developed for the characterisation of some marine microorganisms. Cell and lipid compositions in lipid-rich marine yeasts collected from the Australian coast were characterised in comparison to a commercially available PUFA-producing marine fungoid protist, thraustochytrid. Multivariate classification methods provided good discriminative accuracy evidenced from (i) separation of the yeasts from thraustochytrids and distinct spectral clusters among the yeasts that conformed well to their biological identities, and (ii) correct classification of yeasts from a totally independent set using cross-validation testing. The findings further indicated additional capability of the developed FPA-FTIR methodology, when combined with partial least squares regression (PLSR) analysis, for rapid monitoring of lipid production in one of the yeasts during the growth period, which was achieved at a high accuracy compared to the results obtained from the traditional lipid analysis based on gas chromatography. The developed FTIR-based approach when coupled to programmable withdrawal devices and a cytocentrifugation module would have strong potential as a novel online monitoring technology suited for bioprocessing applications and large-scale production.
1 Introduction
In recent years, omega-3 (n − 3) polyunsaturated fatty acids (PUFAs), particularly the n − 3 docosahexaenoic acid (DHA; 22:6n − 3) and eicosapentaenoic acid (EPA; 20:5n − 3), have become increasingly popular in the nutraceutical arena due to their important roles in brain function and prevention of cardiovascular diseases as well as maintaining good health.1 This has led to a rapid increase in PUFA consumption, based mainly on fish. Other potential alternative resources not relying on fish stocks have been the subject of active research. Many studies in marine biology have found high lipid accumulation, particularly for PUFAs, in a broad range of marine microorganisms including micro-algae, bacilli, fungi, yeasts and protists.2–5 As a consequence, these marine microbes have received extensive attention in the fields of biotechnology and biodiesel due to their potential to form the basis of a viable industry to supply a large scale of vegetative biomass containing oils rich in PUFAs.In particular, recent studies have shown that pigmented marine-derived yeasts of the genus Rhodotorula are capable of accumulating high lipid content, including essential PUFAs,6 and of growing at a high rate under optimised culture conditions, thus providing a rapid increase in biomass.7 Such characteristics are crucial for a large-scale production and therefore the yeasts promise to play key roles in modern biotechnology. In this study, Rhodotorula species were collected off the coast near Queenscliff (Victoria, Australia), and molecular identification was carried out using 18s rDNA gene sequence analysis after strain isolation.8 Each of the four strains of Rhodotorula sp. selected for this study possesses distinctive colors varying from pale yellow to orange, pink and red tones. The colours arise from pigments, which are produced to screen wavelengths of light that can potentially damage the cell.9 The traditional identification of yeasts is based mainly on the morphology and physiological tests that determine enzyme production profiles and growth characteristics, which involve an intensive use of reagents and are cumbersome as well as time consuming.
Recently, Fourier transform infrared (FTIR) microspectroscopy, combined with chemometric approaches, has emerged as a viable alternative to traditional techniques and has been used extensively in biological and medical fields.10–13 In particular, the ability to use FTIR spectroscopy for taxon specific identification was first demonstrated with bacteria,14,15 and more recently with eukaryotic fungi and yeasts.16–18 Our present study further demonstrates the potential to discriminate strains of novel marine yeasts from thraustochytrids using chemometric approaches developed based on the FTIR spectral data. The technique is fast, non-destructive and requires only minimal sample preparation. In practice, the marine microorganisms can be directly examined as intact cells.10,19 This results in highly accurate analyses of the chemical compositions of the whole cells, which can lead to a better understanding and optimisation of PUFA production in these cultured microorganisms. Focal plane array (FPA) FTIR imaging has proven to be very powerful for the rapid acquisition of thousands of spectra and collection into one spectral image within minutes compared to the hours required for single-point measurements for the same number of spectra. By applying multivariate data analysis to the thousands of spectra collected simultaneously from a monolayer of cells, complex information on the chemical variation within cell populations can be rapidly assessed for identification, classification, and quality control standardization purposes. Furthermore, there is potential for direct quantification of PUFA produced in the cells.
In this study, we report applications of FPA-FTIR microspectroscopy combined with the multivariate data analysis methods, including principal component analysis (PCA), partial least squares discriminant analysis (PLS-DA), soft independent modelling of class analogy (SIMCA) and hierarchical cluster analysis (HCA), for discrimination and classification of the newly isolated Rhodotorula yeast strains in comparison to a commercially available PUFA-producing thraustochytrid that has been used in the commercial production of vegetative n − 3 PUFAs, and is used here as a standard for assessing the potential of these marine yeasts. In addition, partial least squares regression (PLSR) analysis using the FPA-FTIR spectral datasets and their lipid profiles acquired from the traditional gas chromatography (GC) technique was applied to monitor the production of unsaturated fatty acids (UFAs) and total lipids in a Rhodotorula strain grown in an optimised glucose medium. The optimal UFA and lipid contents were then compared to those of the control in a nutrient medium without glucose, and of thraustochytrids grown under a recommended culture condition. The accuracy of the PLSR calibration models was subsequently tested using a cross-validation approach based on two independent replicate datasets, in order to evaluate the capability and the potential of the developed technique as a rapid lipid analyser of cultured cells.
2 Materials and methods
2.1 Materials and reagents
Four marine-derived yeast strains genetically identified as Rhodotorula sp. were collected from the Queenscliff region in Victoria, Australia in August 2010.8 A commercial Thraustochytrium sp. AH-2 (PRA-296™) was procured from ATCC (Manassas, VA, USA) and grown under the recommended optimised condition for this specific thraustochytrid strain according to the product information sheets. All chemicals used in this study were of analytical grade and purchased from Sigma-Aldrich (Australia) and Merck Chemicals (Australia).2.2 Biological methodology
As illustrated in Fig. 1, the isolation and screening of the Rhodotorula sp. were performed simultaneously in five biological replicates, which were prepared in five independent growth cultures (i.e. different cultivation flasks) under the same controlled conditions based on the recently published procedure.8 The growth study and the GC-based fatty acid analysis of each microorganism were performed and observed in triplicate according to the published protocols,8 whilst FPA-FTIR measurements were performed in duplicate using the other two biological replicates.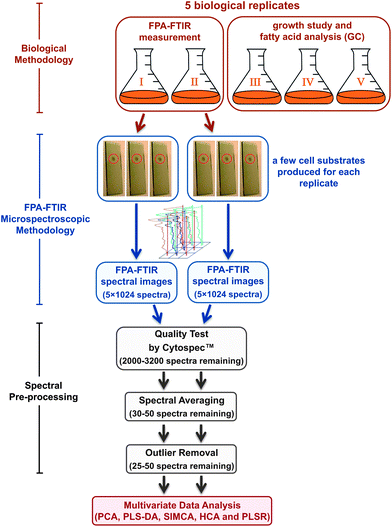 | ||
| Fig. 1 Flow diagram of the experimental procedure used in the study including biological and FPA-FTIR microspectroscopic methodologies followed by spectral pre-processing, prior to the multivariate data analysis. The number of spectra mentioned in the figure represents the total number of spectra remaining after each processing step. | ||
In brief, the yeast samples from the original sea water and sediments were directly placed into 50 mL polyethylene Falcon tubes containing penicillin and streptomycin (300 mg L−1 each), and kept in ice prior to laboratory use. Suspensions were spread on Petri plates containing an agar medium prepared using 1 g yeast extract, 1 g peptone, 2 g glucose and 10 g agar in 1 L of Instant Ocean™ artificial seawater (Aquarium Systems, Mentor, OH) and the same combination of the antibiotics (i.e. 300 mg L−1 penicillin and streptomycin) prior to incubation at 25 °C for 5 days. After that, the colonies were picked and sub-cultured on agar plates to ensure purity.
To grow the yeast isolates for lipid production, an optimised liquid medium was prepared by adding 10 g yeast extract, 15 g peptone and 30 g glucose to 1 L of the artificial seawater. A nutrient medium without glucose to be used as a control was prepared by adding 15 g beef extract, 15 g yeast extract, 5 g peptone to 1 L of the artificial seawater. The prepared growth media were autoclaved at 121 °C for 20 min and then subsequently brought to room temperature prior to use. Cultures of the four yeast strains, labelled as AMCQ10C, AMCQ12C, AMCQ1D and AMCQ8A, were then inoculated from their agar plates into autoclaved 250 mL Erlenmeyer flasks containing 50 mL of the sterile media. The culture solutions of each yeast isolate were collected on a daily basis and the growth was observed in terms of cell concentration using a Bright-Line™ haemocytometer (Sigma-Aldrich, New South Wales, Australia). The harvested yeast cells were subsequently preserved in 5% formalin in an isotonic saline solution. The onset of the stationary phase, at which optimal lipid accumulation was observed in a broad range of marine eukaryotic and prokaryotic cells,20 was found to occur on day 5 for these yeast isolates after the cells were sub-cultured into the liquid media.
Thraustochytrium sp. AH-2 (PRA-296™) was grown in 50 mL of ATCC recommended medium #2673 prepared by adding 1 g yeast extract, 15 g peptone and 20 g glucose to 1 L of the artificial seawater. The medium was autoclaved and allowed to cool to room temperature. Thraustochytrids were then grown and harvested at the onset of the stationary phase, found to occur on day 7 of their growth following inoculation of the culture according to observation of the cell concentration and the optical density (OD) at 600 nm using a UV-visible absorption spectrophotometer (Model UV-1800, Shimadzu Scientific Instruments, Japan) performed at regular time intervals. Similarly, the harvested thraustochytrium cells were preserved in 5% formalin in an isotonic saline solution prior to use for FPA-FTIR measurements.
2.3 FPA-FTIR microspectroscopy methodology
The formalin solution was initially discarded from the preserved cells by decanting the supernatant after centrifugation of the cell suspension at 5000 × g and 4 °C. The cell pellet was then washed with sterile isotonic saline solution and centrifuged twice before being deposited using a cyto-centrifuge (Cytospin-III, Thermo Fischer Scientific, MA, USA) to produce monolayers of cells on IR reflective glass slides (MirrIR slides, Kevley Technologies, OH, USA). The films were left to dry in a desiccator containing silica gel beads for ca. 30 min to dehydrate the cells, prior to FTIR spectral data collection. The unique advantage of using this specific cytocentrifugation instrument is due particularly to the use of disposable filter pads that adsorb the salt solution from the cell suspension during deposition of the cell monolayer, thereby no residual salt crystals that can lead to strong scattering artefacts in collected spectra were observed in this study.In addition, it should be noted that the main purpose of using the formalin fixation method is to preserve and thus to minimise degradation of the cell content, particularly the PUFAs that are prone to oxidation, from the time the cells were harvested until the FTIR spectral datasets were acquired. There are, of course, macromolecular changes especially those associated with cross-linking in proteins produced by the fixation in this step. However, previous studies have shown that these changes are largely confined to the amide I modes, with little or no effect on lipid modes.21
FPA-FTIR spectra were collected using a FTIR microscope (Model 600 UMA, Agilent Technologies, Santa Clara, CA, USA), equipped with a liquid-N2 cooled 64 × 64 element Stingray FPA detector (Agilent Technologies) and a 15× objective lens, coupled to a FTIR spectrometer (Model FTS 7000, Agilent Technologies). Spectra were collected in reflectance mode in the 4000–800 cm−1 spectral region as a single FTIR image covering a sampling area of 350 × 350 μm2. Each FTIR spectral image consisted of a 32 × 32 array of spectra resulting from binning the signal from each square of 4 detectors on the 64 × 64 element FPA array. As a consequence, a single spectrum contained in a FTIR image represented molecular information acquired from ca. 10.9 × 10.9 μm2 area on the sample plane, which was equivalent to the average size of one single yeast cell (i.e. 10 μm diameter), whilst a few single spectra could be obtained from the same thraustochytrium cell because their size was on average twice the size of the yeast cells. For each biological replicate, at least five high-quality FTIR spectral images were collected at 8 cm−1 resolution, 128 co-added scans, Blackman–Harris 3-Term apodization, Power-Spectrum phase correction and a zero-filling factor of 2 using Resolution Pro™ IR imaging software (Agilent Technologies). Background measurements were performed prior to each sample spectral image measurement, by focusing on a clean unused surface of the substrate using the same acquisition parameters.
2.4 Spectral pre-processing
FTIR spectra embedded in each spectral image were first extracted and quality-screened using CytoSpec™ v. 1.4.02 (Cytospec Inc., Boston, MA, USA). Two criteria were selected for the quality screening test. The first involved an appropriate sample thickness, which was assessed according to the absorbance over the 950–1750 cm−1 spectral range, to remove spectra with the maximum absorbance less than 0.2 or greater than 0.8. The second criterion aimed for high-quality spectra was based upon a minimum signal-to-noise (S/N) ratio of 150 measured using the signal and the noise over the spectral ranges of 1600–1760 and 1800–1900 cm−1, respectively. The absorbance and S/N ratio figures used in these two criteria were set based on the previous experience in our laboratory with spectra acquired from monolayers of biological cells using the FPA-FTIR microspectroscopy in a similar optical setting. From our experience, these cut-off values have been shown to eliminate noisy spectra and those from regions of the sample where the substrate may be only partially covered with cells, as well as those possessing very high absorbance outside the linearity range of the detector. As a result, the quality-screening procedure ensured that only spectra of high quality (i.e. good S/N ratio) were used for further analysis.After the quality test, averaging of every 64 spectra was performed on the raw spectra that passed the prior quality-test screening criteria to further improve the quality of the spectra and to produce spectra most representative of the sample population, before spectral pre-treatment and further analysis. It should be noted that although the spectral averaging procedure reduces the spatial discriminatory features among spectra in the same image set, the trade-off is the improvement of the model robustness and classification performance as a result of high quality spectral input. In this light, the FPA-FTIR technique provides a key advantage over single-point data acquisition, through its unique capability of efficient spectral selection to remove spectra of poor quality including those possessing low S/N ratio, signal saturation and scattering artefacts, and subsequently for the generation of pristine average spectra.
In each cell strain, the representative average spectra (approximately 30–50 spectra) from the two replicates were combined and converted to 2nd derivatives using a 9-point Savitzky–Golay algorithm to eliminate the broad baseline offset and curvature.22 The resultant derivative spectra were corrected by the extended multiplicative scatter correction (EMSC) method23 in the spectral regions 3100–2800 and 1780–965 cm−1 that contain the molecular information relevant to most biological samples (i.e. protein, lipid, carbohydrate and nucleic acid signals). In essence, the EMSC algorithm removes light-scattering artefacts and normalises the spectra accounting for pathlength differences. The EMSC pretreatment often yields a better interpretability, more robust calibration models, and thereby an improved predictive accuracy as the EMSC-corrected spectra respond more linearly to the analyte concentration when compared to those obtained from untreated spectra.
2.5 Multivariate data analysis
Multivariate data analyses including PCA,24 PLS-DA,25,26 SIMCA,27 HCA,14 and PLSR25 were performed using The Unscrambler® 10.1 software package (CAMO Software AS, Oslo, Norway). The PCA approach was first applied to individual groups of the four yeast isolates and the thraustochytrids containing two replicates, in order to identify and eliminate outliers among samples in the same class. This initial PCA outlier assessment additionally revealed a good consistency in the spectral variation between the two biological replicates in each cell group. In fact, the outliers represented less than 5% of the total dataset in all cases, and approximately 95% of the total spectra from both replicates in each class were selected from the main cluster of spectra in influence (residual versus leverage) plots with low levels of the residual variance and model leverage and hence most representative of the PCA models.After the selection of representative spectra from each cell group, the EMSC-corrected 2nd derivative spectral datasets of all the yeast isolates and the thraustochytrids were combined into one single set. PCA was subsequently performed on the entire combined set in order to investigate similarities and differences between the cell groups. Note that due to the good consistency of the data previously observed within the same class, the duplicate datasets of each cell class were presented as a single set in the PCA and HCA analyses in order to simplify and provide a better clarity for the presentation of the results.
Classification of spectra using PLS-DA and SIMCA, on the other hand, was performed by keeping the replicate datasets separate following the outlier removal. The spectral datasets of each replicate from every yeast isolate and the thraustochytrids were subsequently combined to form replicate I and II sets including 81 and 79 spectral samples, respectively. A similar data pre-processing procedure as described previously including 2nd derivatisation and the EMSC approach was applied to spectra in each replicate set individually within the sets. Initially, the pre-processed replicate I and II sets were used to perform as training and independent validation (test) sets, respectively. Spectra in the training set were then used to construct PCA-based regression or local models, while samples in the independent validation (test) set were set aside for subsequent classification. After acquiring the classification results of the first model, the role of the two replicate sets was reversed in the second model by using the replicate II dataset as the training set and replicate I spectra as the independent test samples. The classification results obtained from the two cross-validation models were later compared. The cross-validation employing independent biological replicates was used to investigate the influence of each dataset on the model robustness and predictive accuracy. The classification performance was estimated from the number of correctly classified samples in each validation (test) set, whereas the discriminative capability particularly in the HCA was assessed based on the good correlation between the biological identity of the samples and the dendrogram structure.
2.6 Quantitative analysis of lipid contents produced during the growth
Initially, semi-quantitative analysis of the lipid content accumulated in the cells was performed in terms of %UFAs per total lipids, using integrated areas under EMSC-corrected 2nd derivative bands. Peaks centred at 3006 (or 3014 for thraustochytrids) and 1743 cm−1 were used as representatives of UFAs and total lipids, respectively. The %UFA values were thus calculated as the percentage ratio of the integrated area covering the band centered at 3006 (or 3014) cm−1 to that covering the band centered at 1743 cm−1.Consecutively, quantitative determination of %UFAs was performed using PLSR analysis by combining the EMSC-corrected 2nd derivative FPA-FTIR spectra of the replicate I dataset and their corresponding reference %UFA values obtained from the GC technique,8 in order to construct an initial PLSR calibration model. The validation was subsequently conducted on the pre-processed replicate II spectral dataset to obtain predicted %UFA values. Similar to PLS-DA and SIMCA, the cross-validation approach was implemented by reversing the roles of the replicate datasets to cross-check and compare the model performance and the predictive accuracy obtained from the two cross-validation models, in relation to the reference values derived from the GC data.
3 Results and discussion
3.1 FTIR spectral comparison of yeast and thraustochytrium cells
Fig. 2 presents the representative EMSC-corrected absorbance and 2nd derivative spectra of the yeasts Rhodotorula sp., in comparison to that of thraustochytrids, obtained by averaging the spectra in each of the 5 datasets. The detailed assignments of the minima found in 2nd derivative spectra and the references for these are given in Table 1. Typically, spectral features in the range of 3100–2800 cm−1 are characteristic of the C–H stretching vibrations of lipids.28,29 The C–H stretching band of olefinic C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CH– chains observed either at 3014 or 3006 cm−1 (for thraustochytrids and the Rhodotorula yeasts, respectively) is a result of UFAs produced inside the cells, and is thereby used for examining the degree of unsaturation in lipids and oils.30,31 The bands at 2960/2872 and 2925/2852 cm−1 are attributable to asymmetric/symmetric C–H stretching vibrations of –CH3 and aliphatic –CH2 functional groups, respectively. The other prominent peak relevant to the lipid moiety occurs in the lower wavenumber region at ∼1743 cm−1 assignable to ν(CO) stretches of ester functional groups from lipid triglycerides and FAs,28 and therefore represents total lipids in the cells.
CH– chains observed either at 3014 or 3006 cm−1 (for thraustochytrids and the Rhodotorula yeasts, respectively) is a result of UFAs produced inside the cells, and is thereby used for examining the degree of unsaturation in lipids and oils.30,31 The bands at 2960/2872 and 2925/2852 cm−1 are attributable to asymmetric/symmetric C–H stretching vibrations of –CH3 and aliphatic –CH2 functional groups, respectively. The other prominent peak relevant to the lipid moiety occurs in the lower wavenumber region at ∼1743 cm−1 assignable to ν(CO) stretches of ester functional groups from lipid triglycerides and FAs,28 and therefore represents total lipids in the cells.
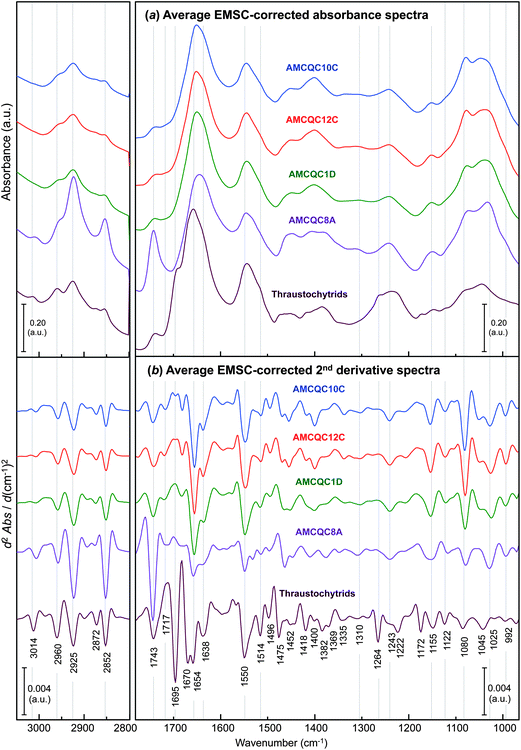 | ||
| Fig. 2 Comparisons of the average EMSC-corrected (a) absorbance and (b) 2nd derivative spectra of the four Rhodotorula yeast isolates and the thraustochytrids (PRA-296™) taken at the onset of the stationary phase. Note that the EMSC-corrected 2nd derivative spectra were processed by 2nd derivatisation and then EMSC in a similar order used throughout the manuscript. | ||
| Wavenumber values (cm−1) | Band assignmenta | Reference |
|---|---|---|
| a νas = asymmetric stretch; νs = symmetric stretch; δs = symmetric in-plane deformation (bend); δas = asymmetric in-plane deformation (bend); γ = out-of-plane deformation.b Representative bands for UFAs.c Representative bands for total lipids.d Bands are sensitive to the orthorhombic-like to hexagonal packing transition of the –CH2 groups in the phospholipid bilayers.e Often present with ν(C–O) of the dimeric ring at 1300 cm−1. | ||
| ∼3014 (3006)b | ν(C–H) of cis CCH– |
28 |
| ∼2960 | νas(C–H) from methyl (–CH3) groups of lipids | 29 |
| ∼2925 | νas(C–H) from methylene (–CH2) groups of lipids | 29 |
| ∼2872 | νs(C–H) from methyl (–CH3) groups of lipids | 29 |
| ∼2852 | νs(C–H) from methylene (–CH2) groups of lipids | 29 |
| ∼1743c | ν(CO) of esters from lipid triglycerides and fatty acids |
28 |
| ∼1717 | ν(CO) of free fatty acids and α,β-unsaturated esters |
28 |
| ∼1695 | ν(CO) of α,β-unsaturated aldehydes |
29 |
| Amide I: aggregated β-sheet | 34 | |
| ∼1670 | Amide I: β-turn | 34 |
| ∼1654 | Amide I: α-helix | 34 |
| ν(CC) of disubstituted cis-olefins |
28 | |
| ∼1638 | Amide I: antiparallel β-sheet | 34 |
| ν(CO) of carboxylate and ν(CC) of aromatic compounds |
43 | |
| ∼1550 | Amide II: perpendicular modes of the α-helix and antiparallel β-sheet | 50 |
| ∼1514 | Amide II: parallel mode of the α-helix | 50 |
| ∼1496 | ν(CC) of phenyl rings from the aromatic amino acid phenylalanine (Phe) |
34 |
| ∼1475, 1465d | δscissor(CH2) from methylene (–CH2) groups in acyl chains of lipid bilayers in orthorhombic packing | 28, 41 and 42 |
| ∼1452 | δas(CH3) of proteins (possibly in DNA and RNA) | 44 |
| ∼1441 | ν(C–N) of the pyridine ring | 51 |
| ∼1418 | δrock(CH2) of disubstituted cis-olefins | 28 |
| δ(O–H) of the dimeric carboxyl groupe | 45 | |
| ∼1400 | νs(COO−) associated with δs(CH3) of proteins | 44 and 52 |
| ∼1382 | δs(CH3) and δs(CH2) of lipids and proteins | 28 |
| ∼1369 | δs(CH3) from methyl groups of cholesteryl and fatty acid radicals | 45 |
| ∼1335 | γwag(CH2) of α-CH2 groups in polymethylene chains | 45 |
| ∼1310 | Amide III: α-helix | 35 |
| ∼1264 | νs(C–O) and/or δ(O–H) possibly of carboxylic acids | 31 and 43 |
| ∼1243 | Amide III: β-sheet | 35 |
| ∼1222 | νas(PO2−) of the phosphodiester backbone of nucleic acids (DNA and RNA) and phospholipids | 44 |
| ∼1172 | νs(C–O–C) from esters | 34 |
| ∼1155 | νas(CO–O–C) of glycogen and nucleic acids (DNA and RNA) | 34 |
| ∼1122 | νs(C–O) at the 2′-OH group of ribose rings in RNA | 36 and 53 |
| ∼1080 | νs(PO2−) of the phosphodiester backbone of nucleic acids (DNA and RNA) and phospholipids | 42 and 44 |
| ∼1065 | νs(R–O–P–O–R′) from ring vibrations of carbohydrates | 42 |
| ∼1045 | ν(C–O) coupled with δ(C–O) of C–OH groups of carbohydrates | 44 |
| ∼1025 | ν(C–C)skeletal coupled with δ(CH2) of α-CH2 in –CH2OH groups of polysaccharides | 44 and 45 |
| ∼992 | γ(CH) of conjugated trans,trans isomers |
45 |
| Ribose-phosphate main chain vibration involving the 2′-OH group of ribose rings in RNA | 36 and 53 | |
Of note is the peak representing UFAs observed at 3014 cm−1 for thraustochytrids, but red-shifted to 3006 cm−1 in the yeast spectra. The difference between the mean positions of these UFA band minima in 2nd derivative spectra for the thraustochytrid and the yeasts was found to be highly significant statistically (i.e. 3014 ± 0.14 cm−1 and 3006 ± 0.11 cm−1, respectively, with P < 0.001 by ANOVA). In accordance with the fact that the higher the number of olefinic (CCH–) double bonds the higher the wavenumber of the peak maximum,32 the shift of this peak maximum to a lower wavenumber suggests a lower degree of unsaturation in the yeast oil compared to that produced by thraustochytrids. The intensities of the band at 1743 cm−1 suggests that the yeast AMCQ8A produced the highest amount of the total lipids among the other cells. The GC-based FA composition profile from the oil extracted from the yeast isolate in our recently published results8 revealed three types of UFAs present inside the cells consisting mainly of mono-unsaturated oleic acid (C18:1n − 9) with di-unsaturated linoleic acid (C18:2n − 6) and tri-unsaturated α-linolenic acid (C18:3n − 3) present to a lesser extent. In contrast to this, our recent GC results from the thraustochytrium oil reported a number of UFAs with higher numbers of olefinic bonds. The first five PUFAs with highest % total fatty acids are docosahexaenoic acid (DHA, C22:6n − 3; 34.67 ± 2.07%), docosapentaenoic acid (osbond acid, 22:5n − 6; 9.73 ± 0.42%), eicosapentaenoic acid (EPA, C20:5n − 3; 3.75 ± 0.09%), docosapentaenoic acids (DPA, C22:5n − 3; 1.63 ± 0.07%) and eicosatetraenoic acid (ETA, C20:4n − 3; 1.26 ± 0.05%) (see the ESI S1† for the complete list of the fatty acid composition of the thraustochytrids). As a consequence, these highly unsaturated FAs in the oil produced by thraustochytrids with at least four CC bonds in the structures are consistent with the shift of the band to a higher wavenumber as compared to those observed for the yeast cells. Nevertheless, the value of the yeasts for fatty acid production is indicated by the formation of high levels of linoleic and α-linolenic acids – two essential FAs that cannot be synthesised in mammals, but play a crucial role as precursors in an enzymatic conversion to convert into DPA, EPA and DHA in the human body.33 Together with the advantages of fast growth, high biomass and high total lipid content, the Rhodotorula yeast particularly for the isolate AMCQ8A shows potential as an alternative resource of essential FAs suitable for large-scale vegetative oil production in both the biotechnology and biodiesel fields.
The bands in the ranges of 1680–1630, 1560–1510 and 1260–1220 cm−1 arise due to amide I, II and III modes in proteins, respectively. Among these spectral regions, amide I and III spectral bands have been found to be the most sensitive to the variations in secondary structure folding of peptides and proteins.34,35 In particular, the amide I modes, which primarily represent CO stretching vibrations of amide groups, are most often used and by far best characterised for types of secondary protein structures due to their strong absorbance. Accordingly, the amide I bands were primarily used in this study to determine differences in protein conformation present in the two types of cells. Specifically, the amide I bands found in the yeast isolates have a distinct peak at 1654 cm−1 between two weaker bands around 1638 and 1670 cm−1, suggesting the dominance of α-helical proteins in the yeast cells with substantially smaller contributions from β-sheet and β-turn protein conformers in respective order. In contrast, proteins in the thraustochytrium strain are prominently in α-helices and β-turns combined with β-sheets to a lesser extent as evidenced by the doublets observed at 1670 and 1654 cm−1 with a weaker band at 1638 cm−1. The amide III bands present at 1310 and 1243 cm−1, albeit relatively weak, further support the presence of characteristic α-helix and β-sheet protein conformations, respectively, in the thraustochytrium strain.
Of interest is the presence of the sharp peak at 1695 cm−1 observed only for thraustochytrids. Although a band at this position is commonly attributed to CO stretching vibrations of the nucleic acid bases in single-stranded DNA,36 the intensity of the peak is far stronger than those normally found for DNA components and the majority of nuclear DNA in cells will rather be double stranded.37,38 Due to the thraustochytrium cells being very rich in PUFAs, the band is more likely due to CO stretching modes in isoprostanes as well as α,β-unsaturated aldehydes and ketones, which are the end products of spontaneous lipid peroxidation through a free radical mechanism.33 Since this lipid peroxidation predominantly occurs with PUFAs or their esters that contain three or more CC bonds, it explains why such a strong CO band is observed only for the PUFA-rich thraustochytrids, and not for the yeast cells that produced only fatty acids of a low degree of unsaturation. The formation of aldehyde products through lipid peroxidation is very common and certain aldehyde species have been used as biomarkers to measure the level of oxidative stress in an organism in vivo.39 The presence of an additional peak within the amide III region at 1264 cm−1 in the thraustochytrid spectrum further supports the existence of lipid peroxidation in the cells as this feature corresponds to C–O stretching and/or O–H in-plane bending vibration, which was previously used as evidence of peroxidative damage in model phospholipids and human erythrocytes.31
According to our on-going experiment using synchrotron FTIR microspectroscopy to examine live thraustochytrium cells (see ESI S2†), the 2nd derivative synchrotron FTIR spectra of the live cells revealed prominent bands around 1695, 1638 and 1264 cm−1 similar to those observed for the formalin-fixed cells using a laboratory-based FPA-FTIR microspectroscope. However, these bands which represent oxidative moieties (e.g. aldehydes, ketones, carboxylic and carboxylate species) are present at substantially lower intensities than seen in the spectra of the dehydrated (formalin-fixed) cells as described above. Because polyunsaturated acyl chains of membrane phospholipids are particularly sensitive to lipid peroxidation that is self-propagating in the cellular membrane,33 the prolonged period of time spent for cell fixation increases the likelihood of the cell membrane being exposed to atmospheric conditions, and this is speculated to be the main factor influencing the larger amount of peroxidation products in the formalin-fixed cells. The presence of spectral bands indicative of lipid peroxidation in the live cells was presumably due to oxidative stress promoted by the environmental conditions experienced in the IR wet cell used in the current experiments,40 which did not include temperature control and was not a flow-through design. Further FTIR experiments should aim at following the lipid peroxidation process in extracted thraustochytrium oils under UV exposure and subjected to anti-oxidants to gain a better understanding of lipid chemistry in the thraustochytrids.
3.2 Classification of cells by multivariate analysis
O stretches of oxidative products and β-turn protein conformers that are predominant in thraustochytrids. The other influential component involves the negative loading at 1475 cm−1 suggesting that the preferred orientation of methylene groups in the phospholipid bilayers exists more as orthorhombic (rather than hexagonal) packing in the thraustochytrium cells.28,41,42 As expected, the loading plot also reveals a substantial negative loading at 1264 cm−1 attributed to stretching vibrations of C–O bonds possibly in carboxylic acids,31,43 which further supports the existence of lipid peroxidation in the PUFA-rich thraustochytrids as discussed above. Other differences with a considerable impact on classification are indicated by the negative loadings at 1222 and 1172 cm−1 (i.e. asymmetric phosphate stretching modes of phosphorylated moieties44 and symmetric stretches of C–O–C bonds in esters,34 respectively), as well as the positively loaded peaks present at 1065 and 1025 cm−1 due to C–O stretching vibrations from carbohydrates.42,44,45 The PC2 loading plot, on the other hand, reveals major components that set the yeast isolate AMCQ8A apart from the other isolates. While the heavily loaded peaks at 2925, 2852 and 1743 cm−1 are accounted for by differences in lipid composition, the positive loadings at 1654 and 1550 cm−1 suggest differences involving α-helical conformations of proteins in the different strains. The loadings at 1078 and 1065 cm−1 further indicate contributions from the stretching vibrations of phosphorylated molecules and carbohydrates.42,44
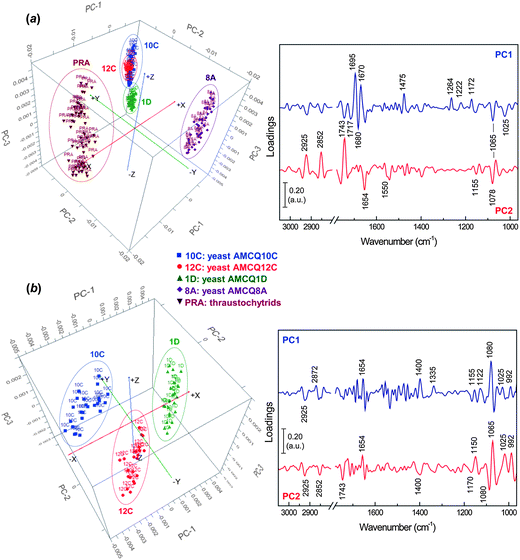 | ||
| Fig. 3 PCA score (left) and loading (right) plots showing projections against the first 3 PCs that explain the majority of the spectral variation with the inclusion of the datasets from (a) all yeast isolates and a thraustochytrium strain and (b) yeast isolates AMCQ10C, AMCQ12C and AMCQ1D, alone. | ||
Subsequently, PCA was performed with only the datasets of three yeast isolates AMCQ10C, AMCQ12C and AMCQ1D, where the spectral clusters were previously located close to each other in the PCA score plot. The results in Fig. 3b clearly show distinct separation of spectral clusters on score plots from the three isolates explained by strong loadings at 1080 and 1065 cm−1 for phosphate and carbohydrate moieties. The other substantial negative loadings at 1025 and 992 cm−1 are attributed respectively to major functional groups in polysaccharides and conjugated trans,trans isomers.44,45
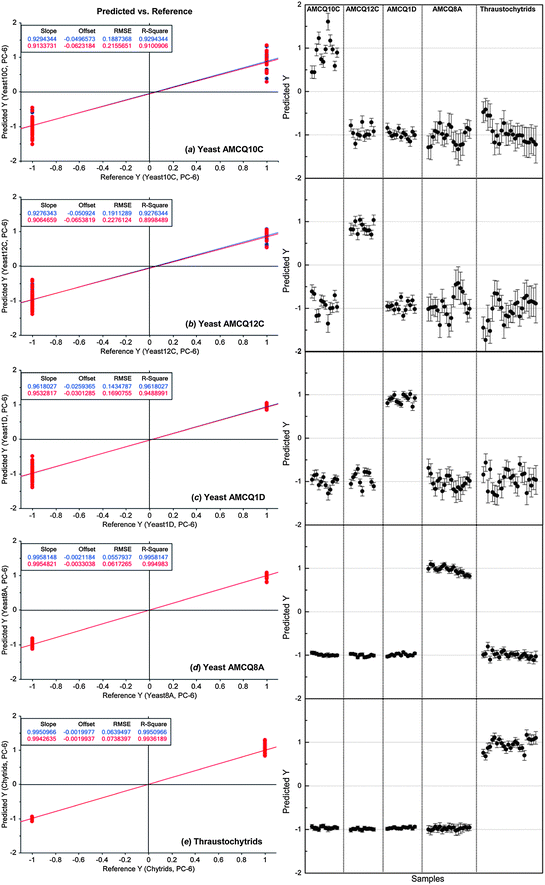 | ||
| Fig. 4 PLS-DA results showing linear regression models of individual yeast isolates and the thraustochytrid trained by using the replicate I spectral dataset (left) and their corresponding prediction results to identify the yeast/thraustochytrid samples in the replicate II set as independent validation samples (right). The nominated Y values of +1/−1 in the prediction represent yes/no classification decisions, respectively, showing that 100% of samples in the independent validation set were correctly classified. Note that the numbers of the cell samples included in replicate I and II sets are 81 and 79, respectively. | ||
Next, SIMCA was applied to test the robustness and discrimination power of different classification methods using the same cross-validation approach. Although both PLS-DA and SIMCA are based upon PCA, the main difference between the two classification methods is the criterion used to build models – SIMCA computes individual models based on PCA to identify variations within each class, but the PLS-DA identifies directions in the data space that discriminate classes directly and due to the number of variables PLS-DA was performed in this study in order to model several Y-variables simultaneously. The prediction results obtained by SIMCA according to the cross-validation approach are presented in Table 2 and ESI S4,† showing that classification of some of the test samples that belonged to the yeast isolates AMCQ10C and AMCQ12C were confounded. This typically occurs for SIMCA when the inter-cluster distance becomes close, which was true for these two yeast isolates since their PCA clusters were observed to be overlapped in the score plot as depicted in Fig. 3a. Because of this, the test samples that belong to the yeasts AMCQ1D, AMCQ8A and the thraustochytrids of which the PCA clusters are well isolated were all correctly classified by SIMCA.
| Samples class membership 5% | Yeasts Rhodotorula sp. | Thraustochytrids | |||
|---|---|---|---|---|---|
| AMCQ10C | AMCQ12C | AMCQ1D | AMCQ8A | ||
| 10C-R2_04 | * | * | |||
| 10C-R2_06 | * | * | |||
| 10C-R2_07 | * | ||||
| 10C-R2_10 | * | ||||
| 10C-R2_15 | * | * | |||
| 10C-R2_16 | * | ||||
| 10C-R2_19 | * | ||||
| 10C-R2_23 | * | ||||
| 10C-R2_24 | * | ||||
| 10C-R2_27 | * | ||||
| 10C-R2_29 | * | * | |||
| 10C-R2_30 | * | ||||
| 12C-R2_11 | * | ||||
| 12C-R2_12 | * | ||||
| 12C-R2_13 | * | ||||
| 12C-R2_17 | * | ||||
| 12C-R2_19 | * | ||||
| 12C-R2_20 | * | ||||
| 12C-R2_21 | * | ||||
| 12C-R2_23 | * | ||||
| 12C-R2_25 | * | ||||
| 12C-R2_27 | * | ||||
| 12C-R2_29 | * | ||||
| 1D-R2_03 | * | ||||
| 1D-R2_05 | * | ||||
| 1D-R2_06 | * | ||||
| 1D-R2_09 | * | ||||
| 1D-R2_11 | * | ||||
| 1D-R2_12 | * | ||||
| 1D-R2_16 | * | ||||
| 1D-R2_18 | * | ||||
| 1D-R2_19 | * | ||||
| 1D-R2_20 | * | ||||
| 1D-R2_21 | * | ||||
| 1D-R2_23 | * | ||||
| 1D-R2_25 | * | ||||
| 8A-GC5-R2_12 | * | ||||
| 8A-GC5-R2_13 | * | ||||
| 8A-GC5-R2_18 | * | ||||
| 8A-GC5-R2_20 | * | ||||
| 8A-GC5-R2_21 | * | ||||
| 8A-GC5-R2_24 | * | ||||
| 8A-GC5-R2_27 | * | ||||
| 8A-GC5-R2_29 | * | ||||
| 8A-GC5-R2_30 | * | ||||
| 8A-GC5-R2_33 | * | ||||
| 8A-GC5-R2_35 | * | ||||
| 8A-GC5-R2_36 | * | ||||
| 8A-GC5-R2_39 | * | ||||
| 8A-GC5-R2_40 | * | ||||
| 8A-GC5-R2_41 | * | ||||
| 8A-GC5-R2_43 | * | ||||
| 8A-GC5-R2_44 | * | ||||
| 8A-GC5-R2_47 | * | ||||
| 8A-GC5-R2_49 | * | ||||
| PRA-R2_02 | * | ||||
| PRA-R2_03 | * | ||||
| PRA-R2_06 | * | ||||
| PRA-R2_08 | * | ||||
| PRA-R2_09 | * | ||||
| PRA-R2_12 | * | ||||
| PRA-R2_15 | * | ||||
| PRA-R2_17 | * | ||||
| PRA-R2_18 | * | ||||
| PRA-R2_20 | * | ||||
| PRA-R2_23 | * | ||||
| PRA-R2_24 | * | ||||
| PRA-R2_26 | * | ||||
| PRA-R2_27 | * | ||||
| PRA-R2_29 | * | ||||
| PRA-R2_30 | * | ||||
| PRA-R2_33 | * | ||||
| PRA-R2_34 | * | ||||
| PRA-R2_37 | * | ||||
| PRA-R2_38 | * | ||||
| PRA-R2_40 | * | ||||
| PRA-R2_43 | * | ||||
| PRA-R2_45 | * | ||||
| PRA-R2_46 | * | ||||
Considering the fact that every test sample was correctly classified by PLS-DA and only 5 in a total of 160 test samples from both models (i.e. ca. 3% of the total population) were falsely classified into two classes, the differentiation between four yeast and thraustochytrid strains was quite distinct as evidenced by the ability to classify them at a high level of sensitivity and specificity using these two totally different classification methods (i.e. PLS-DA and SIMCA). Therefore, both multivariate data analysis approaches particularly PLS-DA demonstrated satisfactory linearity, robustness and predictive accuracy suitable for classification of the specific marine microbes used in this study. It should also be emphasised that our cross-validation approach based on the use of separate replicates for different roles was designed in order to ensure that the test samples used for the validation purpose are totally independent of those involved in the model construction because each replicate came from different cultivations and was pre-processed individually within the set. In addition to providing a fair assessment of the model performance, the approach also imitates a realistic practice in an actual experimental setting in a way that a model is initially built and optimised by a standard set prior to the validation step to identify unknown samples from different cultivations.
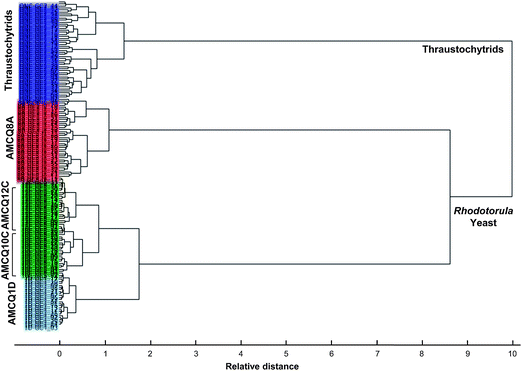 | ||
| Fig. 5 HCA dendrogram obtained by Ward's algorithm and squared Euclidean distance measure criterion, using the entire dataset that included the four Rhodotorula yeast isolates and thraustochytrids harvested at the onset of the stationary phase. | ||
3.3 Monitoring lipid production in the yeast isolate AMCQ8A
The FPA-FTIR technique was further applied to monitoring lipid accumulation during the growth period of the yeast isolate AMCQ8A grown in the optimised glucose medium. In this medium, the highest amount of total lipids in the cells was achieved, in comparison to that produced by the same isolate grown in a control medium without glucose. Our initial qualitative analysis using the same PCA approach with the same spectral windows and 4 PCs produced the results displayed in Fig. 6, which shows discrete clustering of spectra from the yeast isolate grown in the glucose media at different harvest periods as well as a separate cluster of spectra acquired from cells grown in the control medium. The corresponding loading plots reveal the main influence of the separation to be lipid moieties explained by the strong negative loadings at 2925, 2852 and 1743 cm−1, in association with a strong positive loading at 990 cm−1, which is due to the characteristic olefinicCH deformation modes in conjugated trans,trans UFAs and esters. Other loaded bands observed at 1654, 1080 and 1150 cm−1 suggest additional contributions from α-helical proteins, phosphate groups (in nucleic acids and phospholipids), and stretching vibration related to the structure CO–O–C found mainly in glycogen and nucleic acids, respectively.
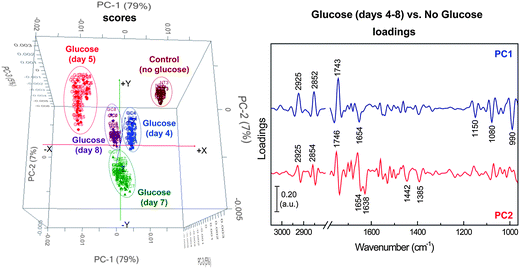 | ||
| Fig. 6 PCA score (left) and loading (right) plots of the yeast isolate AMCQ8A grown in the optimised glucose medium (days 4–8), in comparison to that of a control medium (without glucose) collected at the onset of the stationary phase (day 3). | ||
Fig. 7a further demonstrates the average spectra of the yeast isolate AMCQ8A grown in the glucose medium that were collected on a daily basis, and of those grown in the control medium harvested at the onset of the stationary phase in which an optimal amount of total lipids was found according to the FTIR and GC data as follows. For an initial semi-quantitative purpose, band areas of total lipids and UFAs were measured after the individual spectra were converted to 2nd derivatives and EMSC-corrected. These pre-processed spectra were subsequently offset over two spectral ranges within 3025–2990 and 1760–1725 cm−1, to cover the peaks centred around 3006 and 1743 cm−1 under which the integrated areas directly represent the proportions of UFAs and total lipids, respectively. The total lipids and the ratio of UFAs per total lipids in terms of %UFAs, based on the semi-quantitative band area approach, are plotted in Fig. 7b along with the cell concentration (million cells per mL) as a function of time. Note that the absence of the lipid data for the yeast AMCQ8A on day 0 (inoculation day) and days 1–3 (cultivation days) was due to insufficient cell density in the medium that resulted in a failure to produce good continuous monolayers of cells on an IR substrate for the FPA-FTIR measurements. As anticipated from previous studies and supporting literature,20 the optimal amounts of total lipids produced in the yeast isolate AMCQ8A were also achieved at the onset of the stationary phase of its growth. By comparison, the yeast isolate AMCQ8A grown in the glucose medium produced significantly more total lipids than the others throughout the growth phase, even though the proportions of the UFAs were found to be substantially lower than that observed under the optimised conditions of the thraustochytrids and the same yeast isolate grown in the control medium without glucose, respectively. It is interesting to note that the FTIR results do not take into account the degree of unsaturation of the FAs produced. However, the results are apparently in good agreement with the GC-derived FA profile (see ESI S1†), indicating that the FAs produced in the thraustochytrids are of a higher degree of unsaturation including mainly DHA and EPA – the two essential FAs highly in demand by industry.
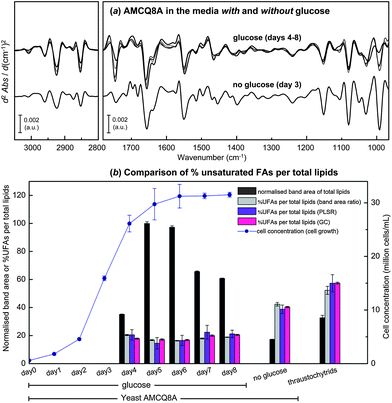 | ||
| Fig. 7 (a) Average EMSC-corrected 2nd derivative spectra of the yeast isolate AMCQ8A grown in the glucose and the control media. (b) Cell concentration plotted together with the normalised 2nd derivative band area of total lipids, and %UFAs per total lipids observed for the yeast AMCQ8A in the media with glucose (days 4–8) and without glucose (day 3), in comparison to that of thraustochytrids (day 7). Three different methods were used to obtain %UFAs including (i) percentage ratio of integrated 2nd derivative band areas, (ii) PLSR analysis, and (iii) GC technique. | ||
To achieve a higher level of accuracy in determining %UFAs in these yeast cells, PLSR analysis was conducted using a similar cross-validation approach to those used in PLS-DA and SIMCA by using each replicate dataset to individually perform as calibration (training) and validation (test) sets. The pre-processed spectral data used for the PCA in Fig. 6 were then transferred and input into the PLSR analysis together with their corresponding %UFA values obtained from the GC technique. By using the same spectral windows that contain biological information about the cells (i.e. 3100–2800 and 1800–965 cm−1) and 2 latent factors, optimised PLSR calibration models with good linearity were produced as indicated by good values of coefficient of determination R2 ≥ 0.92 for both cross-validation models (see ESI S5†). It should be emphasised that although the optimal number of latent factors was initially found to be 4 factors based on the two commonly accepted criteria of (i) the minimum explained variance and root mean standard error of calibration (RMSEC) and (ii) the correlation coefficient R2 close to 1, we have chosen for this study a conservative approach to present the results obtained with 2 latent factors in order to avoid the possibility of model over-fitting and to make sure that only chemical information was employed in the model optimisation rather than random or spurious correlations.47,48 To support the claim, a comparison of the PLSR results obtained using different number of latent factors and their corresponding regression coefficients was made according to the same cross-validation approach (see the ESI S5 and S6†). The model performance and the predictive accuracy achieved with only 2 latent factors, as compared to those of 4 latent factors, appeared to still be in an acceptable range for both cross-validation models. The respective regression coefficients additionally revealed only spectral features relative to the 2nd derivative spectra of the cells, providing strong indication that the calibrations were based on genuine chemical features and not on noise contributions. Accordingly, the %UFAs obtained from the PLSR analysis as illustrated in Fig. 7b reflect the results obtained using only 2 latent factors. As a result, the two complementary PLSR models led to highly accurate predictions of %UFA values according to (i) good linear fittings (R2 ≥ 0.93) obtained in the plots of predicted versus reference %UFA values, and (ii) low root mean square errors of prediction (RMSEP = 3.99% and 3.96%) in both cases with the reference %UFAs in the cell samples over the range of 17–60%. To evaluate the model performance and predictive accuracy of the developed PLSR approach, the predicted %UFA values of the cell samples that were harvested on the same day were averaged and plotted along with their corresponding %UFA values previously obtained from the band area ratio approach and those acquired from the GC technique in triplicate,8 as illustrated in Fig. 7b. By comparison, the %UFA values acquired from the FTIR-based methods (i.e. band area ratio and PLSR analysis) were found to be in good agreement with their GC counterparts, suggesting a high accuracy of the method and thus a strong potential of the combined FTIR and PLSR approaches for lipid monitoring purposes. These investigations additionally provide insights into the UFA production in these marine microorganisms, showing an invariable change in the UFA level from the exponential stage until reaching the end of the stationary phase of the yeast isolate AMCQ8A grown in the glucose medium. Although these yeast cells produced substantially higher total lipids throughout their growth period, the results from the three different analysis methods further indicated that the optimum %UFAs were rather achieved at significantly higher levels in the thraustochytrids and the same yeast isolate were grown in the control medium, respectively, than those in the glucose medium. Such findings point out the advantage of the yeast isolates in terms of total lipid production suited for biodiesel applications, for example.
In summary, our present results based on the independent biological replicates that were prepared simultaneously under the same controlled conditions suggest the potential application of the FPA-FTIR technique for classification and rapid monitoring of lipid production, both in terms of total lipids and %UFAs, in these marine cells. However, it should be emphasised that, further from the preliminary investigation of this nature, prospective testing of models across independent experiments will be needed in order to gain the best measure of the model performance and a more accurate assessment of the developed approach towards its use in actual routine practice.
Although the GC technique can provide the details of FA species and their actual quantities, the technique involves invasive cell processing as well as time-consuming and tedious sample preparation in order to convert the lipids into free FAs, which could take a day or more before an accurate measurement is achieved and therefore cannot be considered to be a rapid monitoring technique. FTIR microspectroscopy, on the other hand, requires minimal simple sample preparation to transfer the preserved intact cells onto an IR substrate as a monolayer with subsequent removal of water through desiccation prior to the spectral data collection, resulting in a fast analysis. With advances in bioprocessing technology, it is possible to couple programmable withdrawal devices to a cytocentrifugation module to obtain the cultured cells from a bioreactor for the acquisition of the spectral datasets, which can be subsequently transferred to an automated spectral processing unit for further analysis based on the developed multivariate data analysis approach. Such an implementation will further lead to an automated lipid analysis platform that is suitable for online monitoring purposes.
Furthermore, the ‘speed’ advantage of the FPA-FTIR imaging technique over a conventional single-point FTIR microspectroscopic measurement should be emphasised because, with the same number of scans per spectrum, an acquisition of 32 × 32 array of FPA-FTIR spectra (i.e. 1024 spectra in total) takes approximately the same period of time as acquiring one single-point spectrum using a single-point detector. This is due to the fact that each element on the 32 × 32 array FPA detector works as a single-channel detector, and thus processes the data collection simultaneously. Although a previous study has indicated better spectral quality, in terms of S/N ratio, of a single-point spectrum than a FPA-FTIR spectrum,49 the ‘speed’ advantage of the FPA-FTIR approach far outweighs the differences in the spectral quality between the two measurement systems, given the still acceptable spectral S/N ratio obtained using the FPA-FTIR technique. Moreover, with the large spectral resources acquired from each spectral image, spectral averaging as used in this study can provide a solution for improving the quality of the spectral input before further analysis. Although such a practice may compromise the ‘speed’ advantage of the technique, the quality-screening procedure can be easily performed using computer-programming software in a rapid or even automated fashion, and still requires less time to obtain a satisfactory number of high-quality spectra compared with acquiring single-point measurements.
4 Conclusion
A technique based on FTIR microspectroscopy and multivariate data analysis to discriminate and classify lipid-rich marine microbes including four yeast strains in Rhodotorula sp. and thraustochytrids has been developed. Rapid FPA-FTIR data collection with minimal sample preparation, in conjunction with the powerful multivariate data analysis methodologies including PCA, PLS-DA, SIMCA, HCA and PLSR, demonstrated combined attributes for rapid, low-cost, online monitoring of lipid production in these marine microorganisms, which have strong commercial potential. The techniques in combination are shown to be capable of probing differences in cellular composition between the diverse cell types examined. The results from PLS-DA, SIMCA and HCA indicated satisfactorily high accuracy in identification, model robustness and strong performance in classifying and discriminating among the Rhodotorula yeast strains and thraustochytrids into classes that are closely correlated with their classification based on morphology and genotyping. The FTIR technique with the PLSR approach was additionally shown to possess the potential for online quantitative monitoring of total lipids and UFAs produced in the cells during the growth period.Acknowledgements
The authors gratefully acknowledge financial support from the Alfred Deakin Postdoctoral Research Fellowship (Project ID. RM22134) for JV, the Centre for Chemistry and Biotechnology (CCB) at Deakin University for bio-processing research, and the Centre of Biospectroscopy at Monash University for provision of the FPA-FTIR microspectroscopic instrumentation. The ESI S2† regarding the synchrotron FTIR microspectroscopic experiment of the live thraustochytrium cells was acquired at the Australian Synchrotron (Victoria, Australia) through the merit-based access program (Project ID. AS121/IRM/4398) for the provision of the synchrotron beamtime at the IR microspectroscopic beamline.References
- A. P. Simopoulos, Am. J. Clin. Nutr., 1991, 54, 438–463 CAS.
- J. P. Wynn and C. Ratledge, in Bailey's Industrial Oil and Fat Products, ed. F. Shahidi, John Wiley & Sons, Hoboken, New Jersey, 6th edn, 2005, vol. 3, pp. 121–153 Search PubMed.
- O. P. Ward and A. Singh, Process Biochem., 2005, 40, 3627–3652 CrossRef CAS.
- C. G. Carter, M. P. Bransden, T. E. Lewis and P. D. Nichols, Mar. Biotechnol., 2003, 5, 480–492 CrossRef CAS.
- E. Molina Grima, J. A. Sánchez Pérez, F. Garcia Camacho, A. Robles Medina, A. Giménez Giménez and D. López Alonsot, Process Biochem., 1995, 30, 711–719 CAS.
- C. Saenge, B. Cheirsilp, T. T. Suksaroge and T. Bourtoom, Process Biochem., 2011, 46, 210–218 CrossRef CAS.
- Y. Li, Z. Zhao and F. Bai, Enzyme Microb. Technol., 2007, 41, 312–317 CrossRef CAS.
- A. Gupta, J. Vongsvivut, C. J. Barrow and M. Puri, J. Biosci. Bioeng., 2012, 114, 411–417 CrossRef CAS.
- J. A. Barnett, R. W. Payne and D. Yarrow, Yeasts: Characteristics and Identification, Cambridge University Press, Cambridge, UK, 2nd edn, 1990 Search PubMed.
- P. Heraud, B. R. Wood, J. Beardall and D. McNaughton, in New Approaches in Biomedical Spectroscopy, ed. K. Kneipp, R. Aroca, H. Kneipp and E. Wentrup-Byrne, American Chemical Society, Washington, DC, 2007, vol. 963, pp. 85–106 Search PubMed.
- P. Heraud and M. J. Tobin, Stem Cell Res., 2009, 3, 12–14 CrossRef.
- B. R. Wood and D. McNaughton, in Spectrochemical Analysis Using Infrared Multichannel Detectors, ed. R. Bhargava and I. Levin, Blackwell, UK, 2005, pp. 204–233 Search PubMed.
- B. R. Wood, T. Chernenko, C. Matthaus, M. Diem, C. Chong, U. Bernhard, C. Jene, A. A. Brandli, D. McNaughton and M. J. Tobin, Anal. Chem., 2008, 80, 9065–9072 CrossRef CAS.
- D. Naumann, H. Labischinski and P. Giesbrecht, in Modern Techniques for Rapid Microbiological Analysis, ed. W. H. Nelson, VCH Verlag Chemie, Weinheim, 1990 Search PubMed.
- D. Naumann, D. Helm and H. Labischinski, Nature, 1991, 351, 81–82 CrossRef CAS.
- T. Udelhoven, D. Naumann and J. Schmitt, Appl. Spectrosc., 2000, 54, 1471–1479 CrossRef CAS.
- K. Maquelin, C. Kirschner, L. P. Choo-Smith, N. A. Ngo-Thi, V. T. van, M. Stammler, H. P. Endtz, H. A. Bruining, D. Naumann and G. J. Puppels, J. Clin. Microbiol., 2003, 41, 324–329 CrossRef CAS.
- N. A. Ngo-Thi, C. Kirschner and D. Naumann, J. Mol. Struct., 2003, 661–662, 371–380 CrossRef CAS.
- A. P. Dean, D. C. Sigee, B. Estrada and J. K. Pittman, Bioresour. Technol., 2010, 101, 4499–4507 CrossRef CAS.
- M. Wältermann and A. Steinbüchel, J. Bacteriol., 2005, 187, 3607–3619 CrossRef.
- E. Ó. Faoláin, M. B. Hunter, J. M. Byrne, P. Kelehan, M. McNamara, H. J. Byrne and F. M. Lyng, Vib. Spectrosc., 2005, 38, 121–127 CrossRef.
- A. Savitzky and M. J. E. Golay, Anal. Chem., 1964, 36, 1627–1639 CrossRef CAS.
- A. Kohler, C. Kirschner, A. Oust and H. Martens, Appl. Spectrosc., 2005, 59, 707–716 CrossRef CAS.
- A. Kohler, N. K. Afseth and H. Martens, in Applications of Vibrational Spectroscopy in Food Science, ed. E. Li-Chan, P. R. Griffiths and J. M. Chalmers, John Wiley & Sons, Chichester, UK, 2010, vol. 1, pp. 89–108 Search PubMed.
- P. Geladi and B. R. Kowalski, Anal. Chim. Acta, 1986, 185, 1–17 CrossRef CAS.
- S. Chevallier, D. Bertrand, A. Kohler and P. Courcoux, J. Chemom., 2006, 20, 221–229 CrossRef CAS.
- S. Wold and M. Sjostrom, in Chemometrics Theory and Application, American Chemical Society Symposium Series 52, ed. B. R. Kowalski, American Chemical Society, Washington, DC, 1977, pp. 243–282 Search PubMed.
- M. D. Guillen and N. Cabo, J. Am. Oil Chem. Soc., 1997, 74, 1281–1286 CrossRef CAS.
- G. Socrates, Infrared and Raman Characteristic Group Frequencies, John Wiley & Sons, Chichester, UK, 3rd edn, 2001 Search PubMed.
- F. Severcan, G. Gorgulu, S. T. Gorgulu and T. Guray, Anal. Biochem., 2005, 339, 36–40 CrossRef CAS.
- R. H. Sills, D. J. Moore and R. Mendelsohn, Anal. Biochem., 1994, 218, 118–123 CrossRef CAS.
- S. Yoshida and H. Yoshida, Biopolymers, 2004, 74, 403–412 CrossRef CAS.
- P. J. H. Jones and S. Kobow, in Modern nutrition in health and disease, ed. M. E. Shils, J. A. Olson, M. Shike and A. C. Ross, Williams & Wilkins, Baltimore, MD, 9th edn, 1999, pp. 67–94 Search PubMed.
- U. Bocker, R. Ofstad, Z. Y. Wu, H. C. Bertram, G. D. Sockalingum, M. Manfait, B. Egelandsdal and A. Kohler, Appl. Spectrosc., 2007, 61, 1032–1039 CrossRef CAS.
- S. Cai and B. R. Singh, Biophys. Chem., 1999, 80, 7–20 CrossRef CAS.
- M. Banyay, M. Sarkar and A. Gräslund, Biophys. Chem., 2003, 104, 477–488 CrossRef CAS.
- H.-Y. N. Holman, H. A. Bechtel, Z. Hao and M. C. Martin, Anal. Chem., 2010, 82, 8757–8765 CrossRef CAS.
- D. R. Whelan, K. R. Bambery, P. Heraud, M. J. Tobin, M. Diem, D. McNaughton and B. R. Wood, Nucleic Acids Res., 2011, 39, 5439–5448 CrossRef CAS.
- D. Del Rio, A. J. Stewart and N. Pellegrini, Nutr., Metab. Cardiovasc. Dis., 2005, 15, 316–328 CrossRef.
- M. J. Tobin, L. Puskar, R. L. Barber, E. C. Harvey, P. Heraud, B. R. Wood, K. R. Bambery, C. T. Dillon and K. L. Munro, Vib. Spectrosc., 2010, 53, 34–38 CrossRef CAS.
- D. G. Cameron, H. L. Casal and H. H. Mantsch, Biochemistry, 1980, 19, 3665–3672 CrossRef CAS.
- D. C. Lee and D. Chapman, Biosci. Rep., 1986, 6, 235–256 CrossRef CAS.
- E. Smidt, K.-U. Eckhardt, P. Lechner, H.-R. Schulten and P. Leinweber, Biodegradation, 2005, 16, 67–79 CrossRef CAS.
- P. T. T. Wong, R. H. Wong, T. A. Caputo, T. A. Godwin and B. Rigas, Proc. Natl. Acad. Sci. U. S. A., 1991, 88, 10988–10992 CrossRef CAS.
- D. Chapman, 15th Annual Summer Program Symposium on Quantitative Methodology in Lipid Research: Part II, Pennsylvania State University, PA, USA, 1965 Search PubMed.
- J. H. Ward, J. Am. Stat. Assoc., 1963, 58, 236–244 CrossRef.
- M. Kansiz, K. C. Schustera, D. McNaughton and B. Lendl, Spectrosc. Lett., 2005, 38, 677–702 CrossRef CAS.
- N. K. Afseth, H. Martens, A. Randby, L. Gidskehaug, B. Narum, K. Jørgensen, S. Lien and A. Kohler, Appl. Spectrosc., 2010, 64, 700–707 CrossRef CAS.
- P. Heraud, S. Caine, G. Sanson, R. Gleadow, B. R. Wood and D. McNaughton, New Phytol., 2007, 173, 216–225 CrossRef.
- T. Miyazawa and E. R. Blout, J. Am. Chem. Soc., 1961, 83, 712–719 CrossRef CAS.
- T. J. Johnson, S. D. Williams, N. B. Valentine and Y. F. Su, Appl. Spectrosc., 2009, 63, 908–915 CrossRef CAS.
- D. M. Haaland, H. D. T. Jones and E. V. Thomas, Appl. Spectrosc., 1997, 51, 340–345 CrossRef CAS.
- J. Liquier, A. Akhebat and E. Taillandier, Spectrochim. Acta, Part A, 1991, 47, 177–186 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c3an00485f |
| This journal is © The Royal Society of Chemistry 2013 |