 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Quantifying degradation of collagen in ancient manuscripts: the case of the Dead Sea Temple Scroll†
R.
Schütz
ab,
L.
Bertinetti
a,
I.
Rabin
b,
P.
Fratzl
a and
A.
Masic
*a
aDepartment of Biomaterials, Max Planck Institute of Colloids and Interfaces, Wissenschaftspark Golm, 14424 Potsdam, Germany. E-mail: masic@mpikg.mpg.de; Fax: +49 331 5679402; Tel: +49 331 5679419
bBAM Federal Institute for Materials Research and Testing, Unter den Eichen 44-46, 12203 Berlin, Germany
First published on 18th June 2013
Abstract
Since their discovery in the late 1940s, the Dead Sea Scrolls, some 900 ancient Jewish texts, have never stopped attracting the attention of scholars and the broad public alike, because they were created towards the end of the Second Temple period and the “time of Christ”. Most of the work on them has been dedicated to the information contained in the scrolls' text, leaving physical aspects of the writing materials unexamined. They are, however, crucial for both historical insight and preservation of the scrolls. Although scientific analysis requires handling, it is essential to establish the state of degradation of these valued documents. Polarized Raman Spectroscopy (PRS) is a powerful tool for obtaining information on both the composition and the level of disorder of molecular units. In this study, we developed a non-invasive and non-destructive methodology that allows a quantification of the disorder (that can be related to the degradation) of protein molecular units in collagen fibers. Not restricted to collagen, this method can be applied also to other protein-based fibrous materials such as ancient silk, wool or hair. We used PRS to quantify the degradation of the collagen fibers in a number of fragments of the Temple Scroll (11Q19a). We found that collagen fibers degrade heterogeneously, with the ones on the surface more degraded than those in the core.
Introduction
The Dead Sea Scrolls (DSS) are a collection of some 900 fragmented texts of great religious and historical significance from the late Second Temple period. Discovered between 1947 and 1956 in eleven caves in the vicinity of the ruins of an ancient settlement, Qumran, they have never stopped attracting the attention of scholars and the broad public alike, because they were created in the “time of Christ”. The mystery of their origin is still debated among scholars. Written with carbon inks on parchment, papyrus and leather, some of the scrolls stored in clay vessels have retained their good condition over the last two thousand years. Others, found on the cave floors, survived only in a highly fragmented state. All the scrolls have experienced a complicated and seldom accurately documented discovery and post-discovery history. The Temple Scroll (TS) holds a special place in the collection, since it differs in many ways from the rest of the manuscripts. It contains previously unknown text that is inscribed on the flesh side of the extremely thin, eight-meter-long, light-tainted parchment (Fig. 1). | ||
| Fig. 1 A detail of the Temple Scroll. Photos © The Israel Museum, Jerusalem. | ||
Found by Bedouins in 1956, it reached scholars in 1967 allegedly greatly damaged by moisture. Immediately after the purchase, the unrolling of the tightly wound roll was conducted in accordance with the Plenderleith method.1 This involved humidification of the scroll up to 100% RH, with subsequent short freezing to arrest the gelatinization process. The fragments were furthermore reinforced with paper and a PVA-based adhesive. Thus, the first visual description of the scroll, named by Yigael Yadin as “Temple Scroll”, reflects its state after unrolling and the build-up of the previously detached fragments. Yadin remarked that the state of preservation, which varied greatly from sheet to sheet, resulted from a non-uniform treatment of the parchment in antiquity.2 Later it was shown that the production process involved alum tawing that rendered the scroll extremely sensitive to humidity.3 But already in 1980, an X-ray diffraction study of the scrolls indicated that the high degree of degradation of the Temple Scroll contrasted strongly with the apparently good state of preservation.4 Later, scientists from the Getty Institute who analyzed the scrolls within the framework of a preservation project arrived at the same conclusion.5 Therefore, quantitative damage assessment of this scroll seems to be crucial for monitoring its state and for its further preservation.
Parchment is a final product of the processing of animal skin and consists mainly of type I collagen, the most abundant constituent of the dermal matrix. Deterioration of parchment is caused by chemical changes due to gelatinization, oxidation and hydrolysis of the collagen chains.6 Historically, the state of degradation of collagen within parchments has been studied using several physical and chemical methods. Methods such as shrinkage temperature,7 X-ray scattering,8,9 thermal analysis,10AFM,11 and solid state NMR12 provide qualitative information on the physical and chemical integrity of collagen at various levels of the hierarchical structure. However, they are usually invasive, i.e. require small samples of the original material. This fact, together with the intrinsic heterogeneity of the skin material, explains why none of these methods can be used to monitor the state of preservation of the ancient parchment. Therefore, an efficient and non-invasive technique for quantitative damage assessment has great potential for early warning and monitoring systems.13 Recent attempts to describe the evolution of parchment deterioration use NIR,14 and multispectral imaging techniques.15,16 It seems, however, that low spatial resolution and sensitivity result in inaccuracy that outweighs the advantage of the ease of application. Recently, unilateral and solid state NMR techniques showed potential for determining the state of conservation of collagen indirectly, through the relaxation behavior of water molecules involved in the stabilization of the collagen triple helix.12,17,18
Raman spectroscopy has long been explored as a method for assessing damage to parchments.19–22 Its non-invasive and non-destructive nature made it an attractive candidate for both damage assessment and monitoring. However, small changes in spectral features and the low sensitivity of the method itself prevented the acquisition of sufficiently accurate information to be routinely applied for damage assessment. Recent technical developments in polarized Raman spectroscopy (PRS) showed huge potential in structural studies of collagen-based materials.23,24 The possibility of obtaining both chemical (molecular interactions) and structural (orientation) information25–28 in a single experiment opens the way to the use of PRS for studies on orientation changes induced by deterioration. The methodology is based on the sensitivity of Raman scattering bands (associated with distinct molecular vibrations) to the polarization direction of the incident laser light. The method can be applied in order to map fibrous materials within other heterogeneous tissues and, in principle, it is possible to concurrently map the distribution of other chemical components associated with it.29,30
In this study, we applied PRS to study the collagen fibers within the parchment of the Temple Scroll at two different levels of organization (molecular and fibrous). Quantitative damage assessment methodology has been established based on the specific polarization response of Raman amide bands. To this end, we have compared native (highly structured) and gelatinized (randomly oriented collagen molecules) rat-tail tendon (RTT), freshly prepared parchments and collagen fibers of the TS.
Materials and methods
Samples studied
Two regions of interest (ROI1 and ROI2) on two analogue fragments (S1 and S2) of ca. 5 × 5 mm2 of the TS were analyzed. The exact original positions of the fragments inside the scroll are not known. The fragments were found detached from the heavily damaged part of the TS. Additionally, fibers of specifically prepared reference samples have been investigated. As a reference for a sample with high orientation anisotropy, a rat-tail tendon (RTT) was stretched by 10% of its length and analyzed in the dry state. In addition, fibers of recently manufactured limed new parchment (NP) were studied. As a reference for the isotropic collagen network, gelatin was prepared from RTT and NP by a thermal treatment (80 °C) in water for 4 hours.Theoretical background
The PRS approach is a consolidated methodology and has been frequently applied on systems with uniaxial symmetry in the last decade. Since a theoretical description of the technique has been presented in detail by several authors, e.g. Turrell,31 Bower,32 Rousseau et al.33 and Sourisseau,34 a compact guide to the basic concepts that have been applied in this study is presented in the ESI.†Confocal PRS
A continuous laser beam was focused down to a micrometer-sized spot on the sample through a confocal Raman microscope (CRM200, WITec, Ulm, Germany) equipped with a piezo-scanner (P-500, Physik Instrumente, Karlsruhe, Germany). The diode-pumped 785 nm near-infrared (NIR) laser excitation (Toptica Photonics AG, Graefelfing, Germany) was used in combination with a 100× (Olympus MPlan IR, NA = 0.95) microscope lens. The linearly polarized laser light was rotated using a half-wave plate, and the scattered light was filtered, introducing a further polarizer (analyzer) before the confocal microscope pinhole. The spectra were acquired using an air-cooled CCD (PI-MAX, Princeton Instruments Inc., Trenton, NJ, USA) behind a grating (300 g mm−1) spectrograph (Acton, Princeton Instruments Inc., Trenton, NJ, USA) with a spectral resolution of 6 cm−1. Forty to sixty accumulations with an integration time of 1 s were used for four polarization adjustments of a single spot analysis.Data acquisition and processing
In the TS fragment, the collagen fibers are characterized by a diameter in the range of 5 to 10 μm. The size of the focal spot of the used 100× lens lies in the range of 1 μm2 laterally and around 2 μm along the optical axis. The confocal setup ensures that the measured spectra are representative of the single collagen fiber and not of several overlapping collagen fibers.A chosen fiber was pre-adjusted in the focus of the lens parallel to the z-axis by means of the integrated light microscope. The adjustment is achieved by rotating the whole fragment, so that neither fiber sampling nor harm is done to the fragment. The z-axis corresponds to the direction of polarization 0° of the laser on the sample (x-axis corresponds to the 90° laser polarization). Finally, Raman overall intensity was maximized by adjusting the focus along the y-axis (optical beam axis).
To determine the intensity ratios R1 = I0–90/I0–0 and R2 = I90–0/I90–90, the analyzer for polarization has been placed in the path of the backscattered light. The analyzer orientation is indicated by the second indices of the intensity (I0–90; 0°-polarizer and 90°-analyzer regarding the fiber orientation (z-axis)). Four experiments are needed to calculate the orientation parameters33 and one additional control measurement at the 0–0° position is performed on the specific spot. The control measurement is used to check the stability of the spot position on the selected fiber during the 4 min needed for complete measurement.
The spectra were acquired with the WITec Program 1.94® and then processed in three steps with OPUS 6.0®:
1. Cut the region around the amide I band (1517–2000 cm−1).
2. Baseline correction.
3. Integration over the spectral range between 1600 and 1720 cm−1.
The laser power at the sample is kept no higher than 4 mW. This is of particular importance when measuring the precious TS fragment, which has to be investigated non-destructively.
The signal-to-noise ratio of the spectra under these conditions is not optimal, especially for the cross-polarized configuration (0–90 and 90–0) where the intensities are very close to the spectral noise level. For such spectra, neither peak fitting nor intensity determinations can be performed and therefore, an integral over the amide I wavenumber region was used.
For the calculation of Legendre polynomials P2 and P4 (for details see the ESI†) the ratio Riso = 0.20 ± 0.015 (gelatin) of an isotropic reference material was used. For the distribution function of C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O vibrational units, a Gaussian shape was assumed, where the angle of the maximum θmax and the corresponding FWHM (ω) can be determined by the obtained P2 and P4 coefficients.
O vibrational units, a Gaussian shape was assumed, where the angle of the maximum θmax and the corresponding FWHM (ω) can be determined by the obtained P2 and P4 coefficients.
Results and discussion
From the macroscopic morphological observation of the surface of the analyzed TS fragments, it was possible to distinguish between two areas. The first area (ROI1), with clearly visible intact collagen fiber network, was originally covered by another layer. The remnants of the missing surface layer can be still found on the adjacent ROI2 of the fragment. This smooth layer consists of gypsum and alum supported by an extended collagen fiber network and originated from the manufacturing of the scroll.3 The collagen fibers of the surface layer appear to be more damaged than those in ROI1. Therefore, the intact fibers initially protected by the surface layer correspond to the core parchment. Though a simple visual inspection demonstrates the heterogeneity of the collagen degradation, its degree cannot be established in a simple way. Furthermore, the heterogeneous state of degradation leads to a different response of the various parts of the scroll to conservation treatments and environmental storage conditions (such as humidity and temperature) and therefore a quantitative estimation of the fibers' integrity is crucial. For damage quantification, we first considered two borderline cases, namely (i) fresh collagen fibers from rat-tail tendon and new parchment, and (ii) completely gelatinized collagen (gelatin).In Fig. 2 the polarized Raman spectra of RTT, gelatin and NP fiber samples are presented. Red spectra are acquired by placing polarizer and analyzer parallel to the fiber axis (ZZ), whereas blue spectra are acquired with the polarizer and analyzer perpendicular (XX). Grey and orange spectra are cross-polarized configurations (XZ and ZX) useful for calculating orientation parameters. Longitudinally stretched RTT was used to obtain perfectly aligned collagen fibers as a calibration standard (Fig. 2, top). In this study we focus on the collagen backbone vibrational modes, and in this context, an intense band centered at 1665 cm−1 is observed, corresponding to the amide I vibration, which is mainly due to CO stretching vibration. Further bands can be seen at ∼1450 and ∼1250 cm−1 and are assignable to C–H bending and amide III vibrations respectively. It is clearly visible band anisotropy (differences in scattered intensities that are related only to the orientation of the laser polarization), particularly in the amide I region (evidenced with the dashed box in the figure). Because collagen carbonyl groups are oriented mainly perpendicularly to the collagen molecular axis, the amide I band is more intense in the direction that is perpendicular to the collagen molecular axis (blue lines in Fig. 2).28 Very similar anisotropic behavior is found for collagen fibers within the NP sample (Fig. 2, bottom), whereas the anisotropy of all bands completely vanishes in the case of gelatin (Fig. 2, middle).
![Polarized Raman spectra of rat tail tendon fiber (top), gelatin (middle) and newly prepared parchment fiber (bottom). The dashed box highlights the amide I region used for evaluating orientation parameters. Amide I anisotropy is evident [comparing XX (blue) and ZZ (red) configurations] for RTT and new parchment fibers, but completely vanishes for the gelatin sample.](/image/article/2013/AN/c3an00609c/c3an00609c-f2.gif) | ||
| Fig. 2 Polarized Raman spectra of rat tail tendon fiber (top), gelatin (middle) and newly prepared parchment fiber (bottom). The dashed box highlights the amide I region used for evaluating orientation parameters. Amide I anisotropy is evident [comparing XX (blue) and ZZ (red) configurations] for RTT and new parchment fibers, but completely vanishes for the gelatin sample. | ||
The latter result is to be expected, considering the complete loss of the supramolecular organization of collagen molecules in the gelatin sample and the resulting random orientation of the molecular units in the analyzed volume.
Fig. 3 presents PR spectra from two characteristic fibers selected from the different areas of a single TS fragment.
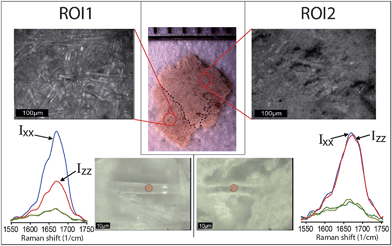 | ||
| Fig. 3 Stereo- and optical microscopy images (top) of two ROIs of the Temple Scroll fragment examined in this study. Collagen fibers can be found in both ROIs. The bottom part shows single collagen fibers in both ROIs and polarized Raman spectra in the amide I region performed on the fibers in correspondence of red spots. A clear difference in anisotropic behavior can be observed. | ||
Using an optical microscope, we located suitable fibers in both the aforementioned ROI1 and ROI2 and applied the PRS approach. From a simple comparison of the anisotropic responses of the amide I band, it can be clearly concluded that the fiber from the ROI1 (Fig. 3, left) shows a very similar anisotropic pattern to that found in the new parchment, whereas the fiber from the ROI2 (Fig. 3, right) matches better the gelatin spectral features. This simple experimental evidence is extremely important because it shows that TS, commonly considered heavily gelatinized, locally contains collagen fibers that are in a relatively good state of preservation. Furthermore, it shows that some fibers, although lacking any order at the molecular level of the structure, can preserve its macroscopic fibrous morphology.
Starting from this spectroscopic evidence, and assuming that deterioration of collagen material will naturally lead to a general disorder of collagen structural units, we can now design a new approach for damage assessment of the collagen fibers. The methodology applied here is based on the fact that the orientation of a molecular unit within a biomolecule can be quantified from Raman band anisotropy measurements, if the Raman band tensor of the relevant molecular unit is known.35,36 In the case of the amide I band, the tensor properties are well documented in the literature and can be used for this purpose. From parameters provided by the PRS (see ESI†), it is possible to determine the arrangement or orientation of the CO vibrational units with respect to the fibrous system.30,37–39
Table 1 shows the calculated averaged values of two main orientation parameters (Legendre polynomials P2 and P4) relative to the amide I band for RTT, NP, gelatin and several fibers within TS samples.
| Sample | P2 | P4 |
|---|---|---|
| RTT | −0.35 ± 0.02 | 0.10 ± 0.02 |
| New parchment | −0.29 ± 0.01 | 0.06 ± 0.01 |
| Gelatin | −0.00 ± 0.01 | 0.00 ± 0.01 |
| TS (S1 and S2) ROI1 | −0.18 ± 0.07 | −0.08 ± 0.09 |
| TS (S1 and S2) ROI2 | −0.09 ± 0.06 | −0.01 ± 0.07 |
The average values of the order parameters vary significantly among different samples. As expected, values obtained for the RTT and new parchment are in the range of highly ordered molecular units, whereas the P2 and P4 close to zero found for gelatin indicate complete disorder. In the case of TS, P2 values lie between the two just discussed limits, with ROI1 apparently more organized than ROI2. However, P4 values are relatively far away from the values obtained for RTT and new parchment. This discrepancy can be explained by the fact that there is no simple relationship between order parameters and the distribution function of the vibrational units. Therefore, here we propose a more efficient way of quantifying disorder in collagen fibers.
By approximating molecular distribution of vibrational units around the fiber axis by a Gaussian curve, it is possible to calculate the associated P2 and P4 values for any distribution. In this way, for any P2 and P4 there will be a corresponding distribution characterized by given angular maximum (θmax) and full width at half maximum (FWHM) (ω). The angular maximum corresponds to the most likely orientation of collagen units within the volume illuminated by the laser spot and FWHM is qualitatively linked to the variability of the orientation. Plotting the iso-θmax and iso-ω lines in the P2–P4 space (Fig. S2†) enables a simple correlation between the order parameters and the distribution of the vibrational units and therefore allows the quantification of the order–disorder in a fiber. For instance, in the case of collagen, the results show a distribution always centered on the θmax = 90° but characterized by different ω. These results can be depicted in the P2–P4 space spread along the θmax = 90° line. Fig. 4 (top) shows such a plot, where P2 and P4 values obtained for all investigated samples are superimposed, as well.
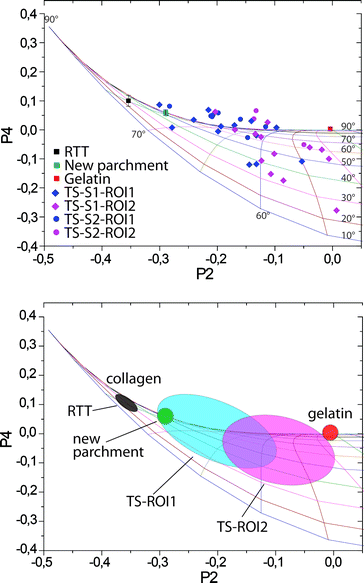 | ||
| Fig. 4 Top: P2–P4 plot of orientation parameters relative to all samples analyzed in this study. Bottom: characteristic regions found for different samples indicative of different states of preservation of collagen. | ||
It appears that the main difference between various collagen samples (RTT, new parchment, gelatin, TS) lies in the width of the distribution function. Thus, a well-preserved collagen fiber will have a width of 30–40°, while a strongly gelatinized one will have a width larger than 120°. The highly organized collagen of the stretched RTT was found to have a FWHM of 40°. The Gaussian distribution function of the results of the NP has a FWHM of 60°. This is supposed to correspond to the best state of preservation of collagen in a parchment treated with lime, particularly in historical parchments. The broadening of the distribution function with respect to that of the RTT indicates a slight disorganization of collagen in freshly prepared parchments, probably induced by treatments necessary for depilating and finishing.
The results of the P2 and P4 coefficients for both areas of the TS fragment display a wider scattering around the θmax = 90° line than in the case of NP or RTT. The reason for the stronger variations from the θmax = 90° line is likely to be found in the weak and noisy signal intensity in the spectra of the 2000-year-old collagen fibers. Nevertheless a tendency can be recognized in the distribution in the P2–P4 space (Fig. 4). The collagen fibers of the formerly covered ROI1 show P2 values that are significantly different from those of the ROI2 (statistically significant difference between groups as determined by one-way ANOVA (F(1.19) = 6.233, P = 0.021)). Spread over a large range from 60°, the FWHM values found for ROI1 are equivalent to a well-preserved modern parchment, with some points going up to 120°, which is very close to strongly disorganized collagen in gelatin (Fig. 4). The FWHM values related to the ROI2 lie in the range between 80° and 120° of width, indicating a greater degradation of the collagen in this area than in the ROI1. It is worth repeating that previous studies on TS fragments show very high levels of gelatinization. In contrast, we find that several fibers in the ROI1 are relatively well-preserved. The methodology described here is based on a single parameter, i.e. FWHM of the distribution function, and results in a reliable and simple way to quantify the amount of degradation that leads to the disorder of molecular units of collagen in ancient parchments. The disorder, however, can be induced by several factors, such as gelatinization, hydrolysis, oxidation, etc. Orientation parameters defined in this work reflect global structural changes induced by all of these degradation processes and do not carry any information about the cause of the disorder. However, through an analysis of the spectral features of the Raman bands it would be possible to address these points. We believe that the combination of this latter approach with the methodology presented here could provide further insights into deterioration pathways. Future work will focus on applying this strategy to artificially aged and other ancient parchments.
Conclusions
In this study, we developed a methodology based on PRS that is capable of establishing the degree of degradation of collagen in historical parchments. The approach is non-invasive and non-destructive and allows a simple quantification of the degree of disorder (which can be related to the degradation) of protein molecular units in collagen fibers. Given its nature, the method can be applied to other protein-based fibrous materials, such as ancient silk, wool or hair. We used this method to study the preservation of fibers in the fragments of the TS, one of the most important documents of the Dead Sea Scrolls collection. We found that extremely heterogeneous degradation of collagen fibers can be correlated with the fibers' location in the parchment. Contrary to the common opinion, a relatively good state of preservation was found for the core part of the Temple Scroll's parchment.Acknowledgements
P.F. and A.M. are grateful for support from the Alexander von Humboldt Foundation and the Max Planck Society in the framework of the Max Planck Research Award funded by the Federal Ministry of Education and Research.References
- H. J. Plenderleith, in Discoveries in the Judean desert I, ed. D. Barthélemy and J. T. Milik, Clarendon Press, Oxford, 1955, pp. 39–40 Search PubMed.
- Y. Yadin, Biblical Archaeol., 1967, 30, 135–139 CrossRef.
- I. Rabin, R. Schütz, E. Kindzorra, U. Schade, O. Hahn, G. Weinberg and P. Lasch, presented in part at the Multidisciplinary Conservation: a Holistic View for Historic Interiors, Rome, 2010 Search PubMed.
- S. Weiner, Z. Kustanovich, E. Gil-Av and W. Traub, Nature, 1980, 287, 820–823 CrossRef CAS.
- W. S. Ginell, unpublished work.
- C. J. Kennedy and T. J. Wess, Restaurator, 2003, 24, 61–80 CrossRef CAS.
- N. S. Cohen, M. Odlyha and G. M. Foster, Thermochim. Acta, 2000, 365, 111–117 CrossRef CAS.
- C. J. Kennedy, J. C. Hiller, D. Lammie, M. Drakopoulos, M. Vest, M. Cooper, W. P. Adderley and T. J. Wess, Nano Lett., 2004, 4, 1373–1380 CrossRef CAS.
- C. A. Maxwell, T. J. Wess and C. J. Kennedy, Biomacromolecules, 2006, 7, 2321–2326 CrossRef CAS.
- E. Badea, G. Della Gatta and P. Budrugeac, J. Therm. Anal. Calorim., 2011, 104, 495–506 CrossRef CAS.
- L. Bozec and M. Odlyha, Biophys. J., 2011, 101, 228–236 CrossRef CAS.
- A. E. Aliev, Biopolymers, 2005, 77, 230–245 CrossRef CAS.
- I. Rabin and O. Hahn, Restaurator, 2012, 33, 101–121 Search PubMed.
- A. Možir, M. Strlic, T. Trafela, I. Kralj Cigic, J. Kolar, V. Deselnicu and G. de Bruin, Appl. Phys. A: Mater. Sci. Process., 2011, 104, 211–217 CrossRef.
- E. Marengo, M. Manfredi, O. Zerbinati, E. Robotti, E. Mazzucco, A. Gosetti, G. Bearman, F. France and P. Shor, Anal. Chim. Acta, 2011, 706, 229–237 CrossRef CAS.
- E. Marengo, M. Manfredi, O. Zerbinati, E. Robotti, E. Mazzucco, A. Gosetti, G. Bearman, F. France and P. Shor, Anal. Chem., 2011, 83, 6609–6618 CrossRef CAS.
- E. Badea, L. Miu, P. Budrugeac, M. Giurginca, A. Masic, N. Badea and G. Della Gatta, J. Therm. Anal. Calorim., 2008, 91, 17–27 CrossRef CAS.
- A. Masic, M. Chierotti, R. Gobetto, G. Martra, I. Rabin and S. Coluccia, Anal. Bioanal. Chem., 2011, 1–7 Search PubMed.
- A. Facchini, C. Malara, G. Bazzani and P. L. Cavallotti, J. Colloid Interface Sci., 2000, 231, 213–220 CrossRef CAS.
- M. Bicchieri, M. Monti, G. Piantanida, F. Pinzari and A. Sodo, Vib. Spectrosc., 2011, 55, 267–272 CrossRef CAS.
- E. Mannucci, R. Pastorelli, G. Zerbi, C. E. Bottani and A. Facchini, J. Raman Spectrosc., 2000, 31, 1089–1097 CrossRef CAS.
- H. G. M. Edwards and F. R. Perez, J. Raman Spectrosc., 2004, 35, 754–760 CrossRef CAS.
- M. Kazanci, H. D. Wagner, N. I. Manjubala, H. S. Gupta, E. Paschalis, P. Roschger and P. Fratzl, Bone, 2007, 41, 456–461 CrossRef CAS.
- M. Kazanci, P. Roschger, E. P. Paschalis, K. Klaushofer and P. Fratzl, J. Struct. Biol., 2006, 156, 489–496 CrossRef CAS.
- M. Janko, P. Davydovskaya, M. Bauer, A. Zink and R. W. Stark, Opt. Lett., 2010, 35, 2765–2767 CrossRef CAS.
- N. Gierlinger, S. Luss, C. Konig, J. Konnerth, M. Eder and P. Fratzl, J. Exp. Bot., 2010, 61, 587–595 CrossRef CAS.
- T. Lefevre, M. E. Rousseau and M. Pezolet, Biophys. J., 2007, 92, 2885–2895 CrossRef CAS.
- A. Masic, L. Bertinetti, R. Schuetz, L. Galvis, N. Timofeeva, J. W. C. Dunlop, J. Seto, M. A. Hartmann and P. Fratzl, Biomacromolecules, 2011, 12, 3989–3996 CrossRef CAS.
- M. Raghavan, N. D. Sahar, R. H. Wilson, M.-A. Mycek, N. Pleshko, D. H. Kohn and M. D. Morris, J. Biomed. Opt., 2010, 15, 037001 CrossRef.
- M. Tsuboi, J. Biomed. Opt., 2002, 7, 435–441 CrossRef CAS.
- G. Turrell, J. Raman Spectrosc., 1984, 15, 103–108 CrossRef CAS.
- D. Bower, J. Polym. Sci., Polym. Phys. Ed., 1972, 10, 2135–2153 CAS.
- M. E. Rousseau, T. Lefevre, L. Beaulieu, T. Asakura and M. Pezolet, Biomacromolecules, 2004, 5, 2247–2257 CrossRef CAS.
- C. Sourisseau, Chem. Rev., 2004, 104, 3851 CrossRef CAS.
- M. Tsuboi, K. Nakamura, M. Aida, Y. Kubo, J. M. Benevides and G. J. Thomas, Biophys. J., 2002, 82, 457A–458A Search PubMed.
- M. Tsuboi, Y. Kubo, K. Akahane, M. Pezolet, T. Lefevre and G. J. Thomas, Biophys. J., 2005, 88, 559A Search PubMed.
- M. Tsuboi, Y. Kubo, K. Akahane, J. M. Benevides and G. J. Thomas, J. Raman Spectrosc., 2006, 37, 240–247 CrossRef CAS.
- S. A. Overman, M. Tsuboi and G. J. Thomas, Biophys. J., 1998, 74, A73 Search PubMed.
- L. Rintoul, E. A. Carter, S. D. Stewart and P. M. Fredericks, Biopolymers, 2000, 57, 19–28 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c3an00609c |
| This journal is © The Royal Society of Chemistry 2013 |