DOI:
10.1039/C2BM00087C
(Paper)
Biomater. Sci., 2013,
1, 213-223
A facile fabrication of upconversion luminescent and mesoporous core–shell structured β-NaYF4:Yb3+, Er3+@mSiO2 nanocomposite spheres for anti-cancer drug delivery and cell imaging†
Received 7th July 2012, Accepted 9th September 2012
First published on 17th October 2012
Abstract
Upconversion luminescent β-NaYF4:Yb3+, Er3+ nanoparticles (UCNPs) were encapsulated with uniform mesoporous silica shells, which were further modified with poly(ethylene glycol) (PEG) and cancer-targeting ligand folic acid (FA), resulting in the formation of water-dispersible and biologically functionalized core–shell structured UCNPs@mSiO2 nanoparticles with an overall average size of around 80 nm. The obtained multifunctional nanocomposite spheres can be performed as an anti-cancer drug delivery carrier and applied for cell imaging. It is found that anti-cancer drug doxorubicin hydrochloride (DOX) can be absorbed into UCNPs@mSiO2-PEG/FA nanospheres and released in a pH-sensitive pattern. In vitro cell cytotoxicity tests on cancer cells verified that DOX-loaded UCNPs@mSiO2-PEG/FA nanospheres exhibited greater cytotoxicity with respect to free DOX and DOX-loaded UCNPs@mSiO2-PEG at the same concentrations, owing to the increase of cell uptake of anti-cancer drug delivery vehicles mediated by the FA receptor. Moreover, the upconversion luminescence image of UCNPs@mSiO2-PEG/FA taken up by cells shows green emission under 980 nm infrared laser excitation, making the UCNPs@mSiO2-PEG/FA nanocomposite spheres promising candidates as bioimaging agents. These findings highlight the promise of the highly versatile multifunctional nanoparticles for simultaneous imaging and therapeutic applications.
Introduction
The design and synthesis of multifunctional nanomedical platforms that integrate multiple nanomaterials with different properties into a single nanosystem provide unparalleled opportunity for simultaneous diagnosis and therapy of diseases.1 In particular, the construction of core–shell structured multifunctional nanocomposites composed of mesoporous silica and rare-earth doped upconversion nanoparticles (UCNPs) has been a research hotspot in the forefront of materials science.2 This is because this kind of nanocomposite combines the special advantages of a mesoporous structure and UCNPs. On the one hand, the high surface area/pore volume of mesoporous silica nanoparticles (MSNs) can be used as an effective nanocarrier to provide compartmental reservoirs for multiple components such as anti-cancer drugs and photodynamic molecules so as to realize the applications in drug delivery and photodynamic therapy (PDT). Furthermore, the MSNs have an easily modifiable and functionalized surface and significant biocompatibility.3 On the other hand, UCNPs can emit higher-energy visible photons after being excited by lower-energy near-infrared (NIR) photons. Compared to conventional downconversion luminescence materials, UCNPs have many attractive merits including narrow emission peak, long lifetimes, large Stokes shifts, superior photostability, and low toxicity.4,5 More importantly, UCNPs are excited by NIR light, which holds such advantages as an absence of photodamage to live organisms, a very low autofluorescence background, a high signal-to-noise ratio and detection sensitivity, and a greater tissue penetration depth in biological tissues. Therefore, UCNPs, especially β-NaYF4:Yb3+, Er3+, have been used for the imaging of cells, tissues and small animals.6 Thus, the design and development of the nanocomposites combining mesoporous silica with UCNPs is undoubtedly of great importance for simultaneous bioimaging and therapeutic applications.Although there are some reports on composite materials consisting of UCNPs and mesoporous silica, these materials either tend to agglomerate significantly or have a larger particle size.2 The synthesis of these materials typically requires the intermediate coatings of solid silica followed by further deposition of a mesoporous silica layer, which leads to the complicated and multistep procedures. Moreover, silane coupling agent octadecyltrimethoxysilane (C18TMS) was generally used as a pore-generating template, which needs to be removed by calcination treatment in order to produce mesopores.7 However, calcination will cause irreversible and considerable particle aggregation, which makes the materials difficult to disperse again in water, which is essential for biomedical applications.8
In the field of mesoporous silica-based drug delivery systems, in order to increase the water solubility and the biocompatibility, and to reduce undesired interactions between the biological environment and the nanocarriers, the porous nanoparticles could be coated by soft organic macromolecules such as poly(ethylene glycol) (PEG).9 In addition, during the drug delivery, targeting by utilizing antibodies or specific ligands to selectively bind to the cell surface to trigger receptor-mediated endocytosis is especially attractive for cancer therapies, as most of the commonly used anti-cancer drugs have serious side-effects due to nonspecific action on healthy cells.10
Recently, we have reported the synthesis of up-conversion luminescent (UCL) and porous NaYF4:Yb3+, Er3+@SiO2 fiber composites by the electrospinning process and investigated their drug delivery and cell imaging properties.11 It seems that the as prepared multifunctional NaYF4:Yb3+, Er3+@SiO2 nanocomposite fibers (several tens of micrometers long and several hundred nanometers thick) may be too large for direct use in biomedical areas. As a continuation of and great improvement on our previous work, here in this paper we report the direct encapsulation of upconversion β-NaYF4:Yb3+, Er3+ nanoparticles with a uniform mesoporous silica shell, which is modified with hydrophilic and biocompatible polymer poly(ethylene glycol) (PEG) and cancer-targeting ligand folic acid (FA) to create water-dispersible and biologically functionalized nanocomposite spheres. By using cetyltrimethylammonium bromide (CTAB) as a pore-producing agent, uniform, monodisperse and upconversion core–mesoporous silica shell structured UCNPs@mSiO2 nanoparticles with a smaller size of around 80 nm were obtained in a very short time (10 min). Compared to the previous methods,2 the current methodology is more facile and effective because it avoids the utilization of the intermediate solid silica layer and overcomes the hurdles of the aggregation of nanoparticles by calcination. We found that the nanocomposite spheres were highly biocompatible in vitro, capable of loading anti-cancer drug doxorubicin (DOX) and selectively targeting cancerous cells, and amenable to upconversion luminescence imaging.
Experimental
Chemicals and materials
All the chemical reagents are used as received without further purification. Rare earth oxides of Y2O3, Yb2O3 and Er2O3 were purchased from the Science and Technology Parent Company of the Changchun Institute of Applied Chemistry (China). Octadecene (90%, technical grade), folic acid (FA, ≥97%), N-hydroxysuccinimide (NHS, 98%), N-(3-dimethylaminopropyl)-N-ethylcarbodiimide hydrochloride (EDC, 98%), and (3-aminopropyl)triethoxysilane (APS, ≥98%) were purchased from Aldrich. 2-[Methoxy-(polyethyleneoxy)propyl]trimethoxysilane (PEG500–silane, Mw = 460–590, tech-90) was purchased from Gelest. Oleic acid, tetraethylorthosilicate (TEOS), cetyltrimethylammonium bromide (CTAB, ≥99%), ammonium nitrate (NH4NO3, ≥99.0%), NaOH (reagent grade, ≥98%) and ammonium fluoride (NH4F) were purchased from Beijing Yili Fine Chemical Regent Company (China). Doxorubicin hydrochloride (DOX) was purchased from Nanjing Duodian Chemical Limited Company (China). The rare earth chlorides YCl3, YbCl3 and ErCl3 were prepared by dissolving the corresponding rare earth oxides in hydrochloric acid at elevated temperature followed by evaporating the solvent under vacuum.Synthesis of UCNPs@mSiO2 nanocomposite spheres
First, 18% Yb3+, 2% Er3+ co-doped β-NaYF4 nanocrystals were prepared according to the literature method.12 Then, UCNPs@mSiO2 nanocomposite spheres were synthesized according to the modified procedures we reported recently.13 2 mL cyclohexane solution (about 5–10 mg mL−1) containing the UCNPs was mixed with 0.1 g CTAB and 20 mL water. The mixture was then stirred vigorously to evaporate the cyclohexane solvent at room temperature, resulting in a transparent and clear UCNPs–CTAB water solution. For coating mesoporous silica shells onto UCNPs, 10 mL of the aqueous CTAB-stabilized UCNPs solution was added to the mixture of 20 mL water, 3 mL ethanol and 150 μL 2 M NaOH solution. The mixture was heated up to 70 °C under stirring. When the temperature was stable, 200 μL tetraethylorthosilicate (TEOS) was added dropwise and the reaction was allowed to proceed to 10 min. The as-synthesized materials were centrifuged and washed with ethanol three times. The surfactants CTAB were removed via a fast and efficient ion exchange method, where the as-synthesized UCNPs@mSiO2 (20 mg) are transferred to 50 mL ethanol containing 0.3 g NH4NO3 and kept at 60 °C for 2 h. Finally, the as-obtained UCNPs@mSiO2 nanocomposite spheres were dispersed in ethanol.Synthesis of UCNPs@mSiO2-PEG
Surface modification of poly(ethylene glycol) (PEG) of UCNPs@mSiO2 was performed according to the method developed previously.14 PEG500–silane (30 μL) was added to a mixture of ethanol (30 mL), deionized water (6 mL), NH4OH (300 μL), and the as-prepared UCNPs@mSiO2 nanoparticles (20 mg). After 24 h of stirring, the samples were centrifuged several times to remove the unreacted chemicals.Synthesis of UCNPs@mSiO2-PEG/FA
20 mg of the UCNPs@mSiO2-PEG nanomaterials were washed with dimethyl sulfoxide (DMSO) and resuspended in 3 mL DMSO to form the NPs–DMSO solution. In a separate flask, FA (1 mg) and APS (5 μL) were mixed in 3 mL DMSO. Subsequently, NHS (1.1 mg) and EDC (1.6 mg) were added into the mixture and stirred for 2 h. Finally, the NPs–DMSO suspension was added and allowed to react with stirring overnight at room temperature. The materials were recovered by centrifugation and washed twice with DMSO and ethanol.Preparation of drug storage/delivery systems
10 mg of the UCNPs@mSiO2-PEG/FA sample dispersed in 2 mL water was mixed with 2 mL DOX aqueous solution (1 mg mL−1). After stirring for 24 h under dark conditions, the DOX-loaded sample was collected by centrifugation and denoted as DOX-UCNPs@mSiO2-PEG/FA. Then, DOX-UCNPs@mSiO2-PEG/FA samples were immersed in 2 mL pH = 7.4, 5.5 and 2.0 phosphoric acidic buffer solutions (PBS) at 37 °C with gentle shaking. At predetermined time intervals, PBS was taken by centrifugation and replaced with an equal volume of fresh PBS. The amount of released DOX in the supernatant solutions was measured by UV-vis spectrophotometer at a wavelength of 480 nm. To evaluate the DOX-loading efficiency, the supernatant and washed solutions were collected and the residual DOX content (RDOX) was obtained by UV-vis measurement at a wavelength of 480 nm. The loading efficiency of DOX can be calculated as follows: [(ODOX − RDOX)/ODOX] × 100%, in which ODOX is the original DOX content.The biocompatibility of the UCNPs@mSiO2, UCNPs@mSiO2-PEG and UCNPs@mSiO2-PEG/FA nanocomposites
To evaluate the biocompatibility of the nanomaterials, MTT cell assay was performed on the L929 cell. MTT is a standard test for screening the toxicity of biomaterials and is carried out in accordance with ASTM standards. Briefly, L929 fibroblast cells were plated out at a density of 5000–6000 cells per well in a 96-well plate and incubated overnight at 37 °C in a humidified atmosphere of 5% CO2 to allow the cells to attach to the wells. The UCNPs@mSiO2, UCNPs@mSiO2-PEG and UCNPs@mSiO2-PEG/FA nanocomposites were sterilized by autoclaving, and then serial concentrations of the nanocomposites were added and incubated for 24 h in 5% CO2 at 37 °C. 5 mg mL−1 stock solution of MTT [3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide] was prepared in PBS and this stock solution (20 μL) was added to each well containing a different amount of the monodisperse UCNPs@mSiO2, UCNPs@mSiO2-PEG and UCNPs@mSiO2-PEG/FA nanocomposites. The plates were incubated at 37 °C for 24 h. The medium and MTT were then removed, and the MTT–formazan crystals were dissolved in 150 μL DMSO and placed on a shaking table at 150 rpm for 5 minutes, to thoroughly mix the formazan into the solvent. The absorbance of the suspension was recorded under a microplate reader (Thermo Multiskan MK3) at 490 nm.In vitro cytotoxicity of DOX-UCNPs@mSiO2-PEG/FA against HeLa cells
HeLa cells were plated out in 96-well plates at a density of 8000 cells per well and were allowed to attach and grow for 24 h to allow the cells to attach. The free DOX, DOX-UCNPs@mSiO2-PEG, and DOX-UCNPs@mSiO2-PEG/FA were added to the medium, and the cells were incubated in 5% CO2 at 37 °C for 24 h. The concentrations of DOX were 0.78125, 1.5625, 3.125, 6.25, 12.5, 25, and 50 μg mL−1, respectively. At the end of the incubation, the medium containing the nanocomposites was removed, and 20 μL MTT solution was added into each cell and incubated for another 24 h. The supernatant in each well was aspirated and 150 μL DMSO was added to each well before the plate was examined using a microplate reader (Thermo Multiskan MK3) at a wavelength of 490 nm.Cellular uptake of DOX-UCNPs@mSiO2-PEG/FA nanospheres
Cellular uptake by HeLa cells was examined using flow cytometry, confocal laser scanning microscopy (CLSM) and ICP-OES, respectively. For flow cytometry studies, HeLa cells (1 × 106) were seeded in 6-well culture plates and grown overnight. The cells were then treated with DOX-UCNPs@mSiO2-PEG/FA (containing DOX of 20 μg mL−1) at 37 °C for 30 min, 3 h and 6 h. A single cell suspension was prepared consecutively by trypsinization, washing with PBS, and filtration through 35 mm nylon mesh. Thereafter, the cells were lifted using a cell stripper (Media Tech. Inc.), and analyzed using a FACSCalibur flow cytometer (BD Biosciences) for DOX. The excitation wavelength and emission wavelength were 488 nm and 575 nm, respectively.For CLSM, the HeLa cells were seeded in 6-well culture plates (a clean cover slip was put in each well) and grown overnight as a monolayer, and were incubated with DOX-UCNPs@mSiO2-PEG, DOX-UCNPs@mSiO2-PEG/FA, and DOX-UCNPs@mSiO2-PEG/FA + free FA at 37 °C for 1 h. Thereafter, the cells were rinsed with PBS three times, fixed with 2.5% formaldehyde at 37 °C for 10 min, and then rinsed with PBS three times again. For nucleus labeling, fixed cells were incubated with Hoechst 33![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 342 solution (from Molecular Probes, 20 mg mL−1 in PBS, 1 mL per well) for 10 min and then washed with PBS three times. The cover slips were placed on a glass microscope slide, and the samples were analyzed using CLSM for DOX.
342 solution (from Molecular Probes, 20 mg mL−1 in PBS, 1 mL per well) for 10 min and then washed with PBS three times. The cover slips were placed on a glass microscope slide, and the samples were analyzed using CLSM for DOX.
For ICP-OES measure, 2 × 106 HeLa cells were seeded with 2 mL Dulbecco's Modified Eagle Medium (DMEM, Invitrogen) 6-well plates. These cells were then treated with UCNPs@mSiO2-PEG (100 μg) and UCNPs@mSiO2-PEG/FA (100 μg) incubated at 37 °C for 5 h. After washing with PBS three times, cells were lysed by cell lysis buffer. Yttrium contents in the cell lysis solution were determined by ICP-OES.
Upconversion luminescence imaging of UCNPs@mSiO2-PEG/FA
The instrument of up-conversion luminescence microscopy (UCLM) was rebuilt on an inverted florescence microscope (Nikon Ti-S), and an external CW 980 nm laser diode was illuminated onto the samples. Fir UC luminescence (UCL) imaging, HeLa cells (5 × 104 per well) were seeded in 6-well culture plates and grown overnight as a monolayer, and were incubated with UCNPs@mSiO2-PEG/FA at 37 °C for 30 min, 3 h, and 6 h. Finally, the cells were washed with PBS three times, fixed with 2.5% formaldehyde at 37 °C for 10 min, and then washed with PBS three times again.Characterization
Powers X-ray diffraction (XRD) measurements were performed on a D8 Focus diffractometer (Bruker), with the use of Cu-Kα radiation (λ = 0.15405 nm). The UV-vis adsorption spectral values were measured on a U-3310 spectrophotometer. The morphology of the samples was inspected using a field emission scanning electron microscope (Philips XL 30). A transmission electron microscopy (TEM) micrograph was obtained from a FEI Tecnai G2 S-Twin transmission electron microscope with a field emission gun operating at 200 kV. Nitrogen adsorption/desorption analysis was measured using a Micromeritics ASAP 2020 M apparatus. The specific surface area was determined by the Brunauer–Emmett–Teller (BET) method using data between 0.05 and 0.35. The pore volume was obtained from the T-plot method. The UC emission spectra were obtained using a 980 nm laser diode as the excitation source. The emission spectra were dispersed by a monochromator of the Acton SpectraPro-2758 equipped with R928 PMT, and the data were recorded from 400 to 700 nm. Confocal laser scanning microscopy (CLSM) images were observed by confocal laser scanning microscopy (Olympus, FV 1000). The photos of up-conversion luminescence were obtained digitally on a Nikon multiple CCD camera (DS-Ri1). Flow cytometry (FCM, BD Biosciences), with an excitation wavelength of 488 nm, was used to quantify the uptake efficiency of each sample. Inductively Coupled Plasma Optical Emission Spectrometry (ICP-OES) was taken on an iCAP 6300 of Thermo scientific.Results and discussions
Protocol of the synthesis, surface modification and application of core–shell structured β-NaYF4:Yb3+, Er3+@mSiO2 nanocomposites (labeled as UCNPs@mSiO2)
The overall goal of this work was to build a facile and effective approach to the direct encapsulation of mesoporous silica onto UCNP cores to yield water-dispersible, biocompatible, up-conversion luminescent nanocomposite particles for controlled anti-cancer drug delivery and cell imaging. As shown in Scheme 1, first the oleic acid stabilized UCNP core was synthesized by a high boiling solvent process developed by Zhang's group.12 Then the hydrophobic UCNPs dissolved in cyclohexane were transferred into an aqueous medium by using CTAB as the capping molecules, which also act as the surfactants for the subsequent sol–gel reaction to form mesoporous silica. The removal of CTAB surfactants resulted in the formation of mesoporous silica nanospheres embedded with a single upconversion nanoparticle. In this process, CTAB could be used not only as a phase transfer agent but also as an organic template for mesopore generation for uniform nanoparticles encapsulated in a mesoporous silica shell, similar to those of mesoporous silica coated magnetic nanoparticles (such as Fe3O4 and MnO).14,15 Then for drug delivery, the surfaces of UCNPs@mSiO2 nanospheres were modified with polyethylene glycol (PEG) in order to increase colloidal stability and water dispersion. Moreover, folic acid (FA) as a targeting component was grafted to the surface of silica by covalent linking to enhance the probability of targeting delivery of drugs to the cancer cells based on the specific recognition and high affinity between FA and folate receptors on the surface of cancer cells. Finally, the obtained PEGylated and FA modified UCNPs@mSiO2-PEG/FA nanocomposites act as an effective multifunctional platform for upconversion imaging and anti-cancer drug doxorubicin (DOX) delivery.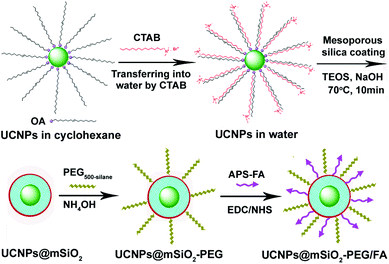 |
| | Scheme 1 Schematic illustration for the synthesis of UCNPs@mSiO2-PEG/FA composite nanospheres. | |
Fabrication and characterization of UCNPs@mSiO2-PEG/FA
The XRD pattern of as-obtained oleic acid stabilized UCNPs is shown in Fig. 1a. All the diffraction peaks can be ascribed to the pure hexagonal phase structure known from bulk β-NaYF4 (JCPDS 16-0334). The typical TEM image (Fig. 2A) indicates that as-obtained upconversion nanocrystals are composed of uniform and nearly spherical nanoparticles with a mean diameter of 30 nm. Dynamic light scattering (DLS) data further demonstrates that the nanocrystals have a narrow size distribution (Fig. S1a, ESI†). The corresponding HRTEM image (inset in Fig. 2A) clearly shows the clear lattice fringes with interplanar spacing of 0.30 nm ascribed to the (110) plane of β-NaYF4. After transferring into aqueous water by simple stirring at room temperature, CTAB capped UCNPs are still well-dispersed and homogenous with unchanged size (Fig. 2B). As disclosed by the HRTEM image (inset in Fig. 2B), the distance of the adjacent lattice fringes is 0.30 nm, also consistent with the (110) plane of β-NaYF4. In this process, the hydrophobic tail of the CTAB surfactant interacts strongly with the hydrophobic oleic acid ligand on the surface of UCNPs,16 and the hydrophilic charged head group of CTAB makes the nanocrystals water-soluble, as shown in Scheme 1. The transfer of the UCNPs to water using CTAB was a key step for the successful growth of mesoporous silica coatings on individual UCNPs. Moreover, this transformation process is very effective because there were no visible precipitates or aggregates in the solution, which can be directly seen from the digital photographs of UCNPs dispersed in cyclohexane and water. Moreover, under the excitation of a 980 nm laser with a power of 1.5 W, UCNPs with the same concentration of 0.5 mg mL−1 before and after transferring exhibit green and yellow emissions, respectively, as presented in Fig. 2C and D. Note that the same magnification was used for these photographs (Fig. S2, ESI†). From their corresponding upconversion luminescence spectra (Fig. 2E and F), it can be clearly seen that the profiles of the emission spectra for these two samples are similar. Namely, there are three emission peaks centered at 521, 542, and 654 nm, which can be assigned to the 2H11/2 → 4I15/2, 4S3/2 → 4I15/2, and 4F9/2 → 4I15/2 transitions of Er3+ respectively.17 However, the relative ratios of red and green emission bands (Ired/Igreen) show an obvious difference for UCNPs before and after transferring, namely, Ired/Igreen of UCNPs in water and cyclohexane was 0.96 and 0.2, respectively, resulting in different luminescence colors. This can be attributed to the large vibrational modes of the H2O solvent molecules (∼3600 cm−1,) which produce an increased nonradiative relaxation of the 4I11/2 level to the 4I13/2 level.18 This gives rise to a relative increase of the red emission relative to the green emission. Liu et al. also demonstrated the effect of surface quenching on upconversion luminescence. A marked decrease in UV/Vis to NIR emission ratios for the NaGdF4:Yb3+/Tm3+ (25/0.3 mol%) nanoparticles was observed as the concentration of water molecules increased.19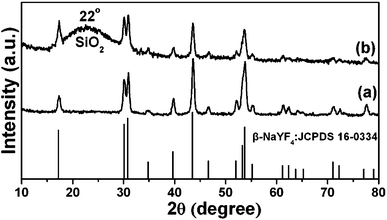 |
| | Fig. 1 XRD patterns of β-NaYF4:Yb3+, Er3+ nanoparticles (UCNPs) (a) and UCNPs@mSiO2 (b). The standard card of β-NaYF4 was given as a reference. | |
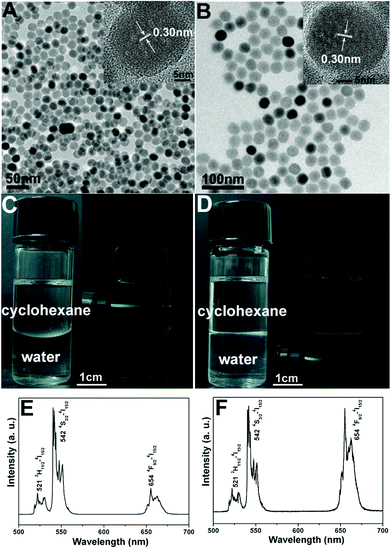 |
| | Fig. 2 TEM images of UCNPs (0.5 mg mL−1) dispersed in cyclohexane (A) and transferring into water (B), their corresponding digital photographs and luminescence photographs dispersed in cyclohexane (C) and water (D) and their upconversion emission spectra of UCNPs dispersed in cyclohexane (E) and water (F) under the excitation of a 980 nm laser with a power of 1.5 W. | |
In the subsequent sol–gel reaction, CTAB-stabilized UCNPs serve as seeds for the formation of spherical mesoporous silica shells via base-catalyzed hydrolysis of tetraethylorthosilicate (TEOS) and condensation of silica. After removing the CTAB templates from the as-synthesized materials by an ion exchange method in ethanol solution containing ammonium nitrate (NH4NO3), core–shell structured mesoporous UCNPs@mSiO2 nanocomposite spheres were obtained. In this core–shell structure, the silica–CTAB layer is formed around the CTAB–UCNP nanoparticles under basic conditions through an electrostatic interaction between the cationic (CTAB) and anionic (silicate) species.14,20 The XRD pattern (Fig. 1b) of the as-obtained UCNPs@mSiO2 nanocomposites shows the amorphous silica structure peak at 2θ = 22° besides the characteristic peaks of β-NaYF4 nanocrystals, indicating the successful coating of the silica shell. TEM images demonstrated clearly that UCNPs@mSiO2 nanocomposites are composed of well dispersed and uniform spherical nanoparticles with an average diameter of around 80 nm (Fig. 3A). From high-magnification TEM images (Fig. 3B), it can be clearly seen that the uniform and homogenous mesoporous silica shell layer possesses a wormhole-like channel with a thickness of about 25 nm. According to DLS data (Fig. S1b, ESI†), the core–shell structured nanomaterials have a narrow size distribution. The N2 adsorption/desorption isotherm and pore-size distribution of UCNPs@mSiO2 composite nanospheres are shown in Fig. 4. The Brunauer–Emmett–Teller (BET) surface area and total pore volume were estimated to be 288 m2 g−1 and 0.67 cm3 g−1, respectively. The isotherm could be classified as a type-IV isotherm, indicating the presence of textural mesopores. The average pore size was calculated to be 2.2 nm using the Barrett–Joiner–Halenda (BJH) method, as shown in the inset of Fig. 4. The upconversion emission spectrum of UCNPs@mSiO2 nanoparticles dispersed in water was shown in Fig. S3 (ESI†). The ratio of Ired/Igreen of UCNPs@mSiO2 was estimated to be 0.73, which is smaller than that of CTAB-stabilized UCNPs in water. This suggests that the coating of mesoporous SiO2 may have an effect on the upconversion behavior of UCNPs to some extent.
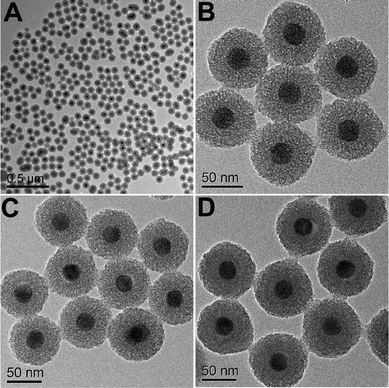 |
| | Fig. 3 TEM images of UCNPs@mSiO2 (A, B), UCNPs@mSiO2-PEG (C) and UCNPs@mSiO2-PEG/FA (D). | |
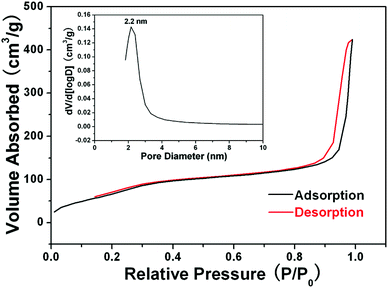 |
| | Fig. 4 N2 adsorption–desorption isotherms and mesopore size distribution (inset) of the as-synthesized UCNPs@mSiO2 nanocomposites. | |
The as-obtained UCNPs@mSiO2 mesoporous nanospheres were subsequently modified by silane-functionalized PEG and cancer-targeting agent FA on the outermost surfaces (Scheme 1). Note that the surface modification has a small effect on the morphology and size of nanoparticles, as has been presented in the corresponding TEM images of UCNPs@mSiO2-PEG (Fig. 3C) and UCNPs@mSiO2-PEG/FA (Fig. 3D). Furthermore, the mesoporous structure of the shell is always clearly visible before and after surface modification, which is important in loading the anti-cancer drugs. FT-IR spectrometry was used to further characterize the successful modification. As shown in Fig. 5a, the unmodified UCNPs@mSiO2 only showed the typical absorption bands of siliceous materials related with Si–O–Si (1084 and 806 cm−1), Si–OH (950 cm−1), and Si–O (453 cm−1).21 This also confirms the removal of CTAB surfactants because the C–H stretch (2850–3000 cm−1) peaks from the CTAB disappeared after ion exchange procedures. For the as-obtained UCNPs@mSiO2-PEG, the new band assigned to CH3 stretching 2926 cm−1 and CH2 stretching (2926 cm−1) is apparent besides the characteristic absorption peaks of SiO2 (Fig. 5b), confirming the successful grafting of the PEG chains on the surface of UCNPs@mSiO2.22 Compared to UCNPs@mSiO2-PEG, the FT-IR spectrum (Fig. 5c) of the UCNPs@mSiO2-PEG/FA shows the well-resolved vibrational peak at 1607 cm−1 assigned to the N–H bending vibration of the CONH group and another obvious peak at 1700 cm−1 attributed to the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O amide stretching of the α-carboxyl group from the FA molecules.23 Here we note that the PEG chains would increase the colloidal stability of nanoparticles in water, which can be directly seen from the digital photographs of nanoparticles with and without PEG modification dispersed in water (Fig. S4, ESI†). Without PEG, the obvious precipitates in the solution can be observed in 1 minute (Fig. S4a†), but in the presence of PEG the nanoparticles can be well-dispersed in the water to form a stable colloidal solution (Fig. S4b and c, ESI†).
O amide stretching of the α-carboxyl group from the FA molecules.23 Here we note that the PEG chains would increase the colloidal stability of nanoparticles in water, which can be directly seen from the digital photographs of nanoparticles with and without PEG modification dispersed in water (Fig. S4, ESI†). Without PEG, the obvious precipitates in the solution can be observed in 1 minute (Fig. S4a†), but in the presence of PEG the nanoparticles can be well-dispersed in the water to form a stable colloidal solution (Fig. S4b and c, ESI†).
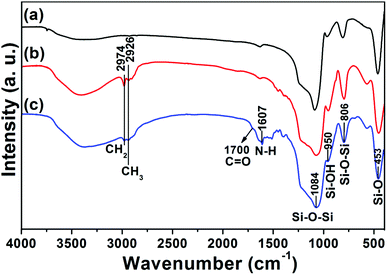 |
| | Fig. 5 FT-IR spectra of UCNPs@mSiO2 (a), UCNPs@mSiO2-PEG (b) and UCNPs@mSiO2-PEG/FA (c). | |
In vitro cytotoxicity and drug delivery properties
The investigation of the cytotoxicity of nanocarrier materials is an important prerequisite and crucial factor for drug delivery systems.24Fig. 6 shows the effects of UCNPs@mSiO2 (labeled as NPs), NPs-PEG and NPs-PEG/FA on the cell viabilities of L929 cells measured by MTT assay. For UCNPs@mSiO2 nanospheres, although the cell maintains >80% cell viability after 24 h treatment at the concentration of the samples as high as 250 μg mL−1, the cell viabilities decrease gradually with the increase in concentration of the sample. This may be caused by the trace CTAB templates that were not removed completely from the surfaces of mesoporous silica. It has been reported that CTAB is hazardous and less biocompatible,25 so it can produce cell toxicity even in the presence of trace CTAB. With PEG and FA modification, the two samples NPs-PEG and NPs-PEG/FA exhibit better biocompatibility with L929 cells up to a concentration of 250 μg mL−1 than the counterpart with unmodified NPs. This indicates that PEGylation not only increases the dispersion stability and avoids aggregation of NPs, but also enhances the biocompatibility of the resultant materials.26 Therefore, the PEGylated and folate-conjugated UCNPs@mSiO2 could be used as drug delivery vehicles for biological applications.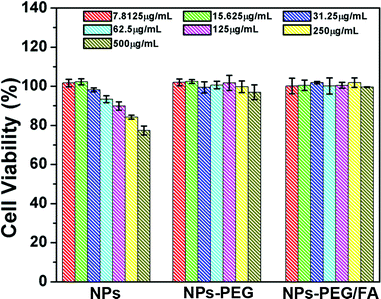 |
| | Fig. 6 Cell viabilities of UCNPs@mSiO2 (NPs), NPs-PEG and NPs-PEG/FA to L929 cells measured by MTT assay. | |
It is commonly accepted that mesoporous materials possess sustained drug release properties.27 A water-soluble chemotherapeutic drug, doxorubicin hydrochloride (DOX) was chosen as a model drug to evaluate the loading capability and release behaviors of UCNPs@mSiO2-PEG/FA composite nanospheres as a drug delivery system. The loading efficiency could reach 67% and the drug loading content is 12%, as determined by UV-vis characterization. The in vitro release profiles of DOX from DOX-loaded UCNPs@mSiO2-PEG/FA nanospheres over a 96 h period in PBS buffer solution of three different pH values (7.4, 5.5 and 2.0) at 37 °C are shown in Fig. 7. The drug release rate of DOX-loaded UCNPs@mSiO2-PEG/FA was obviously pH-dependent and increased with decreasing pH. Only 6% DOX was released from the composite nanospheres even after 48 h at pH 7.4. With an increase of acidity to pH 5.5, the DOX release rate reached to 21% after 48 h and more than 34% of DOX was released within 4 h in PBS of pH 2.0. This trend was ascribed to the increased hydrophilicity and higher solubility of DOX at lower pH caused by increased protonated –NH2 groups on DOX. It is well known that the extracellular pH of many solid tumors is lower than normal tissues and presents an acidic microenvironment.28 The pH-sensitive DOX release might be beneficial at the reduced pH values in intracellular lysosomes, endosomes and certain cancerous tissues for targeted release and controlled therapy at the pathological sites.29 Furthermore, regardless of the pH value, the DOX-loaded nanospheres obviously have a sustained release property. A fast release happened within 8 h and a sustained release followed until the end of the assay. Therefore, combined with the high loading capacity of DOX and simultaneous sustained drug release property, UCNPs@mSiO2-PEG/FA nanospheres could be used in chemotherapy for cancer treatment with continuous therapy without frequent interval medication administrations.30
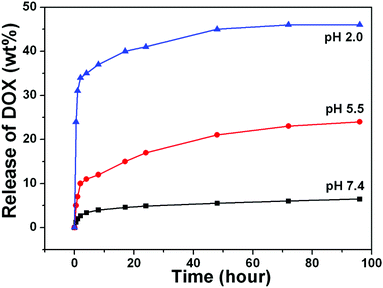 |
| | Fig. 7 (A) Cumulative DOX release from UCNPs@mSiO2-PEG/FA nanocomposite spheres at pH 7.4 (black line), pH 5.5 (red line) and pH 2.0 (blue line) PBS buffer. | |
Cellular uptake and targeted capability of nanomaterials
Cellular-uptake characteristics of DOX-loaded UCNPs@mSiO2-PEG/FA nanospheres were investigated by using flow cytometry and upconversion luminescence imaging. First, the cell uptake degree of the nanocomposites was quantified with flow cytometry by determining the signal of DOX originating from DOX-UCNPs@mSiO2-PEG/FA nanospheres treated with HeLa cells. As shown in Fig. 8, after incubation of HeLa cells with the samples for different times (30 min, 3 h, and 6 h), the median fluorescence levels in the PE–A histograms indicate that DOX-UCNPs@mSiO2-PEG/FA nanospheres were taken up by HeLa cells compared to the controlled cells, and the cell uptake of the sample increases gradually with extension of the incubation time. Besides this, in order to further verify that UCNPs@mSiO2-PEG/FA nanomaterials can be internalized by the cells, the upconversion optical bioimaging was done by utilizing modified inverted fluorescence microscopy equipped with infrared laser excitation at 980 nm. After UCNPs@mSiO2-PEG/FA (200 μg mL−1) were incubated with HeLa cells at 37 °C for 30 min, 3 h, and 6 h, the obvious UCL signal can be observed from the HeLa cells under the excitation of a 980 nm laser (Fig. 9). Meanwhile, more and more UCNPs@mSiO2-PEG/FA nanospheres were internalized into the cells with increasing time. This indicates that UCNPs@mSiO2-PEG/FA can be used as an excellent luminescence probe for cell imaging and monitoring the cell endocytosis process.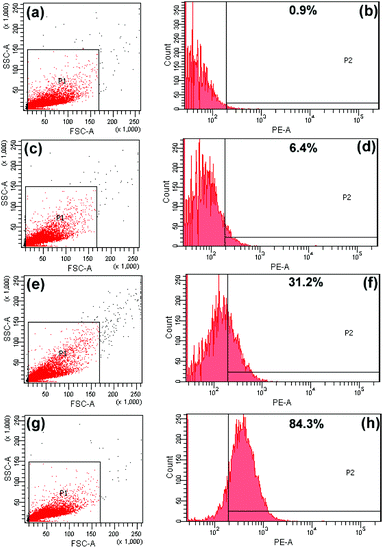 |
| | Fig. 8 Flow cytometry analysis of the control cells (a, b) and HeLa cells incubated with DOX-loaded UCNPs@mSiO2-PEG/FA nanocomposite spheres for 30 min (c, d), 3 h (e, f), and 6 h (g, h). | |
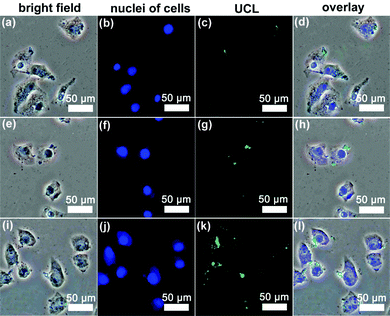 |
| | Fig. 9 Inverted florescence microscope images of HeLa cells incubated with UCNPs@mSiO2-PEG/FA nanospheres for 30 min (a–d), 3 h (e–h) and 6 h (i–l) at 37 °C. Each series can be classified to the bright-field image, nuclei of cells (being dyed in blue by Hoechst 33342 for visualization), upconversion luminescent image (UCL) and the overlay of the three above, respectively. | |
To improve the therapeutic efficacy of our nanoplatform, the targeted delivery of UCNPs@mSiO2-PEG/FA to cancer cells is of paramount significance. Folate-conjugated particles can selectively bind to the cancer cells, and can then be delivered to the interior of a given type of cell via the receptor-mediated endocytosis, which is different from the particles without modification.31 First, to quantitatively evaluate the cell uptake of UCNPs@mSiO2-PEG and UCNPs@mSiO2-PEG/FA nanospheres in HeLa cells, ICP-OES was used to measure the concentration of Y3+ internalized by HeLa cells, which is calculated to be 29.94 and 15.8 μg mL−1 for UCNPs@mSiO2-PEG/FA and UCNPs@mSiO2-PEG spheres, respectively, as shown in Fig. S5 (ESI†). Obviously, the uptake amount of UCNPs@mSiO2-PEG/FA spheres is almost 2 times as high as that of the latter in HeLa cells. Additionally, T-test results also indicate that there is an extremely significant discrepancy (P < 0.01) between them. This confirms that FA grafting is favorable for the cell uptake of UCNPs@mSiO2-PEG spheres. In addition, laser-scanning confocal microscopy (CLSM) was used to further confirm the targeting recognition capability of the FA modified sample. HeLa cells were incubated with DOX-UCNPs@mSiO2-PEG/FA, DOX-UCNPs@mSiO2-PEG and DOX-UCNPs@mSiO2-PEG/FA + free FA for 1 h at 37 °C under otherwise identical conditions. As shown in the CLSM images (Fig. 10), the cells incubated with DOX-UCNPs@mSiO2-PEG/FA (Fig. 10a–c) showed particles distributed primarily throughout the cellular cytoplasmic space and displayed the strongest luminescence signals, compared to the cells treated with DOX-UCNPs@mSiO2-PEG (Fig. 10d–f) and DOX-UCNPs@mSiO2-PEG/FA + free FA (Fig. 10g–i), suggesting that a more folate-modified drug carrier system was taken up specifically by HeLa cells. The above results indicate that the DOX-UCNPs@mSiO2-PEG/FA are preferentially internalized by the cells via a receptor-mediated endocytosis process.32
 |
| | Fig. 10 Confocal laser scanning microscopy (CLSM) of HeLa cells incubated with DOX-UCNPs@mSiO2-PEG/FA (a–c), DOX-UCNPs@mSiO2-PEG (d–f) and DOX-UCNPs@mSiO2-PEG/FA + free FA (g–i) for 1 h at 37 °C. Each series can be classified to the nuclei of cells (being dyed in blue by Hoechst 33342 for visualization), the samples, and the merging of the two channels of both above, respectively. | |
In vitro cytotoxicity of DOX-loaded UCNPs@mSiO2-PEG/FA nanospheres against cells
To verify DOX delivery efficacy of folate-conjugated UCNPs@mSiO2-PEG nanospheres, the cytotoxic effects of DOX-UCNPs@mSiO2-PEG and UCNPs@mSiO2-PEG/FA nanospheres on HeLa cells were tested. HeLa cells were incubated in a culture medium containing free DOX, DOX-UCNPs@mSiO2-PEG nanospheres, and DOX-UCNPs@mSiO2-PEG/FA nanospheres for 24 h at different DOX concentrations. After carefully washing the cells with PBS to remove unbound nanoparticles, the cell viability of the samples was tested by MTT assay. As presented in Fig. 11, the drug-loaded nanospheres caused observable cytotoxicity to HeLa cells. More importantly, DOX-UCNPs@mSiO2-PEG/FA has the highest cytotoxicity in these cases. This may be attributed to the fact that the DOX-UCNPs@mSiO2-PEG/FA nanospheres can be taken up via receptor-mediated endocytosis by HeLa cells and release DOX inside to induce cell death. Furthermore, HeLa cells can take up more UCNPs@mSiO2-PEG/FA nanospheres than unmodified UCNPs@mSiO2-PEG nanospheres, which results in more DOX molecules released in cells. Based on these results, the UCNPs@mSiO2-PEG/FA can potentially be used as a vehicle to store and deliver anti-cancer drugs, target cancer cells and enhance the efficacy of anti-cancer drug delivery.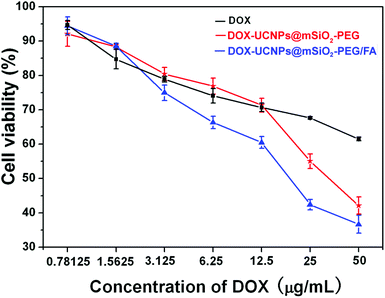 |
| | Fig. 11 In vitro HeLa cell viabilities cultured for 24 h with free DOX (black line), DOX-UCNPs@mSiO2-PEG (red line), and DOX-UCNPs@mSiO2-PEG/FA (blue line) at different DOX concentrations. | |
Conclusions
The uniform, monodisperse and core–shell structured drug delivery nanocarriers UCNPs@mSiO2 composed of mesoporous silica coated upconversion luminescence nanocrystals were fabricated successfully by a facile and fast methodology. The PEG modification on the nanomaterials is able to increase colloidal stability, biocompatibility and water dispersion, while folic acid (FA) as a targeting component can enhance the targeting delivery of drugs to the cancer cells. The anti-cancer drug DOX in UCNPs@mSiO2-PEG/FA nanospheres released is pH-sensitive. The DOX-loaded UCNPs@mSiO2-PEG/FA nanospheres exhibited greater cytotoxicity compared with free DOX and DOX-loaded UCNPs@mSiO2-PEG nanospheres at the same concentration. Moreover, upconversion luminescence image of UCNPs@mSiO2-PEG/FA taken up by cells shows green emission, making the UCNPs@mSiO2-PEG/FA nanocomposite spheres promising candidates for use as bioimaging agents. Thus, these kinds of composite nanoparticles provide a platform for a variety of biomedical applications ranging from upconversion bioimaging to drug delivery. Furthermore, this synthetic strategy can be extended to fabricate other multifunctional nanocarriers for simultaneous bioimaging and therapeutic applications.Acknowledgements
This project is financially supported by National Basic Research Program of China (2010CB327704), National High Technology Program of China (2011AA03A407), the National Natural Science Foundation of China (NSFC 51272248, 51172228, 21101149, 20901074, 51202239), Science Technology Development Program of Jilin Province for Youths (20100106).Notes and references
-
(a) J. Kim, Y. Z. Piao and T. Hyeon, Chem. Soc. Rev., 2009, 38, 372 RSC;
(b) P. P. Yang, S. L. Gai and J. Lin, Chem. Soc. Rev., 2012, 41, 3679 RSC.
-
(a) H. S. Qian, H. C. Guo, P. C.-L. Ho, R. Mahendran and Y. Zhang, Small, 2009, 5, 2285 CrossRef CAS;
(b) J. P. Yang, Y. H. Deng, Q. L. Wu, J. Zhou, H. F. Bao, Q. Li, F. Zhang, F. Y. Li, B. Tu and D. Y. Zhao, Langmuir, 2010, 26, 8850 CrossRef CAS;
(c) X. J. Kang, Z. Y. Cheng, C. X. Li, D. M. Yang, M. M. Shang, P. A. Ma, G. G. Li, N. Liu and J. Lin, J. Phys. Chem. C, 2011, 115, 15801 CrossRef CAS;
(d) Y. F. Yang, Y. Q. Qu, J. W. Zhao, Q. H. Zeng, Y. Y. Ran, Q. B. Zhang, X. G. Kong and H. Zhang, Eur. J. Inorg. Chem., 2010, 33, 5195 CrossRef;
(e) Z. Liu, L. N. Sun, F. Y. Li, Q. Liu, L. Y. Shi, D. S. Zhang, S. Yuan, T. Liu and Y. N. Qiu, J. Mater. Chem., 2011, 21, 17615 RSC;
(f) S. L. Gai, P. P. Yang, C. X. Li, W. X. Wang, Y. L. Dai, N. Niu and J. Lin, Adv. Funct. Mater., 2010, 20, 1166 CrossRef CAS;
(g) F. Zhan, G. B. Braun, A. Pallaoro, Y. C. Zhang, Y. F. Shi, D. X. Cui, M. Moskovits, D. Y. Zhao and G. D. Stucky, Nano Lett., 2012, 12, 61 CrossRef;
(h) J. N. Liu, W. B. Bu, S. J. Zhang, F. Chen, H. Y. Xing, L. M. Pan, L. P. Zhou, W. J. Peng and J. L. Shi, Chem.–Eur. J., 2012, 18, 2335 CrossRef CAS.
-
(a) I. I. Slowing, B. G. Trewyn, S. Giri and V. S. Y. Lin, Adv. Funct. Mater., 2007, 17, 1225 CrossRef CAS;
(b) S. Yang, X. Zhou, P. Yuan, M. Yu, S. Xie, J. Zou, G. Q. Lu and C. Yu, Angew. Chem., Int. Ed., 2007, 46, 8579 CrossRef CAS;
(c) M. Liong, S. Angelos, E. Choi, K. Patel, J. F. Stoffart and J. I. Zink, J. Mater. Chem., 2009, 19, 6251 RSC;
(d) S.-H. Wu, Y. Hung and C.-Y. Mou, Chem. Commun., 2011, 47, 9972 RSC.
-
(a) J. Shen, L. D. Sun and C. H. Yan, Dalton Trans., 2008, 5687 RSC;
(b) F. Wang and X. G. Liu, Chem. Soc. Rev., 2009, 38, 976 RSC;
(c) M. Hasse and H. Schäfer, Angew. Chem., Int. Ed., 2011, 50, 5808 CrossRef;
(d) H. S. Mader, P. Kele, S. M. Saleh and O. S. Wolfbeis, Curr. Opin. Chem. Biol., 2010, 14, 582 CrossRef CAS;
(e) L. Y. Ang, M. E. Lim, L. C. Ong and Y. Zhang, Nanomedicine, 2011, 6, 1273 CrossRef;
(f) A. Kar and A. Patra, Nanoscale, 2012, 4, 3608 RSC.
-
(a) D. Q. Chen, Y. L. Yu, F. Huang, P. Huang, A. P. Yang and Y. S. Wang, J. Am. Chem. Soc., 2010, 132, 9976 CrossRef CAS;
(b) D. Q. Chen, Y. L. Yu, F. Yang and Y. S. Wang, Chem. Commun., 2011, 47, 2601 RSC;
(c) D. Q. Chen, Y. L. Yu, F. Huang, P. Huang, A. P. Yang, Z. X. Wang and Y. S. Wang, Chem. Commun., 2011, 47, 11083 RSC;
(d) D. Q. Chen, P. Huang, Y. L. Yu, F. Huang, A. P. Yang and Y. S. Wang, Chem. Commun., 2011, 47, 5801 RSC;
(e) D. Q. Chen, L. Lei, A. P. Yang, Z. X. Wang and Y. S. Wang, Chem. Commun., 2012, 48, 5898 RSC;
(f) D. T. Tu, L. Q. Liu, Q. Ju, Y. S. Liu, H. M. Zhu, R. F. Li and X. Y. Chen, Angew. Chem., Int. Ed., 2011, 50, 6306 CrossRef CAS;
(g) D. Q. Chen, Y. L. Yu, F. Huang, H. Lin, P. Huang, A. P. Yang, Z. X. Wang and Y. S. Wang, J. Mater. Chem., 2012, 22, 2632 RSC.
-
(a) J. Zhou, Z. Liu and F. Y. Li, Chem. Soc. Rev., 2012, 41, 1323 RSC;
(b) M. Nyk, R. Kumar, T. Y. Ohulchanskyy, E. J. Bergey and P. N. Prasad, Nano Lett., 2008, 8, 3834 CrossRef CAS;
(c) R. Kumar, M. Nyk, T. Y. Ohulchanskyy, C. A. Flask and P. N. Prasad, Adv. Funct. Mater., 2009, 19, 853 CrossRef CAS;
(d) H. Y. Xing, W. B. Bu, S. J. Zhang, X. P. Zheng, M. Li, F. Chen, Q. J. He, L. P. Zhou, W. J. Peng, Y. Q. Hua and J. L. Shi, Biomaterials, 2012, 33, 1079 CrossRef CAS;
(e) L. Cheng, K. Yang, Y. G. Li, J. H. Chen, C. Wang, M. W. Shao, S.-T. Lee and Z. Liu, Angew. Chem., Int. Ed., 2011, 50, 7385 CrossRef CAS;
(f) H. Xu, L. Cheng, C. Wang, X. X. Ma, Y. G. Li and Z. Liu, Biomaterials, 2011, 32, 9364 CrossRef CAS;
(g) M. Wang, C. C. Mi, W. X. Wang, C. H. Liu, Y. F. Wu, Z. R. Xu, C. B. Mao and S. K. Xu, ACS Nano, 2009, 3, 1580 CrossRef CAS;
(h) J. C. Zhou, Z. L. Yang, W. Dong, R. J. Rang, L. D. Sun and C. H. Yan, Biomaterials, 2011, 32, 9059 CrossRef CAS;
(i) G. Tian, Z. J. Gu, L. J. Zhou, W. Y. Yin, X. X. Liu, L. Yan, S. Jin, W. L. Ren, G. M. Xing, S. J. Li and Y. L. Zhao, Adv. Mater., 2012, 24, 1226 CrossRef CAS;
(j) A. Xia, Y. Gao, J. Zhou, C. Y. Li, T. S. Yang, D. M. Wu, L. M. Wu and F. Y. Li, Biomaterials, 2011, 32, 7200 CrossRef CAS;
(k) Q. Liu, T. S. Yang, W. Feng and F. Y. Li, J. Am. Chem. Soc., 2012, 134, 5390 CrossRef CAS.
- W. R. Zhao, J. L. Gu, L. X. Zhang, H. R. Chen and J. L. Shi, J. Am. Chem. Soc., 2005, 127, 8916 CrossRef CAS.
- Y. H. Deng, D. W. Qi, C. H. Deng, X. M. Zhang and D. Y. Zhao, J. Am. Chem. Soc., 2008, 130, 28 CrossRef CAS.
- V. Cauda, C. Argyo and T. Bein, J. Mater. Chem., 2010, 20, 8693 RSC.
- J. M. Rosenholm, A. Meinander, E. Peuhu, R. Niemi, J. E. Eriksson, C. Sahlgren and M. Lindén, ACS Nano, 2009, 3, 197 CrossRef CAS.
-
(a) Z. Y. Hou, C. X. Li, P. A. Ma, G. G. Li, Z. Y. Cheng, C. Peng, D. M. Yang, P. P. Yang and J. Lin, Adv. Funct. Mater., 2011, 21, 2356 CrossRef CAS;
(b) Z. Y. Hou, C. X. Li, P. A. Ma, Z. Y. Cheng, X. J. Li, X. Zhang, Y. L. Dai, D. M. Yang, H. Z. Lian and J. Lin, Adv. Funct. Mater., 2012, 22, 2713 CrossRef CAS.
- Z. Q. Li, Y. Zhang and S. Jiang, Adv. Mater., 2008, 20, 4765 CrossRef CAS.
- C. X. Li, J. L. Liu, S. Alonso, F. Y. Li and Y. Zhang, Nanoscale, 2012, 4, 6065 RSC.
- Y.-K. Peng, C.-W. Lai, C.-L. Liu, H.-C. Chen, Y.-H. Hsiao, W.-L. Liu, K.-C. Tang, Y. Chi, J.-K. Hsiao, K.-E. Lim, H.-E. Liao, J.-J. Shyue and P.-T. Chou, ACS Nano, 2011, 5, 4177 CrossRef CAS.
-
(a) J. Kim, J. E. Lee, J. Lee, J. H. Yu, B. C. Kim, K. An, Y. Hwang, C. H. Shin, J. G. Park, J. Kim and T. Hyeon, J. Am. Chem. Soc., 2006, 128, 688 CrossRef CAS;
(b) H. Fan, E. W. Leve, C. Scullin, J. Gabaldon, D. Tallant, S. Bunge, T. Boyle, M. C. Wilson and C. J. Brinker, Nano Lett., 2005, 5, 645 CrossRef CAS;
(c) J. Kim, H. S. Kim, N. Lee, T. Kim, H. Kim, T. Yu, I. C. Song, W. K. Moon and T. Hyeon, Angew. Chem., Int. Ed., 2008, 47, 8438 CrossRef CAS;
(d) Y. S. Lin and C. L. Haynes, Chem. Mater., 2009, 21, 3979 CrossRef CAS;
(e) T. Kim, E. Momin, J. Choi, K. Yuan, H. Zaidi, J. Kim, M. Park, N. Lee, M. T. Mcmahon, A. Q. Hinojosa, J. W. M. Bulte, T. Hyeon and A. G. Gilad, J. Am. Chem. Soc., 2011, 133, 2955 CrossRef CAS.
-
(a) L. Zhang, S. Z. Qiao, Y. G. Jin, H. G. Yang, S. Budihartono, F. Stahr, Z. F. Yan, X. L. Wang, Z. P. Hao and G. Q. Lu, Adv. Funct. Mater., 2008, 18, 3203 CrossRef CAS;
(b) M. Liong, J. Lu, M. Kovochich, T. Xia, S. G. Ruehm, A. E. Nel, F. Tamanoi and J. I. Zink, ACS Nano, 2008, 2, 889 CrossRef CAS.
- G. F. Wang, Q. Peng and Y. D. Li, Acc. Chem. Res., 2011, 44, 322 CrossRef CAS.
-
(a) J. C. Boyer, M. P. Manseau, J. I. Murray and F. C. J. M. van Veggel, Langmuir, 2010, 26, 1157 CrossRef CAS;
(b) N. Bogdan, F. Vetrone, G. A. Ozin and J. A. Capobianco, Nano Lett., 2011, 11, 835 CrossRef CAS.
- F. Wang, J. Wang and X. G. Liu, Angew. Chem., Int. Ed., 2010, 49, 7456 CrossRef CAS.
- R. I. Nooney, D. Thirunavukkarasu, Y. Chen, R. Josephs and A. E. Ostafin, Langmuir, 2003, 19, 7628 CrossRef CAS.
- P. P. Yang, Z. W. Quan, Z. Y. Hou, C. X. Li, X. J. Kang, Z. Y. Cheng and J. Lin, Biomaterials, 2009, 30, 4786 CrossRef CAS.
- H. X. Wu, G. Liu, S. J. Zhang, J. L. Shi, L. X. Zhang, Y. Chen, F. Chen and H. R. Chen, J. Mater. Chem., 2011, 21, 3037 RSC.
- Y. F. Zhu, Y. Fang and S. Kaskel, J. Phys. Chem. C, 2010, 114, 16382 CAS.
- D. S. Kohane and R. Langer, Chem. Sci., 2010, 1, 441 RSC.
- N. Lewinski, V. Colvin and R. Drezek, Small, 2008, 4, 26 CrossRef CAS.
- L. Cheng, K. Yang, M. W. Shao, X. H. Lu and Z. Liu, Nanomedicine, 2011, 6, 1327 CrossRef CAS.
- Y. Chen, H. R. Chen, D. P. Zeng, Y. B. Tian, F. Chen, J. W. Feng and J. L. Shi, ACS Nano, 2010, 4, 6001 CrossRef CAS.
-
(a) D. C. Drummond, M. Zignani and J. C. Leroux, Prog. Lipid Res., 2000, 39, 409 CrossRef CAS;
(b) Z. M. Bhujwalla, D. Artemov, P. Ballesterors, S. Cerdan, R. J. Gillies and M. Solaiyappan, NMR Biomed., 2002, 15, 114 CrossRef CAS.
- W. Wei, G. H. Ma, G. Hu, D. Yu, T. Mcleish, Z. G. Su and Z. Y. Shen, J. Am. Chem. Soc., 2008, 130, 15808 CrossRef CAS.
-
(a) T. T. Wang, L. Y. Zhang, Z. M. Su, C. G. Wang, Y. Liao and Q. Fu, ACS Appl. Mater. Interfaces, 2011, 3, 2479 CrossRef CAS;
(b) T. T. Wang, F. Chai, Q. Fu, L. Y. Zhang, H. Y. Liu, L. Li, Y. Liao, Z. M. Su, C. G. Wang, B. Y. Duan and D. X. Ren, J. Mater. Chem., 2011, 21, 5299 RSC.
-
(a) S. Setua, D. Menon, A. Asok, S. Nair and M. Koyakutty, Biomaterials, 2010, 31, 714 CrossRef CAS;
(b) X. Wang, J. Li, Y. X. Wang, L. Koenig, A. Giyrezi, P. Giannakakou, E. H. Shin, M. Tighiouart, Z. Chen, S. M. Nie and S. M. Shin, ACS Nano, 2011, 5, 6184 CrossRef CAS;
(c) J. Lu, M. Liong, Z. X. Li, J. I. Zink and F. Tamanoi, Small, 2010, 6, 1794 CrossRef CAS.
- L. L. Li, R. Zhang, L. L. Yin, K. Z. Zheng, W. P. Qin, P. R. Selvin and Y. Lu, Angew. Chem., Int. Ed., 2012, 51, 6121 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: the size distribution of β-NaYF4:Er3+/Yb3+ core and core–shell structured β-NaYF4:Er3+/Yb3+@mSiO2 by dynamic light scattering (Fig. S1), the digital photograph of UCNPs dispersed in cyclohexane under daylight with a ruler (Fig. S2), the upconversion emission spectrum of UCNPs@mSiO2 nanoparticles dispersed in water (Fig. S3), the digital photographs of UCNPs@mSiO2, UCNPs@mSiO2-PEG, and UCNPs@mSiO2-PEG/FA dispersed in water (Fig. S4) and the concentration of Y3+ internalized in HeLa cells measured by ICP-OES technique after incubation with UCNPs@mSiO2-PEG/FA (FA+) and UCNPs@mSiO2-PEG (FA−) (Fig. S5). See DOI: 10.1039/c2bm00087c |
|
| This journal is © The Royal Society of Chemistry 2013 |
Click here to see how this site uses Cookies. View our privacy policy here.