Ambient temperature synthesis of citrate stabilized and biofunctionalized, fluorescent calcium fluoride nanocrystals for targeted labeling of cancer cells
Sisini
Sasidharan
,
Aswathy
Jayasree
,
Sajid
Fazal
,
Manzoor
Koyakutty
,
Shantikumar V.
Nair
* and
Deepthy
Menon
*
Amrita Centre for Nanosciences and Molecular Medicine, Amrita Institute of Medical Sciences and Research Centre, Amrita Vishwa Vidyapeetham, Kochi-682 041, Kerala, India. E-mail: deepthymenon@aims.amrita.edu; nairshanti@gmail.com; Fax: +91-484-2802020; Tel: +91-484-4008750
First published on 7th November 2012
Abstract
Targeted biological contrast agents are emerging as promising candidates in the field of cancer theragnostics. Herein, we report an ambient temperature synthesis of a nanosized, antibody functionalized lanthanide doped CaF2 biolabel and demonstrate in vitro its potential for cancer cell targeting efficacy and specificity. Monodispersed citrate stabilized lanthanide (Eu3+) doped CaF2 nanoparticles with size ∼25 nm, exhibiting strong fluorescent emission at 612 nm, were prepared using an aqueous wet chemical route at room temperature. Biofunctionalization of the fluorescent nanoparticles using an anti-EGFR antibody through EDC–NHS coupling chemistry enabled targeting of EGFR over-expressing cells. The nanobioconjugates showed preferential binding to EGFR+ve oral epithelial carcinoma cells (KB) and human epidermoid carcinoma cells (A431) with no accumulation onto EGFR−ve non-cancerous NIH 3T3 cells. The fluorescence was maintained after the bioconjugation as well as after attachment to the cancer cells, demonstrating their potential as targeted biolabels. Cytotoxicity evaluation with several cancerous (A431, KB) and non-cancerous (NIH 3T3, L929) cell lines revealed no toxicity at concentrations up to 1 mM. Thus, the fluorescence characteristics and biocompatibility, coupled with the molecular receptor targeting capability, suggest the potential use of CaF2 in the field of bioimaging.
1. Introduction
Advances in bioimaging enabled by nanotechnology are driven primarily by the emergence of new tools that assist manipulations at an atomic scale, as well as new materials with tailorable size, surface chemistry, improved solubility, and multifunctionality.1 The use of nanosized fluorescent biolabels for cancer diagnosis is by now well recognized as having diagnostic value both ex vivo2,3 and in vivo.4,5 Such nanomaterials should necessarily possess good photostability and high quantum yield, as well as non-toxicity, to serve as diagnostic agents ex vivo or in vivo. Conventionally, organic fluorophores, fluorescent silica beads, or quantum dots have been used as fluorescent bio-labeling materials.6,7 The first two suffer from problems associated with photobleaching and small Stokes shift, whereas quantum dots pose toxicity concerns in vivo.8,9 This has stimulated interest in developing biocompatible and highly efficient fluorescent nanomaterials for use as biological labels. Our group has developed a host of inorganic nanoparticles based on ZnS,10,11 Y2O3,12 the biomineral hydroxyapatite,13 and organic protein containing gold molecular clusters2,14,15 as novel contrast agents, and demonstrated success in the targeted labeling of cancer cells.Inorganic luminescent materials based on rare earth ions have gained considerable attention and have become promising alternative biolabels owing to their superior optical properties, such as high quantum yield, narrow bandwidth, long-lived emission, large Stokes shift and ligand-dependent luminescence sensitization, all of which meet the stringent requirements of a biological label.12,13,16 The fluorescence emission from lanthanide ions has been made use of in various optoelectronic applications such as displays, lasers, etc.17,18 However, non-radiative relaxation in the luminescence of Ln3+ ions necessitates an appropriate lattice with a lower phonon energy to host the lanthanide ions.19 Among the various inorganic matrices, fluorides, owing to their high transparency arising from low phonon energy, serve as perfect host materials and have been widely established as a laser material for several years.20–22 Additionally, the optically isotropic structure, high stability, non-hygroscopic behavior, non-toxicity and biocompatibility of CaF2 among all the other fluorides make it an ideal host matrix for biological fluorescent contrast material.23
In this study, we have investigated a CaF2 matrix doped with lanthanide ions as a suitable biolabel. Various researchers have developed this promising system as a good host matrix for lanthanide ions, through several approaches such as hydrothermal, reverse micelle, chemical co-precipitation, etc.;24–30 however, two new objectives are explored here: (a) an ambient temperature processing route for the biolabel, which is expected to provide a water dispersible product and also better retain the luminescence intensity of lanthanide ions, and (b) antibody functionalization of the label with the ability to specifically target cancer cells and retain luminescence when functionalized, as well as when in contact with the target cells. Several investigators have attempted to render functional groups on doped CaF2. Bioconjugation of chitosan coated CaF2:Eu nanoparticles synthesized via the microemulsion method with bovine serum albumin has been previously attempted.31 Oleic acid modified CaF2:Eu nanocrystals well dispersed in chloroform were synthesized by Song et al.32 Likewise, using a single step hydrothermal method, oleate capped lanthanide doped upconverting colloidal CaF2 nanoparticles have been prepared.33 Citrate, a calcium complexing agent, is reported to tailor the size and control the morphology of apatite crystals upon binding to its surface.34,35 This strategy of size and morphology control by citrate, inspired by nature, is exploited for the synthesis of nanomaterials such as lanthanide fluorides36 and apatite nanocrystals.37–40 Citrate ions are reported to have the affinity to inhibit CaF2 crystal growth41 and recently citrate stabilized nanoparticles of Tm3+,Yb3+ and Er3+,Yb3+ doped CaF2 have been synthesized by hydrothermal processing at 190 °C.42 However, the combination of water dispersibility and conjugation with biomolecules has not so far been demonstrated. This is challenging, owing to the extreme reaction conditions such as high temperature and pressure used for the synthesis, and hence the need for an alternative strategy.
We report for the first time the synthesis of citrate stabilized lanthanide doped CaF2 nanoparticles (Cit-CaF2:Ln) via a simple wet chemical route at room temperature, and demonstrate its application as a biolabel. Cit-CaF2:Eu nanoparticles synthesized via aqueous chemistry in the present work were bioconjugated with an epidermal growth factor receptor (EGFR) specific antibody and the target specific imaging has been demonstrated using EGFR+ve cells. The high fluorescence emission, biocompatibility and receptor specific targeting of bioconjugated CaF2 nanoparticles suggest its potential use as an inorganic contrast agent for molecular imaging of cancer.
2. Results and discussion
2.1 Synthesis of bare and citrate stabilized CaF2:Ln nanoparticles
In the synthesis of undoped calcium fluoride particles, although the precursors, viz. CaCl2·2H2O and NH4F, are readily water soluble, the formed product (CaF2), being insoluble in water, precipitates out from aqueous solution. The appearance of turbidity during synthesis indicated the formation of CaF2.44 However, it was found very difficult to control the particle size of CaF2 during growth, owing to the high concentration of Ca2+ and F− ions, leading to enhanced reaction rates. Hence, to limit the availability of ions, in the present synthesis route the precursors were added drop-wise into the reaction mixture.Trivalent lanthanide ions (Tb3+, Eu3+) when doped into the calcium fluoride matrix incorporate into the crystal lattice by substituting divalent calcium ions, resulting in the formation of CaF2:Ln particles. The excess positive charge resulting from this doping is generally compensated for by the incorporation of fluoride ions at interstitial sites, resulting in structural defects in the lattice.24,45 A typical reaction scheme for the preparation of lanthanide doped CaF2 is represented below, with the schematic of the synthesis procedure depicted in Fig. 1a.
| CaCl2·2H2O + NH4F + LnCl3(Ln:Tb, Eu) ⇒ Ca1−xF2:Lnx + NH4Cl + 2H2O |
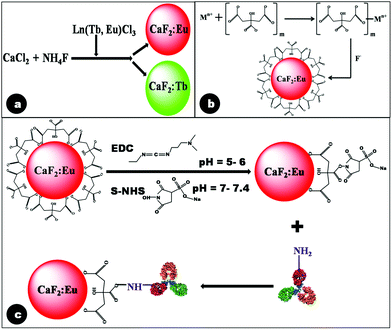 | ||
| Fig. 1 Schematic reaction mechanism depicting (a) formation of lanthanide doped CaF2 particles in the absence of citrate; (b) pre-complexation of citrate ions by Ca2+ followed by formation of Cit-CaF2:Eu nanoparticles in the presence of F− ions at pH 5.5; and (c) antibody conjugation of citrate stabilized CaF2:Eu nanoparticles. | ||
In this reaction, only the product Ca1−xF2:Lnx is the insoluble component, whereas all other reactants and products are water soluble, and hence can be easily removed by centrifugation. Optimization of the sequence of addition of reactants in the above reaction aided efficient incorporation of lanthanide ions into the CaF2 matrix. The different reaction sequences used in preparing highly fluorescent CaF2 particles were: (i) mixing dopant ions with Ca2+ precursors to which the F− precursor (NH4F) was added drop-wise; (ii) mixing the dopant ions with NH4F and adding them drop-wise to the Ca2+ precursor and (iii) adding dopant ions separately while Ca2+ and F− ions react to nucleate CaF2.
It was observed that case (i) and case (iii) resulted in the formation of CaF2 with efficiently incorporated lanthanide ions, as suggested by its high fluorescence emission. However, in case (ii), the dopant ions, upon reacting with NH4F, were found to form white precipitates of lanthanide fluorides (EuF3 or TbF3). Hence, for all further experiments, dopant ions were mixed with Ca2+ precursors and the F− precursor (NH4F) was added drop-wise to this mixture [case (i)].
Additionally, the pH of the solution was found to play a critical role in the formation of CaF2, with a range of 5–6 facilitating a maximum yield. Upon addition of lanthanide chlorides, the pH of the solution decreased below the limit that yielded CaF2 particles. Hence, several trials were made, with variations in pH facilitated by the addition of liquor ammonia. Addition of NaOH to the reaction mixture to maintain the pH can result in the formation of NaF, which decreases the yield of CaF2, and hence was not used in our experiments. Another important observation was that the synthesized lanthanide doped CaF2 nanoparticles were found to agglomerate with time, since no stabilizers were used initially. Use of citrates was additionally able to provide a stable colloid that could be further functionalized with the required biomolecules.
In our experiments, we used trisodium citrate (Na3C6H5O7) as the stabilizer, by drop-wise addition separately with NH4F. Trisodium citrate did not hinder CaF2 formation, as long as it was not mixed with the initial precursors of CaCl2 and NH4F. Further, it was capable of not only providing citrate ions for electrostatic stabilization, but also maintained the required pH for the formation of CaF2, resulting in stable CaF2:Ln nanoparticles. It was noted that a pH of 5.5 yielded nanoparticles with maximum stability.
We hypothesize the mechanism of citrate ion stabilization as follows. Ca2+ ions first form complexes with citrate anions, as has been proposed in the case of oleic acid stabilization.32 Subsequently, citrate complexed metal ions react with F− to form crystalline nuclei, whose growth is terminated in the nanosize range by the association of citrate ions on the growing particle interfaces.46 This proposed mechanism of citrate stabilization of doped CaF2 nanoparticles is illustrated in Fig. 1b. Citrate stabilization of CaF2 nanoparticles also provides the necessary carboxyl functional groups, which readily permit further conjugation with the antibody, using EDC–NHS coupling chemistry.43
Over-expression of EGFR has been observed in human cancers such as head and neck, ovarian, colon, bladder and breast cancer.47,48 Hence, in this study, Epidermal Growth Factor Receptor (EGFR), a transmembrane glycoprotein and a member of the erbB receptor tyrosine kinase family, was chosen as the primary target. Activation of carboxyl groups of citrate molecules by EDC is known to occur efficiently at acidic pH (4.5–6). This reaction results in the formation of an amine reactive O-acylisourea intermediate, which is susceptible to fast hydrolysis in water. Hence this intermediate is stabilized to form amine reactive NHS by the addition of S-NHS. The amine reactive Cit-CaF2:Eu was coupled with the primary amine of the anti-EGFR antibody, resulting in covalent attachment of the antibody to the nanoparticle surface, as schematically represented in Fig. 1c.
2.2 Crystal structure, crystallinity, size, shape and composition
Fig. 2a and 2b depict the SEM images of bare and citrate stabilized CaF2:Eu nanoparticles. The images clearly reveal that citrate stabilization has resulted in well stabilized and monodispersed nanoparticles of size ∼25 nm, in contrast to the agglomerated bare CaF2. The DLS data of citrate stabilized nanoparticles shown in the inset of Fig. 2b confirm this observation, showing a particle size of 20–25 nm. The lack of colloidal stability of bare CaF2:Eu hindered its DLS analysis. The TEM images of Cit-CaF2:Eu nanoparticles shown in Fig. 2c indicate well dispersed spherical nanoparticles with a particle size of ∼25 nm. The HRTEM images shown in Fig. 2d clearly show the ordered crystalline lattice planes of CaF2, with the measured lattice fringe spacing of 0.193 nm agreeing well with the lattice spacing of (220) of cubic CaF2.49 Additionally, few localized defect atomic clusters were seen as dark spots in the HRTEM image. The selected area electron diffraction (SAED) pattern of CaF2 nanocrystallites shown in the inset of Fig. 2d corresponds to the crystalline lattice planes (111), (220), (311) and (400) of CaF2. The virtual circular pattern observed in the diffraction pattern may be due to the presence of several crystallites of CaF2, as in polycrystalline materials. | ||
| Fig. 2 Representative SEM images of (a) bare CaF2:Eu particles; (b) Cit-CaF2:Eu nanoparticles (inset – particle size distribution by DLS); (c) TEM image and (d) HRTEM image (inset: SAED pattern) of Cit-CaF2:Eu nanoparticles. | ||
Citrate stabilized doped CaF2 nanoparticles possessed a moderately high zeta potential value of −22 ± 2.5 mV, with the negative charge due to the carboxyl groups of citrate on the nanoparticle surface. The electrostatic repulsion rendered by the carboxyl groups accounts for its stability in suspension. It was observed that Cit-CaF2:Ln nanoparticles prepared by altering the concentration of citrate ions, thereby changing the solution pH, showed remarkably different colloidal stability. Fig. 3a depicts the changes in zeta potential for samples prepared with varying pH via the addition of different amounts of trisodium citrate. As evident from Fig. 3b, a maximum colloidal stability was obtained at a pH of 5.5.
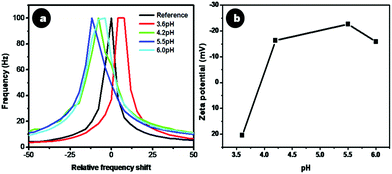 | ||
| Fig. 3 (a) Relative frequency shift and (b) zeta potential of Cit-CaF2:Eu nanoparticles synthesized at different pH. | ||
Crystallinity as well as phase purity of the synthesized doped and undoped CaF2 nanoparticles were analyzed using XRD. Fig. 4a depicts the XRD patterns of CaF2, CaF2:Eu and europium citrate, in comparison with the theoretical JCPDS data of CaF2 (line spectrum). Fig. 4b depicts the XRD of Cit-CaF2:Eu nanoparticles with varying dopant concentrations of Eu. The diffraction patterns of CaF2, CaF2:Eu and Cit-CaF2:Eu can be indexed to the cubic phase of the fluorite-type CaF2 structure (space group: Fm3m). These spectra exhibit the prominent reflections corresponding to (111), (220), (311), (400) and (311), well in accordance with the JCPDS data base (35-0816) of the CaF2 crystal.24 The absence of any characteristic peak of europium citrate in citrate stabilized CaF2:Eu (Fig. 4a) rules out the inclusion of europium citrate in the lattice, thereby confirming the phase purity of the sample. It can be noted from Fig. 4b that with increasing doping concentration of Eu3+ ions, diffraction peaks showed a marked decrease in intensity, with increased full width at half maximum (FWHM), indicating decreased crystallinity with increased disorder. This can be attributed to the structural defects that arise in CaF2 due to the substitution of Ca2+ ions with Eu3+, coupled with the charge compensatory incorporation of the F− ion.18,24 The presence of such lattice defects was evident from the HRTEM image (Fig. 2d). The results collectively suggest that the synthesized Cit-CaF2:Eu nanoparticles are isostructural with the CaF2 crystal, with some disorder and decreased crystallinity.
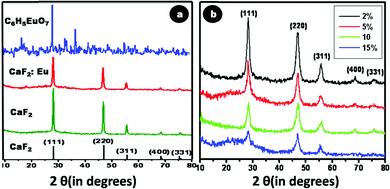 | ||
| Fig. 4 X-ray diffraction spectrum of (a) CaF2 – 35-0816, CaF2, CaF2:Eu, and europium citrate (C6H5EuO7) (b) Cit-CaF2:Eu nanoparticles with varying dopant concentration. | ||
The elemental composition of the synthesized nanoparticles was confirmed through EDX analysis. Fig. 5a shows a representative EDX spectrum of 15% Eu doped Cit-CaF2:Eu, displaying elemental peaks relating to Ca, F and Eu, confirming detectable levels of dopant ions within the CaF2 matrix. A linear increase in the atomic weight percentage of Eu3+ with respect to Ca2+ was noted for increasing Eu content used for synthesis, as depicted in tabular form in Fig. 5b. The values correspond to the experimentally used dopant concentrations, and it was observed that for higher Eu doping concentration, the estimated Eu content was lower, perhaps also due to the precipitation of europium citrate. However, the washed final product did not reveal any peaks of europium citrate, as evident from its XRD pattern (Fig. 4a and 4b).
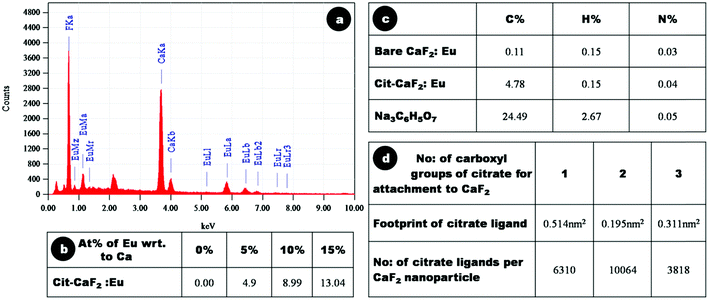 | ||
| Fig. 5 (a) Energy dispersive X-ray analysis spectrum of 15% Eu doped Cit-CaF2:Eu NP, (b) atomic weight percentage of europium with respect to calcium, as determined by EDAX analysis of Cit-CaF2:Eu nanoparticles synthesized with varying dopant concentration, (c) C, H, and N composition by CHN analysis of bare, Cit-CaF2:Eu nanoparticles and trisodium citrate, (d) footprint of citrate ligand and number of citrate ligands per CaF2 nanoparticle for its attachment through 1, 2 and 3 carboxyl groups. | ||
CHN analysis was performed to assess the elemental C, H and N composition of the samples and thereby evaluate the amount of citrate present on the particles. Fig. 5c depicts the tabulated results of elemental analysis of dried Cit-CaF2:Eu (15%), bare CaF2:Eu (15%) and trisodium citrate (Na3C6H5O7). These results are consistent with a mass content of 19.5% of citrate and 80.5% of Eu doped CaF2 in Cit-CaF2:Eu nanoparticles.
In the present study, we have used CHN analysis to assess the mode of attachment of citrate molecules to the surface of nanoparticles, i.e., whether the attachment is via one, two or three of its carboxyl groups. The number of citrate ligands that can attach to spherical CaF2 nanoparticles of size ∼25 nm was deduced by assuming a ligand footprint (corresponding to the area occupied by an anchoring group on the surface) of 0.514, 0.195, 0.311 nm2 for citrate molecule attachment through one, two and three of its carboxyl groups, respectively.50Fig. 5d, in tabulated form, depicts the number of citrate ligands per nanoparticle via one, two and three carboxyl attachments. It was observed that the number of citrate ligands on each nanoparticle (calculated from C % of CHN analysis) coincided with that calculated using a 0.195 nm2 footprint of the citrate molecule, implying that citrate molecules attached to CaF2 nanoparticles via two carboxyl groups. This renders one of the three carboxyl groups of citrate free for further bioconjugation, which is confirmed in the ensuing sections.
2.3 FTIR analysis
Fig. 6 shows the FTIR spectra of trisodium citrate, bare CaF2:Eu, Cit-CaF2:Eu and antibody conjugated nanoparticles. The broad absorption band occurring around 3440 cm−1 is characteristic of H–O–H bending of the water molecules, revealing the presence of hydroxyl groups in all the samples. For CaF2:Eu, the prominent absorbance noted at 1638 cm−1 can be assigned to the O–H bending vibrations of the residual water that is physically adsorbed onto the surface of the particles.31,51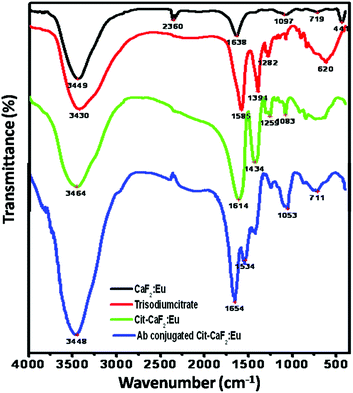 | ||
| Fig. 6 FTIR spectra of CaF2:Eu, trisodium citrate, Cit-CaF2:Eu and antibody conjugated Cit-CaF2:Eu nanoparticles, showing successful citrate functionalization and antibody conjugation on the nanoparticles. | ||
The absorption band at 2360 cm−1 is from the KBr pellets used for recording the FTIR spectrum. The spectra of Cit-CaF2:Eu have absorption bands that correspond to those of trisodium citrate, revealing the presence of the organic functional groups, i.e. citrate anions. Two strong absorption peaks at 1585 and 1394 cm−1 of trisodium citrate, characteristic of the RCO2 symmetric and asymmetric stretches, appear shifted to 1614 and 1434 cm−1, respectively, implying the conjugation of carboxyl groups to the nanoparticle surface (Ca). The very broad absorption band in the region 2500–3500 cm−1 for trisodium citrate and Cit-CaF2:Eu, corresponding to the hydrogen bonded OH stretch of the carboxyl group, obscures other peaks in this region. The typical peaks at 620 cm−1 prove the existence of alkyl groups of citrate.
The conjugation of the antibody to the nanoparticles is confirmed by the emergence of two new bands in the spectrum at 1654 and 1534 cm−1, which are characteristic of amide I and amide II bonds. The absence of carboxyl peaks and the emergence of these two new amide peaks are typical of antibody conjugation.52 Thus, FTIR analysis clearly confirms the presence of citrate ions on the surface of CaF2:Eu, providing it with adequate stabilization and surface functionalization with carboxyl groups, and amide bond formation in the case of antibody conjugated nanoparticles.
2.4 Fluorescence characteristics of doped CaF2
For terbium doped CaF2 nanoparticles, a characteristic green emission with varying intensity was observed when tested initially using a handheld UV lamp at 254 and 365 nm. From spectrofluorimetric analysis, it was deduced that Tb doped CaF2 nanoparticles exhibited characteristic emission peaks at 488, 542, 583 and 620 nm, which correspond to the 5D4 → 7F6, 5D4 → 7F5, 5D4 → 7F4 and 5D4 → 7F3 transitions, respectively, of Tb3+ ions in the host lattice.26 Amongst them, the most intense emission peak lies in the green region at λ = 542 nm, which corresponds to the 5D4 → 7F5 transition.26,27 The excitation peaks for this green emission at 542 nm are characteristic of 7F6 → 5D3, 7F6 → 5GJ and 7F6 → 5L6 transitions of Tb3+ ions for 257, 350 and 368 nm, respectively.26
The influence of doping content on these spectral excitation and emission peaks was also assessed. Fig. 7a and 7b depict a comparison of the excitation and emission spectra of CaF2:Tb for different dopant percentages of Tb (2, 5 and 10%) with respect to atomic weight percentage of calcium. Remarkable differences were noted when the dopant content in the crystalline lattice was varied. The predominant excitation peak for a dopant concentration of 2 and 5% Tb was 257 nm. On the other hand, upon increasing the dopant content to 10%, the excitation spectrum displayed two major peaks at 368 and 378 nm, instead of the prominent peak at 257 nm for low doping concentrations. Such an observation has not been reported earlier on Tb doped CaF2 nanoparticles. However, for 5% Tb doped nanoparticles prepared in anhydrous methanol and re-dispersed in water, Wang et al.26 have observed a green emission at 542 nm for an excitation at 375 nm. On the contrary, nanoparticles co-doped with Ce and Tb (5, 10 mol%) prepared by the polyol-mediated synthesis route, displayed emission at 534 nm for an excitation at 254 nm.27 This implies that electronic transitions are sensitive to dopant ion positions, as well as to their nearest neighbor interactions in the crystal lattice, aside from the crystal symmetry, all of which are influenced by the synthesis route and the dopants used.
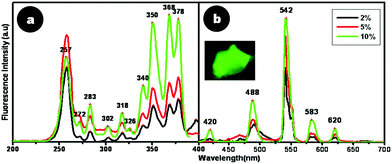 | ||
| Fig. 7 Fluorescence (a) excitation and (b) emission spectra of CaF2:Tb. Inset shows the picture of CaF2:Tb powder when viewed under a fluorescence microscope. | ||
For the highest doping content of 10% Tb used in this study, a marked enhancement in fluorescence emission at 542 nm at a red-shifted excitation wavelength of 378 nm was noticed, which would enable bioimaging applications with better spectral resolution. This implies that an increase in doping concentration enabled better incorporation of Tb3+ into the CaF2 lattice. Thus, from these studies, the optimal dopant concentration of Tb into the CaF2 lattice was found to be 10%, which resulted in high greenish luminescence emission.
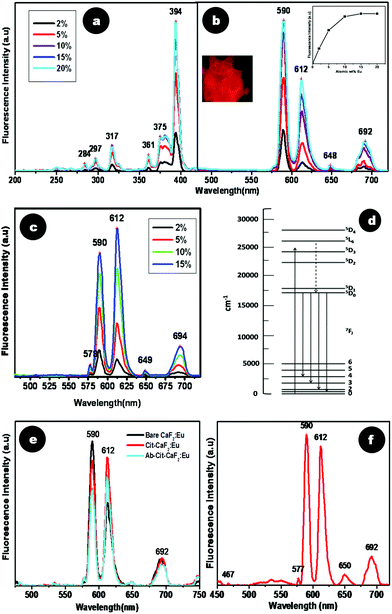 | ||
| Fig. 8 Spectrofluorimetric analysis of CaF2:Eu at varying dopant percentages (a) excitation and (b) emission spectra. Inset of (b) shows a graphical representation of emission (590 nm λex = 394 nm) intensity vs. dopant percentage of CaF2:Eu with saturation at 15%, and a picture of CaF2:Eu powder when viewed under a fluorescence microscope. (c) Emission spectra (λex = 394 nm) of Cit-CaF2:Eu with varying dopant percentages. (d) Intra orbital electronic transitions of Eu3+ ions. (e) Comparative photoluminescence spectra of bare, citrate stabilized and antibody conjugated CaF2:Eu. (f) Emission spectra (λex = 394 nm) of CaF2:Eu synthesized with liquor ammonia, maintaining a pH of 5.5. | ||
As discussed earlier, citrate stabilization of CaF2 yielded nanoparticles with high colloidal stability and monodispersed size. Cit-CaF2:Eu containing Eu at different dopant concentrations (2, 5, 10, and 15 at%) also showed a similar variation in its emission spectra (λex = 394 nm) (Fig. 8c). Similar to the effects observed earlier, increasing concentration of Eu3+ resulted in an enhancement in the emission, indicating higher doping of Eu3+ into the nanocrystal lattice. A doping concentration of 15 at% yielded the maximum fluorescence emission as in bare nanoparticles.
To understand if citrate capping or antibody conjugation in any way altered the pattern of fluorescence emission from Eu doped CaF2, a comparison of the spectra of bare CaF2:Eu and Cit-CaF2:Eu and Ab-Cit-CaF2:Eu was made, as shown in Fig. 8e. Surprisingly, although the excitation spectra of bare and citrate stabilized particles were similar, their emission spectra (λex = 394 nm) appeared distinctly different with regard to the ratios of intensity of the prominent emission transitions, viz.5D0 → 7F1 (590 nm) and 5D0 → 7F2 (612 nm). It is established that the 5D0 → 7F2 electric dipole transition is very sensitive to relatively small changes in the chemical surroundings of Eu3+ ions.32,54,55 The intensity of this transition is markedly affected by the degree of symmetry in the environments around Eu3+ ions. During the preparation of Cit-CaF2:Eu, addition of trisodium citrate alters the pH of the solution. Hence, we hypothesize that this change in pH to 5.5 might have caused variations in the chemical environment of the Eu3+ ions. This in turn might have resulted in the predominance of the 5D0 → 7F2 transition peak (612 nm) in Cit-CaF2:Eu nanoparticles, as compared to 5D0 → 7F1 (590 nm) in bare CaF2:Eu. This influence of pH in regulating the fluorescence emission was further confirmed by the use of liquor ammonia to maintain the pH at 5.5 during the synthesis of CaF2:Eu nanoparticles. The result of this study shown in Fig. 8f depicts the enhancement in 612 nm emission peak for an excitation at 394 nm, in comparison to bare nanoparticles, which always display a predominant 590 nm peak (Fig. 8b). This helps us to conclude that pH has a significant role in influencing the fluorescence emission from CaF2 nanoparticles.
Citrate stabilized CaF2:Eu nanoparticles obtained with citric acid as the precursor for citrate ions and liquor ammonia used to maintain the pH at 5.5 also displayed similar emission/excitation spectra as for nanoparticles prepared using trisodium citrate (data not shown). This implies that the source of citrate does not alter the fluorescence characteristics of CaF2:Eu.
Additionally, as evident from Fig. 8e, citrate stabilized CaF2:Eu nanoparticles, when further conjugated with the antibody, also displayed brilliant fluorescence, although with a slight reduction in intensity. However, the fluorescence intensity measured for Ab-Cit-CaF2:Eu was high enough to enable fluorescent labeling of cells.
2.5 Biological studies
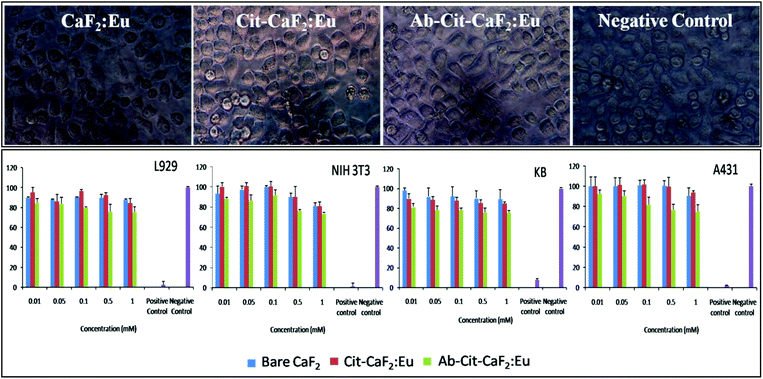 | ||
| Fig. 9 Top panel: representative microscopic images of KB cells incubated with bare CaF2:Eu, Cit-CaF2:Eu, Ab conjugated Cit-CaF2:Eu nanoparticles and negative control for 24 h (magnification: 40×); bottom panel: graphical representation of cell viability tests on L929, NIH 3T3, KB and A431 cells treated with CaF2:Eu nanoparticles (bare, citrate capped and antibody conjugated) at varying concentrations for 24 h. | ||
It is clearly evident from Fig. 9 (top panel) that the cells treated with CaF2 nanoparticles at the highest concentration for 24 hours remained morphologically intact and proliferated well in the culture medium in well plates. The nanomaterial treated cells did not reveal any characteristic features of apoptosis, proving its cytofriendly nature, which was further confirmed by the cytotoxicity studies. In comparison to the negative control, MTT analysis (Fig. 9 bottom panel) showed that more than 80% of cells were viable and metabolically active when tested on four different cell lines using bare, citrate stabilized and antibody conjugated CaF2:Eu nanoparticles at varying doses. These studies confirm that the fluorescent nanobioprobes are cytocompatible, with no adverse effects on their cellular functions, which is an essential requisite for contrast agents. Similar observations on the non-toxic effects of CaF2 upconversion nanophosphors prepared hydrothermally have been reported on HeLa cells for 18 hours of incubation.42 Fluorides are conventionally toxic in vivo.56,57 However, fluoride toxicity is not a concern in the case of CaF2, due to its extreme insolubility, unlike other fluorides such as NaF, BaF2, etc.58 This makes CaF2 an important material for various human applications such as in dentistry, food supplements, etc. Hence, lanthanide doped fluorescent CaF2 nanoparticles, if proposed for in vivo applications, would offer ideal contrast agents for bioimaging, owing to their in vivo biocompatibility.
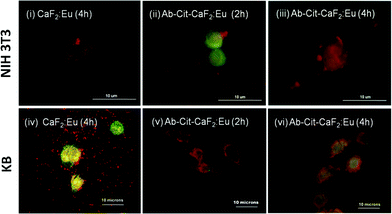 | ||
| Fig. 10 Fluorescent microscopic images showing the interaction of bare CaF2:Eu and antibody conjugated Cit-CaF2:Eu nanoparticles (Ab-Cit-CaF2:Eu) with EGFR−ve NIH 3T3 (top panel) and EGFR+ve KB cells (bottom panel) for different incubation periods. | ||
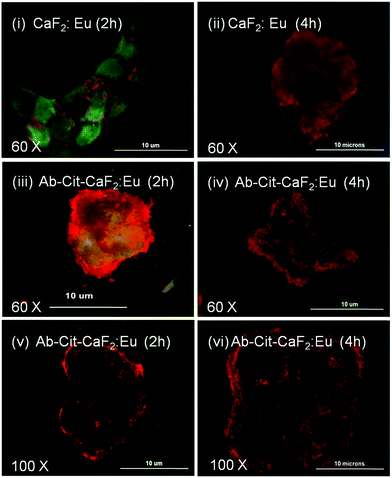 | ||
| Fig. 11 Fluorescent microscopic images showing the interaction of bare CaF2:Eu and antibody conjugated Cit-CaF2:Eu nanoparticles (Ab-Cit-CaF2:Eu) with EGFR over-expressing, human epidermoid carcinoma A431 cells for 2 and 4 hours of incubation, observed at different magnifications. | ||
Similar to the microscopic images shown in Fig. 9, the cells (KB, A431), presenting an enhanced accumulation of nanoparticles on the cell membrane, retained their characteristic morphology and showed good spreading with time, implying that the material does not induce any toxicity to cells. This study clearly indicates the enhanced affinity and specificity of antibody conjugated nanoparticles to EGFR receptors on cancer cells, with minimal attachment to the normal cells, which are essential attributes to any contrast agents suggested for targeted imaging applications.
3. Conclusion
In summary, a biological fluorescent label based on lanthanide doped calcium fluoride on the nanosize scale that can target the EGFR over-expressing cells has been developed in this study. A simple aqueous wet chemical route of synthesis using citrate ions as stabilizers resulted in highly fluorescent lanthanide (europium) doped CaF2 nanoparticles, with a characteristic emission at 612 nm. These nanoparticles of size ∼25 nm and high colloidal stability were conjugated with anti-EGFR antibodies, demonstrating specific targeting of EGFR over-expressing cells and no interaction with normal cells. Cell viability studies on four different cell lines demonstrated its non-toxic nature, even up to a relatively high concentration of 1 mM. Thus a detailed investigation on the synthesis, and characteristic physical, chemical and biological features of citrate stabilized Eu3+ doped calcium fluoride nanoparticles were carried out. Terbium doped CaF2, also developed using similar aqueous chemistry routes, and exhibiting a strong green fluorescence at 542 nm, can be likewise bioconjugated for imaging applications. Hence this study presents the application of biocompatible fluorescent calcium fluoride nanoparticles for targeted imaging of cancer cells. This work opens up the possibilities for extending this strategy to up-conversion nanophosphors in vivo or for multiplexed cancer diagnosis on clinical tissues.4. Materials and methods
4.1 Materials
The starting materials used for the synthesis of doped calcium fluoride nanoparticles include: calcium chloride dihydrate (CaCl2·2H2O) [≥97%, Merck], ammonium fluoride (NH4F) [≥95%, Merck, India], terbium chloride hexahydrate (TbCl3·6H2O) [99.999%, Sigma Aldrich, USA], europium chloride hexahydrate (EuCl3·6H2O) (99.9%, Sigma Aldrich, USA), trisodium citrate dihydrate (99%, Merck, India), citric acid anhydrous (99%, SD Fine Chem Ltd, India) and liquor ammonia (25% NH3, Fischer Scientific, India). EDC [1-ethyl-3-(3-dimethylaminopropyl) carbodiimide hydrochloride] (Thermo Scientific, USA) and S-NHS [sulfo-N-hydroxysulfosuccinimide] (Thermo Scientific, USA) were used for bioconjugation of the nanoparticles with Anti EGFR mouse monoclonal antibody (1 mg ml−1 in PBS, Abcam, UK). All the chemicals were used as received without any further purification.4.2 Synthesis of lanthanide (Ln3+-Tb3+, Eu3+) doped calcium fluoride (CaF2) particles [CaF2:Ln]
An aqueous wet chemical route at room temperature was adopted for the synthesis of bare fluorescent CaF2:Ln particles. In a typical experiment, 5 ml of 0.1 M CaCl2·2H2O was mixed thoroughly with a 0.1 M concentration of the dopant, viz. lanthanide chloride hexahydrate (TbCl3·6H2O or EuCl3·6H2O), at room temperature. To this mixture, 5 ml of 0.2 M NH4F as the precursor for fluorine was added drop-wise and stirred continuously for 2 h, yielding CaF2:Ln particles. To optimize the reaction conditions resulting in the efficient incorporation of lanthanide ions into the CaF2 matrix, the lanthanide precursor was added in three different ways during the synthesis, viz., mixed with the initial precursors CaCl2 or NH4F, or by drop-wise addition separately with NH4F. Highly fluorescent CaF2 particles were obtained under favourable conditions.To obtain different doping concentrations with respect to atomic weight percentage of calcium, varying amounts of dopant ions (0–15 at% for Tb and 0–20 at% for Eu) were employed. The resultant mixture was then centrifuged (24![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 780g) and washed thrice with MilliQ water to remove the reaction by-products and any un-reacted precursors. To visualize the formation of lanthanide doped CaF2, a handheld UV lamp with excitation at 365 nm was used, exhibiting a pale red or green emission for Eu and Tb dopants, respectively. The final product was dispersed in MilliQ water and stored. The resultant colloidal suspension was not stable on storage. The precipitate was also dried at 60 °C for 24 h for further characterization.
780g) and washed thrice with MilliQ water to remove the reaction by-products and any un-reacted precursors. To visualize the formation of lanthanide doped CaF2, a handheld UV lamp with excitation at 365 nm was used, exhibiting a pale red or green emission for Eu and Tb dopants, respectively. The final product was dispersed in MilliQ water and stored. The resultant colloidal suspension was not stable on storage. The precipitate was also dried at 60 °C for 24 h for further characterization.
To evaluate the influence of pH on the fluorescence emission as well as the yield, CaF2:Ln was also synthesized in a similar manner as above by varying the solution pH using liquor ammonia.
4.3 Synthesis of water dispersible citrate stabilized Eu doped calcium fluoride nanoparticles (Cit-CaF2:Eu)
To prepare well stabilized lanthanide doped nanoparticles of CaF2, citrate stabilization was employed. Trisodium citrate dihydrate was used as a stabilizer and pH modifier to prepare stabilized doped CaF2 nanoparticles (Cit-CaF2:Eu) via the same wet chemical route as described earlier. To assess which method would yield the most stable colloidal solution of doped CaF2, citrate was added in three different ways during the synthesis, viz., mixed with the initial precursors CaCl2 or NH4F, or separately with NH4F. Under optimal conditions, a white colloid of well stabilized doped CaF2 nanoparticles was formed, which exhibited brilliant fluorescence under UV excitation. This colloid was then centrifuged and dispersed in water for further characterization. The same protocol can be extended to prepare citrate stabilized Tb doped CaF2 nanoparticles as well.Additionally, a few trials with citric acid instead of trisodium citrate as the precursor for citrate ions were also performed, with liquor ammonia maintaining the desired pH of 5.5.
4.4 Bio-conjugation of Cit-CaF2:Eu with anti-EGFR antibody (Ab) using EDC–NHS coupling chemistry
To obtain fluorescent nanoparticles for bioimaging applications, an appropriate ligand is required to target specific receptors on cancer cells. Here, bio-conjugation of doped Cit-CaF2 nanoparticles was carried out with the anti-EGFR antibody using the conventional EDC–NHS conjugation chemistry to obtain a covalent attachment of the anti-EGFR antibody to the surface of nanoparticles.43 Citrate stabilized nanoparticles bearing free carboxyl groups on their surface were activated by the addition of the zero-length cross-linker EDC and further linked to the amine groups present on the antibody. Briefly, 1 mg Cit-CaF2:Eu was washed and re-dispersed with 10 mM MES buffer and incubated with 5 μM EDC and 7.5 μM S-NHS for 20 min, in the dark, at room temperature. They were then incubated with 50 μl anti-EGFR antibody (1 mg ml−1 in PBS, mouse monoclonal antibody with reactivity towards mouse and human) for 2 h, resulting in the formation of stable antibody conjugated CaF2 nanoparticles (Ab-Cit-CaF2:Eu). Antibody conjugated nanoparticles retained the bright fluorescence emission and were well stabilized.4.5 Particle characterization
Particle size and size distribution of the nanoparticles were measured by the dynamic light scattering technique using NICOMPTM380 Particle size analyzer (PSS, USA). The surface charge and thereby stability of the particles were assessed from the zeta potential measurements using a NICOMPTM380 ZLS zeta potential analyzer. Nanoparticles prepared in aqueous dispersions were used for the measurements in triplicate. Size and surface morphology of bare and citrate stabilized nanoparticles were evaluated by scanning electron microscopy (SEM) (JSM-6490LA, JEOL, Japan). A diluted sample drop-cast on an aluminium stub was sputter-coated with gold before imaging. Transmission electron microscopy (TEM) (JEM-200CX, JEOL, Japan) was employed for deciphering the shape, size and lattice structure of the nanoparticles. An aqueous dispersion of the nanoparticles at appropriate dilution was drop-cast onto a carbon-coated copper grid and air-dried at room temperature before TEM imaging.The phase purity and crystallinity of bulk CaF2, CaF2:Eu and Cit-CaF2:Eu nanoparticles and europium citrate (C6H5EuO7) were studied from X-ray diffraction (XRD) measurements using a Philips X'PERT PRO powder diffractometer fitted with a Cu-Kα source (λ = 1.54056 Å). The spectrum was recorded in the range 10°–80° in steps of 0.02°. Phase identification was done using the standard JCPDS database.
Composition of the nanoparticles, particularly the presence of dopants, was analyzed by Energy Dispersive X-Ray (EDX) spectroscopy (JSM-6490LA, JEOL, Japan). To quantify the amount of citrate molecules, as well as to decipher their mode of attachment, CHN analysis was performed on the samples using a CHN Analyzer (Perkin Elmer 2400, USA) to determine the percentage of carbon (C), hydrogen (H) and nitrogen (N) present in each material.
Spectrofluorimetric measurements of doped CaF2 colloids were recorded using a HORIBA, JOBIN YVON spectrofluorimeter (Model Fluoro Max 4). Fourier transform infrared (FT-IR) spectra of the samples supported on KBr pellets were recorded in the frequency range 4000–400 cm−1 at a resolution of 4 cm−1 using a Spectrum RX1 FT-IR Spectrometer, Perkin Elmer, USA.
4.6 Cell culture
Oral epithelial carcinoma cells (KB) and human epidermoid carcinoma cells (A431), which both over-express EGFR, and normal mouse fibroblast cells (L929) and normal mouse embryonic fibroblast cells (NIH 3T3) that do not have EGFR expression, used in the present study, were purchased from the National Center for Cell Science, Pune, India. L929 and KB cell lines were cultured in a T25 flask in Minimum Essential Medium (MEM) (Sigma-Aldrich, USA) and A431 cell line in Dulbecco's modified Eagles Medium (DMEM) (Sigma Aldrich, USA) supplemented with 10% fetal bovine serum (FBS, Invitrogen, CA, USA), 50 IU ml−1 penicillin and 50 μg ml−1 streptomycin. The cells were maintained in the medium at 37 °C in a humidified environment of 5% CO2.4.7 In vitro cell viability assay
To determine the cell viability upon interaction with calcium fluoride nanoparticles, L929, NIH3T3, A431 and KB cells in the log phase were trypsinized and seeded at a density of 7 × 103 cells per well into 96 well tissue culture plates. MTT assay, a colorimetric assay, based on the ability of viable cells to reduce a soluble yellow tetrazolium salt [3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT)] to purple formazan crystals, was used in the present study. Cells were incubated for 24 h in a humidified 5% CO2 incubator at 37 °C. After 24 h, the culture medium was replaced with media containing five different concentrations (0.01, 0.05, 0.1, 0.5 and 1 mM) of the nanoparticles (bare CaF2:Eu, Cit-CaF2:Eu and antibody conjugated Cit-CaF2:Eu). Cells with Triton X100 (1%) were used as the positive control, while cells maintained in fresh 10% FBS media served as the negative control, all in triplicate. After 24 h of incubation, the wells were washed using PBS and 10 μl of the MTT stock solution supplemented with 100 μl of the medium was added into each well. The cells were incubated further for 4 h, after which 110 μl of the solubilization buffer was added into each well and incubated for 1 h. Optical density of the solution was measured using a microplate reader (BioTek Power Wave XS, USA) at a wavelength of 570 nm and the percentage cell viability was assessed. Cell viability was calculated using the equation:where Intensitysample and Intensitycontrol represent the OD values of cells incubated the with sample and culture medium, respectively.
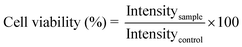
4.8 Cell morphology analysis
The effect of nanoparticle treatment on the cell morphology of normal and cancer cells (L929, NIH 3T3, A431 and KB) was visualized using an inverted microscope (Leica Microsystems, USA). L929, NIH 3T3, and KB cells seeded in MEM medium and A431 cells in DMEM medium were treated with the highest concentration of 1 mM for 24 h and morphological analysis was made by recording their digital live images microscopically.4.9 Targeted cell imaging using fluorescence microscopy
EGFR over-expressing, human epidermoid carcinoma (A431) and oral epithelial carcinoma (KB) cell lines were used to test the cancer specific targeting of antibody conjugated Cit-CaF2:Eu nanoparticles. The EGFR−ve normal mouse embryonic fibroblast NIH 3T3 cell line was kept as the control. The cultured cells were trypsinized and seeded at a density of 5000 cells onto acid etched coverslips placed in 24 well tissue culture plates. After 24 h, adherent cells were washed carefully with PBS and replaced with fresh media containing bare and antibody conjugated nanoparticles of varying concentrations (100 μM, 500 μM, 1 mM). Cells were incubated for two different time durations (2 and 4 h) at 37 °C and further washed twice with PBS, fixed with 2% paraformaldehyde, washed and mounted with DPX mounting medium. Imaging was carried out using an Olympus Fluorescent Microscope (Model BX51) equipped with a CCD camera (Model DP71), using oil immersion objectives. Fluorescence was detected using band pass excitation and emission filters (370–410 nm, >485 nm) and a 400 nm dichroic mirror.Acknowledgements
Authors gratefully acknowledge Department of Science & Technology, Government of India for financial assistance under the projects “Theragnostics, Regenerative Medicine and Stem Cell Research using Nanomaterials” and “PG Training Programs (M. Tech Nanomedical Science) SR/NM/PG-02/08. The authors also thank Mr Sajin P. Ravi for SEM analysis and Amrita Vishwa Vidyapeetham for providing all infrastructural facilities.References
- F. Wang, W. B. Tan, Y. Zhang, X. Fan and M. Wang, Nanotechnology, 2006, 17, R1–R13 CrossRef CAS.
- V. M. Peterson, C. M. Castro, H. Lee and R. Weissleder, ACS Nano, 2012, 6, 3506–3513 CrossRef CAS.
- A. P. Retnakumari, P. L. Hanumanthu, G. L. Malarvizhi, R. Prabhu, N. Sidharthan, M. V. Thampi, D. Menon, U. Mony, K. Menon, P. Keechilat, S. Nair and M. Koyakutty, Mol. Pharmaceutics, 2012 DOI:10.1021/mp300172e.
- X. He, K. Wang and Z. Cheng, Wiley Interdiscip. Rev.: Nanomed. Nanobiotechnol., 2010, 2, 349–366 CrossRef CAS.
- S. Santra and A. Malhotra, Wiley Interdiscip. Rev. Nanomed. Nanobiotechnol., 2011, 3, 501–510 CAS.
- P. Sharma, S. Brown, G. Walter, S. Santra and B. Moudgil, Adv. Colloid Interface Sci., 2006, 123–126(SPEC. ISS.), 471–485 CrossRef CAS.
- W. J. Parak, T. Pellegrino and C. Plank, Nanotechnology, 2005, 16, R9–R25 CrossRef CAS.
- D. Beer and J. Weber, Opt. Commun., 1972, 5, 307–309 CrossRef CAS.
- C. Kirchner, T. Liedl, S. Kudera, T. Pellegrino, A. M. Javier, H. E. Gaub, S. Stolzle, N. Fertig and W. J. Parak, Nano Lett., 2005, 5, 331–338 CrossRef CAS.
- J. Aswathy, S. Sajith, M. Koyakutty, S. Nair and D. Menon, Carbohydr. Polym., 2011, 85, 37–43 CrossRef.
- J. Aswathy, S. Jahnavi, R. Krishna, M. Koyakutty, S. Nair and D. Menon, J. Nanosci. Nanotechnol., 2011, 11, 7611–7620 CrossRef CAS.
- S. Setua, D. Menon, A. Asok, S. Nair and M. Koyakutty, Biomaterials, 2010, 31, 714–729 CrossRef CAS.
- A. Ashokan, D. Menon, S. Nair and M. Koyakutty, Biomaterials, 2010, 1–11 Search PubMed.
- A. Retnakumari, S. Setua, D. Menon, P. Ravindran, H. Muhammed, T. Pradeep, S. Nair and M. Koyakutty, Nanotechnology, 2010, 21, 055103 CrossRef (12pp).
- A. Retnakumari, J. Jayasimhan, P. Chandran, D. Menon, S. Nair, U. Mony and M. Koyakutty, Nanotechnology, 2011, 22, 285102 CrossRef (11pp).
- C. Bouzigues, T. Gacoin and A. Alexandrou, ACS Nano, 2011, 5, 8488–8505 CrossRef CAS.
- S. Cotton, Lanthanides and Actinides, Macmillan Education, Basingstoke, 1991 Search PubMed.
- C. Feldmann, T. Jüstel, C. R. Ronda and P. J. Schmidt, Adv. Funct. Mater., 2003, 13, 511–516 CrossRef CAS.
- Yu. E. Perlin and A. A. Kaminskii, Phys. Status Solidi B, 1985, 132, 11–40 CrossRef CAS.
- D. C. Tran, G. H. Sigel Jr. and B. Bendow, J. Lightwave Technol., 1984, 2, 566–586 CrossRef.
- F. Auzel, Mater. Sci. Forum, 1991, 67–68, 489–502 CrossRef CAS.
- C. A. Klein, J. Appl. Phys., 2006, 100, 083101 CrossRef (1–13).
- P. Maushake, Optik & Photonik, 2008, 2, 46–47 Search PubMed.
- F. Wang, X. Fan, D. Pi and M. Wang, Solid State Commun., 2005, 133, 775–779 CrossRef CAS.
- L. G. Jacobsohn, C. J. Kucera, T. L. James, K. B. Sprinkle, J. R. DiMaio, B. Kokuoz, B. Y. Kukouz, T. A. DeVol and J. Ballato, Materials, 2010, 3, 2053–2068 CrossRef CAS.
- J. Wang, W. Miao, Y. Li, H. Yao and Z. Li, Mater. Lett., 2009, 63, 1794–1796 CrossRef CAS.
- C. Feldmann, M. Roming and K. Trampert, Small, 2006, 2, 1248–1250 CrossRef CAS.
- S. Rungrodnimitchai, Jpn. J. Appl. Phys., 2011, 50, 01BJ17 CrossRef (1–4).
- C. Pandurangappa, B. N. Lakshminarasappa and B. M. Nagabhushana, J. Alloys Compd., 2010, 489, 592–595 CrossRef CAS.
- Z. Quan, D. Yang, P. Yang, X. Zhang, H. Lian, X. Liu and J. Lin, Inorg. Chem., 2008, 47, 9509–9517 CrossRef CAS.
- Y. Tian, J. Yu, X. Qi, X. Wu, R. Hua and S. Fan, J. Mater. Sci.: Mater. Electron., 2009, 20, 439–444 CrossRef CAS.
- L. Song, J. Gao and R. Song, J. Lumin., 2010, 130, 1179–1182 CrossRef CAS.
- M. Pedroni, F. Piccinelli, T. Passuello, M. Giarola, G. Mariotto, S. Polizzi, M. Bettinellia and A. Speghini, Nanoscale, 2011, 3, 1456–1460 RSC.
- Y.-Y. Hu, A. Rawal and K. Schmidt-Rohr, Proc. Natl. Acad. Sci. U. S. A., 2010, 107, 22425–22429 CrossRef CAS.
- B. Xie and G. H. Nancollas, Proc. Natl. Acad. Sci. U. S. A., 2010, 107, 22369–22370 CrossRef CAS.
- A. M. Cross, P. S. May, F. C. J. M. van Veggel and M. T. Berry, J. Phys. Chem. C, 2010, 114, 14740–14747 CAS.
- A. López-Macipe, J. Gómez-Morales and R. Rodríguez-Clemente, Adv. Mater., 1998, 10, 49–53 CrossRef.
- A. Wang, H. Yin, D. Liu, H. Wu, M. Ren, T. Jiang, X. Cheng and Y. Xu, Mater. Lett., 2007, 61, 2084–2088 CrossRef CAS.
- J. M. Delgado-López, M. Iafisco, I. Rodríguez, A. Tampieri, M. Prat and J. Gómez-Morales, Acta Biomater., 2012, 8, 3491–3499 CrossRef.
- Y. Wu, Y. Tseng and J. C. C. Chan, Cryst. Growth Des., 2010, 10, 4240–4242 CAS.
- Z. Amjad, Langmuir, 1993, 9, 597–400 CrossRef CAS.
- N. N. Dong, M. Pedroni, F. Piccinelli, G. Conti, A. Sbarbati, J. E. R. Hernández, L. M. Maestro, M. C. I. Cruz, F. S. Rodriguez, A. Juarranz, F. Chen, F. Vetrone, J. A. Capobianco, J. G. Sole, M. Bettinelli, D. Jaque and A. Speghini, ACS Nano, 2011, 5, 8665–8671 CrossRef CAS.
- G. T. Hermanson, Bioconjugate Techniques, Academic Press Inc., 2nd edn, 2008, ch. 3, 598–599 Search PubMed.
- M. Markovic, S. Takagi, L. C. Chow and S. Frukhtbeyn, J. Res. Natl. Inst. Stand. Technol., 2009, 114, 293–301 CrossRef CAS.
- A. S. Shcheulin, T. S. Semenova, L. F. Koryakina, M. A. Petrova, A. E. Angervaks and A. I. Ryskin, Opt. Spectrosc., 2011, 110, 617–623 CrossRef CAS.
- J. Turkevich, P. C. Stevenson and J. Hillier, Discuss. Faraday Soc., 1951, 11, 55–75 RSC.
- R. S. Herbst, Int. J. Radiat. Oncol., Biol., Phys., 2004, 59, S21–S26 CrossRef.
- D. S. Salomon, R. Brandt, F. Ciardiello and N. Normanno, Crit. Rev. Oncol. Hematol., 1995, 19, 183–232 CrossRef CAS.
- S. Hou, Y. Zou, X. Liu, X. Yu, B. Liu, X. Sun and Y. Xing, Cryst. Eng. Commun., 2011, 13, 835–840 RSC.
- T. Huang, P. D. Nallathamby and X. N. Xu, J. Am. Chem. Soc., 2008, 130, 17095–17105 CrossRef CAS Supporting information.
- M. Falk, J. Raman Spectrosc., 1990, 21, 563–567 CrossRef CAS.
- J. Bandekar, Vib. Spectrosc., 1993, 5, 143–173 CrossRef CAS.
- A. F. Kirby and F. S. Richardson, J. Phys. Chem., 1983, 87, 2544–2556 CrossRef CAS.
- X. Fan, M. Wang and G. Xiong, J. Mater. Sci. Lett., 1993, 12, 1552–1554 CrossRef CAS.
- P. P. Gawryszewska, M. Pietraszkiewicz, J. P. Riehl and J. Legendziewicz, J. Alloys Compd., 2000, 300–301, 283–288 CrossRef CAS.
- B. D. Gessner, M. Beller, J. P. Middaugh and G. M. Whitford, N. Engl. J. Med., 1994, 330, 95–99 CrossRef CAS.
- National Research Council, Fluoride in Drinking Water: A Scientific Review of EPA's Standards, National Academies Press, Washington, DC, 2006 Search PubMed.
- Scientific Opinion of the Panel on Food Additives and Nutrient Sources added to Food (F. Aguilar, U. R. Charrondiere, B. Dusemand, P. Galtier, J. Gilbert, D. M. Gott, S. Grilli, R. Guertler, G. E. N. Kass, J. Koenig, C. Lambré, J.-C. Larsen, J.-C. Leblanc, A. Mortensen, D. Parent-Massin, I. Pratt, I. Rietjens, I. Stankovic, P. Tobback, T. Verguieva and R. Woutersen), Calcium fluoride as a source of fluoride added for nutritional purposes to food supplements, EFSA J., 2008, 882, 1–15 Search PubMed.
- C. M. Stoscheck and G. Carpenter, J. Cell. Physiol., 1984, 120, 296–302 CrossRef CAS.
- H. Miyagi and I. N. Maruyama, Open Spectrosc. J., 2010, 4, 28–31 CrossRef CAS.
- M. C. Willingham, H. T. Haigler, D. J. P. Fitzgerald, M. G. Gallo, A. V. Rutherford and I. H. Pastan, Exp. Cell Res., 1983, 146, 163–175 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2013 |