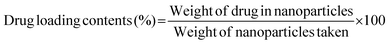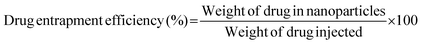DOI:
10.1039/C3BM00007A
(Paper)
Biomater. Sci., 2013,
1, 647-657
Facile preparation of multifunctional hollow silica nanoparticles and their cancer specific targeting effect
Received
6th January 2013
, Accepted 26th February 2013
First published on 22nd March 2013
Abstract
Efficient delivery of therapeutics to tumor cells is one of the key issues in cancer therapy. In the present work, we have established a facile and unique chemical strategy for fabrication of highly biocompatible and water-dispersible multifunctional hollow silica nanoparticles (HSNPs). These mesoporous silica nanoparticles, having ring-like morphology, were fabricated by the sol–gel method followed by selective etching of the inner inorganic core. Further, to accomplish cancer-specific targeting properties, folic acid was tethered to the surface of HSNPs through amide bond formation using the EDC/NHS coupling method. Thereafter, rhodamine B isothiocyanate (RITC) was conjugated to the HSNPs to endow the fluorescent property to the nanoparticles required for biological imaging applications. The successful formation of multifunctional HSNPs was confirmed by XRD, FTIR, zeta potential, TEM, FESEM, and BET surface area measurements. The average particle size of HSNPs was found to be 50 nm to 70 nm from TEM analysis, which is the desired size-range for drug-delivery vehicles. These HSNPs showed good mesoporous behavior and were found to be an excellent candidate for loading and releasing the anticancer drug doxorubicin (DOX). The bioactivity of the HSNPs was verified by biological assay including cell cytotoxicity by MTT assay, intracellular uptake by fluorescence microscopy, cell cycle analysis by fluorescence-activated cell sorting (FACS), and apoptosis study. Besides, the effect of salt concentration on the drug release performance was evaluated. An in vitro biological study revealed that these DOX-loaded folate-targeted HSNPs achieved excellent efficacy for simultaneously targeting and destroying cancer cells.
1. Introduction
Targeted drug delivery systems have recently gained significant attention in pharmaceuticals, owing to several advantages over the established forms of dosage, such as prolonged efficiency time and improved patient compliance.1 Currently, there has been an increasing trend towards designing new nanoparticles for targeted drug delivery systems due to their inherent fine size and biocompatibility.2 The suitability of these nanoparticles for drug delivery applications depends upon factors like stability, functionality, dispensability and nature of interface. In this context, porous silica nanoparticles with their large surface area and appropriate pore structures have been particularly interesting as a potential material for application in the fields of drug delivery,3 catalysis,4 gas adsorption,5 enzyme immobilization,6 and gene delivery.7 Silica based nanomaterials provide an excellent choice for delivery of therapeutics owing to their highly hydrophilic surface, inert properties, and biocompatibility.
Many techniques have been developed to date to synthesize silica based porous nanomaterials.8–11 Most of the synthetic approaches involve the use of surfactant molecules as soft templates which are later removed for the uniform generation of pores. Owing to the ease of surface modification of functionalized mesoporous silica materials, they have been extensively studied as hosts for various guest molecules. In recent years, development of hollow mesoporous silica spheres has gained more prominence than conventional mesoporous silica materials, such as MCM-41, SBA-15, and MCF. Mesoporous silica spheres with hollow cavities are preferred for their extraordinarily high drug and gene loading capacity, and sustained drug release kinetics.12 Such nanomaterials possess a number of beneficial properties like high surface area, low density, thermal and mechanical stability, unique optical, and chemical properties.13 So far, many research groups have reported several controlled drug delivery systems based on the mesoporous silica materials.14,15 Of late, Na and his coworkers reported mesoporous silica nanoparticles with very large pores of diameter 23 nm by employing trimethylbenzene as pore directing agent for an efficient delivery of siRNA.16 In addition, a controlled release and delivery system has been fabricated using mesoporous silica nanoparticles coupled with cadmium sulfide (CdS) nanocrystals as chemically removable caps to encapsulate the maximum amount of pharmaceutical drug molecules and neurotransmitters.17 Moreover, mesoporous silica nanoparticles with hollow cavities as controlled drug release systems have been explored by many groups.18–21 Mesoporous hollow silica materials with diverse morphology have been developed using different methods. In a recent report, Wang et al. have designed raspberry-like monodisperse magnetic and silica hollow hybrid nanospheres using polystyrene (PS) spheres as a template.22 Venkatathri et al. have obtained mesoporous hollow silica utilizing ammonia and organic amines e.g. n-propylamine, diethyl amine, triethylamine, and triethanolamine precursors as templates.23
Recently, our group has established the practical implementation of different functionalized magnetic nanoparticles in targeted drug delivery and MRI contrast agents for the treatment of tumour cells.24–27 Following the same line of thought, we have developed multifunctional designed hollow silica nanoparticles for targeted anticancer drug delivery in the present work. Hollow mesoporous silica nanoparticles (HSNPs) have been prepared through a facile one-pot sol–gel coating method with assistance of tetraethylorthosilicate as silane precursor and iron oxide as template. After that, the nanoparticles were conjugated to folic acid (FA) to achieve tumor-selective targeting since FA functions as an active recognition moiety for folate receptors which are highly expressed in several human tumors e.g. ovarian and breast cancers. Thereafter, the HSNPs were subjected to fluorescent tagging with RITC. Finally, the anticancer drug doxorubicin was encapsulated into the nanoparticles, and the effect of the drug loaded nanoparticles on the toxicity behavior of HeLa cells have been studied and were compared to the effect on normal cells.
2. Experimental section
2.1. Materials
Anhydrous ferric chloride (FeCl3), ferrous sulphate (FeSO4·7H2O), ammonia, concentrated HCl, dimethyl sulphoxide (DMSO), and ethanol were purchased from Merck, Germany. Tetraethylorthosilicate (TEOS), 3-aminopropyltriethoxysilane (APTES), folic acid (FA), N-hydroxysuccinimide (NHS), 1-[3-(dimethylamino)propyl]-3-ethylcarbodiimide hydrochloride (EDC) and doxorubicin hydrochloride (DOX), rhodamine B isothiocyanate (RITC), trinitrobenzene sulfonic acid (TNBS), 4′-6-diamidino-2-phenylindole (DAPI), propidium iodide (PI), RNase and 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) were purchased from Sigma-Aldrich Chemicals, USA. Fetal bovine serum and Minimum Essential Medium (MEM) were procured from Hyclone, USA and Himedia, India, respectively. All the chemicals were reagent grade and used without further purification. Water used throughout the experiment was ultrapure milliQ water.
2.2. Methods
2.2.1. Synthesis of iron oxide (Fe3O4) nanoparticles.
Superparamagnetic Fe3O4 nanoparticles were prepared by a chemical co-precipitation method according to our previously reported procedure.28 For the synthesis of Fe3O4 nanoparticles, 0.324 g FeCl3 (2 mmol) and 0.278 g FeSO4·7H2O (1 mmol) were taken in 40 ml milliQ water in a two necked round bottom flask equipped with a mechanical stirrer. The solution was vigorously stirred and heated to 70–80 °C under an argon atmosphere. Subsequently, 5 ml ammonia solution (25%) was added drop wise into the solution and stirring continued for another 30 min to allow the uniform formation of the nanoparticles. The solution was then cooled to RT and the resulting particles were subjected to magnetic decantation followed by repeated washing with milliQ water and finally dried in a vacuum oven at 70 °C.
2.2.2. Synthesis of hollow silica nanoparticles (HSNPs).
The HSNPs were prepared using iron oxide nanoparticles as template. For the synthesis, 50 mg of the iron oxide nanoparticles were well dispersed in a mixture of 50 ml ethanol, 10 ml milliQ water and 1 ml 28% ammonia solution. Then, 0.5 ml TEOS was added drop wise under vigorous stirring conditions to the above nanoparticle suspension. After continuous stirring for 12 h at RT, the silica coated magnetic nanoparticles were recovered and washed with ethanol. The silica coated material was then treated with 5 ml conc. hydrochloric acid and stirred for 6 h. After treatment with HCl, the colour of solution changed from brown to dark yellow due to the dissolution of the magnetic nanoparticles residing in the core. The resultant solution was precipitated with ethanol and centrifuged at 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 rpm. The precipitates were washed properly with water and recovered. The resultant HSNPs were finally dried in a vacuum oven at 100 °C.
000 rpm. The precipitates were washed properly with water and recovered. The resultant HSNPs were finally dried in a vacuum oven at 100 °C.
Further, the HSNPs were reacted with APTES in order to introduce amine groups on the surface. 100 mg of HSNPs were dispersed in 10 ml toluene, followed by addition of 1 ml APTES. The resulting mixture was allowed to reflux with vigorous stirring at 80 °C for 24 h. Thereafter, the reaction mixture was washed several times with toluene and ethanol followed by repeated centrifugation and finally dried in a vacuum oven.
2.2.3. Preparation of folic acid conjugated hollow silica nanoparticles (FA-HSNPs).
Folic acid (FA) was attached covalently to the aminated HSNPs (NH2-HSNPs) through amide bond formation between –NH2 groups of nanoparticles with carboxylic groups of FA. First, FA was dissolved in 1:1 volume ratio of deionised water and DMSO. To this, EDC and NHS were added to activate the carboxylic groups of FA and the pH of the solution was adjusted to 8.0 with 0.1 M NaOH. The FA activation reaction was carried under dark conditions at RT for 4 h. Afterwards, 20 mg (1 mg ml−1) of an aqueous dispersion of amine functionalized HSNPs was added to the activated FA solution followed by further stirring of the resultant solution at 25 °C for 12 h in the dark. The folate-modified nanoparticles were recovered through centrifugation, washed 4–5 times with DMSO and water, and finally dried under vacuum at RT for 24 h.
2.2.4. Quantification of amine density on the nanoparticle surface.
The amine density and the number of amine groups present on nanoparticle surface were determined by TNBS assay according to the procedure described by us previously.24 For amine quantification the nanoparticles were dispersed with 4% sodium bicarbonate solution (pH 8.5), and then 2 ml of it was treated with 2 ml TNBS solution (0.1% w/v). After 1 h incubation at 40 °C, the nanoparticles were isolated by centrifugation. The supernatant (1 ml) was treated with 0.1 ml aqueous solution of glycine (40 mmol ml−1) followed by incubation at 40 °C for 1 h to calculate the unreacted TNBS. After 1 h incubation the reaction was stopped by the addition of 1 ml of 2N HCl, and the sample was consequently hydrolyzed for 20 min at 40 °C. The absorbance of the resulting solution was measured at 410 nm using a Shimadzu UV-1700 spectrophotometer. The –NH2 group concentration was calculated by means of a graph derived by taking glycine (0–2 mmol) as standard (data not given).
2.2.5. Preparation of fluorescence labeled FA-HSNPs (RITC-FA-HSNPs).
For this study, RITC was conjugated onto the FA modified HSNPs through covalent linkage. 1 mg RITC was dissolved in 5 ml of H2O, pH adjusted to 8.0 with 0.1 M NaOH, and then an aqueous suspension of 10 mg of functionalized FA-HSNPs was added and stirred for 12 h in the dark. The unreacted RITC was removed through repeated centrifugation and washing. Finally, RITC labeled FA-HSNPs were recovered and suspended in PBS buffer for further use.
3. Characterization
The phase analysis of the as-prepared magnetite nanoparticles and HSNPs was performed using a diffractometer (XRD) with Ni-filtered Cu-Kα radiation (l = 1.54 Å). The surface chemistry of nanoparticles was determined from FTIR spectra. Samples for FTIR spectra were prepared in KBr and measured in the range 400–4000 cm−1. The size and morphology of the nanoparticles were observed by high-resolution transmission electron microscopy (HRTEM) (JEOL 3010, Japan) operated at 300 kV. The nanoparticles were thoroughly dispersed in water by ultra-sonication and a drop of the solution was placed on a carbon-coated copper grid and a Phillips CM 200 was used for performing the field emission scanning electron microscopy (FESEM). The average particle size from TEM micrographs was analyzed using image J software. The conjugation of folic acid on HSNPs was recorded by UV-Vis spectrophotometric analysis. The hydrodynamic size (HD) of the particle aggregates was measured by laser light scattering using a Brookhaven 90 Plus particle size analyzer. The surface charge of the nanoparticles was investigated through zeta potential measurements (Zetasizer 4, Malvern Instruments, UK). The surface area, pore volume and pore size distribution were determined by using a N2 adsorption–desorption instrument (Quantachrome Corporation; Quantachrome Autosorb Automated Gas Sorption System). Thermal analysis was done with a thermal analyzer (Pyris Diamond TG/DTA) with a heating rate 8 °C min−1 with in temperature range 50 °C to 1000 °C.
3.1. Cell lines and cytotoxicity study by MTT assay
For in vitro cytotoxicity experiments, human cervical adenocarcinoma (HeLa) and normal fibroblast (L929) cell lines were acquired from the National Centre for Cell Sciences (NCCS), Pune, India and cultured in Minimum Essential Medium (MEM) or Dulbecco's Modified Eagle's medium (DMEM) supplemented with 10% fetal bovine serum, penicillin (100 units per ml), streptomycin (100 mg per ml), and 4 mM L-glutamine at 37 °C in tissue culture flasks with 5% CO2 and 95% air humidified atmosphere. For experimental purposes, trypsinized cells were adjusted to a concentration of 1 × 105 cells per ml and plated in a 96 well flat-bottom culture plate (180 μl per well). Cells were then incubated with and without drug loaded FA-HSNPs at 37 °C in a humidified 5% CO2 incubator for 24 h. Similar studies were carried out with free DOX of different concentration for comparison. The percentage of cell viability was finally estimated by MTT assay.
3.2. Intracellular uptake studies
The internalization of nanoparticles into cells was observed by a fluorescence microscope imaging instrument. For intracellular uptake studies, 5 μg ml−1 of RITC-FA-HSNPs were incubated with cells for 30 minutes, 1 h, 2 h and 4 h respectively to determine the time dependent uptake of the nanoparticles in both HeLa and L929 cells. After incubation, cells were fixed with 4% paraformaldehyde for 15 min and stained with DAPI (1 mg ml−1) for 10 min at 37 °C. The cells were then washed with PBS buffer 4–5 times properly to remove the non-internalized nanoparticles in the medium in order to quench the fluorescence due to the non-internalized nanoparticles. Then cells were observed under Olympus IX 70 fluorescence microscopy.
3.3. DAPI staining for nuclear morphology study
To study the nuclear morphology of the HeLa cells, DAPI staining was performed following the reported procedure.29 HeLa cells were treated with PBS and 5, 10, and 25 μg ml−1 DOX-loaded FA-HSNPs for 24 h in vitro. The cells were fixed with 4% formaldehyde for 15 min, permeabilized with 0.1% Triton X-100 and stained with 1 mg ml−1 DAPI for 10 min. The cells were then rinsed with PBS and examined under fluorescence microscopy (Olympus IX 70).
3.4. Cell cycle analysis
Cell cycle analysis was performed in accordance with the method developed by Fried et al.30 For this, HeLa cells (1 × 105) were incubated with different concentrations of 5, 10, and 25 μg ml−1 DOX-FA-HSNPs for 24 h at 37 °C. The cells were harvested by trypsinisation, fixed with chilled 70% ethanol and stored at −20 °C. Cells were then washed with PBS (10 mM, pH 7.4) and incubated with 20 μl DNase free RNase (10 mg ml−1) and 20 μl of DNA intercalating dye PI (1 mg ml−1) for 1 h at 37 °C in the dark. Apoptotic cells were determined by their hypochromic sub-diploid staining profiles. The distribution of cells in the different cell-cycle phases was analyzed from the DNA histogram using a Becton–Dickinson FACS caliber flow cytometer and Cell Quest software.
3.5. Doxorubicin loading and release study
The present paper delineates the drug loading efficacy of HSNPs. We have chosen doxorubicin (DOX) as an anticancer drug to estimate the loading and release patterns of the nanoparticles. For drug loading studies, 10 mg FA-HSNPs were mixed with 10 ml (0.34 mg ml−1) DOX in milliQ water and stirred for 24 h under dark conditions. Thereafter, the DOX-loaded nanoparticles were recovered by centrifugation. The supernatant after loading was collected to determine the drug loading content and drug encapsulation efficiency (EE) by a spectroscopic method at 482 nm. The in vitro interaction of cells with drug loaded HSNPs were evaluated by different biological techniques has been described earlier.
The potential release of DOX from the DOX-FA-HSNPs was carried out using a dialysis technique. The DOX-loaded nanoparticles were taken in a dialysis membrane with a molecular weight cutoff of 12 kDa and dialyzed against 20 ml acetate buffer pH (5.6) on a shaker at 37 °C in the dark. The DOX release profile was calculated spectrophotometrically by measuring the absorbance of the released DOX solution at 482 nm. At certain time intervals, a 3 ml sample from solution was taken for UV-Vis measurement and returned to the same solution after measurement. The effect of NaCl solution on the drug release process was also studied. The drug loading content and entrapment efficiency were determined by the following formula:
4. Results and discussion
4.1. Synthesis and characterization of multifunctional hollow silica nanoparticles
Silica based nanomaterials with suitable functionalities have been continuously explored in order to develop a biocompatible platform for performing a range of biological applications. Also, it is known that DOX has a high affinity to strongly anchor on a silica surface through hydrogen bonding and electrostatic interaction.31 Hence, the efficacy of DOX-loaded hollow silica nanocarriers could be conveniently studied. Here, we have proposed a simple and cheap chemical method for the preparation of highly biocompatible and water-dispersible porous silica nanoparticles using magnetic iron-oxide as template. The detailed synthesis of multifunctional hollow silica nanocarrier is presented in Scheme 1. At first, the spherical magnetite nanoparticles were prepared by a chemical co-precipitation technique and characterized by XRD and TEM analysis.28 These nanoparticles were then coated with a layer of silica by hydrolysis and sol–gel polymerization of tetraethylorthosilicate (TEOS), followed by concentrated HCl treatment (36%) to generate the hollow cavity by dissolution of the internal magnetite core. It is noteworthy that the etching principally functions at the core of the particles rather than the exterior. Moreover, the etching does not affect the size of nanoparticles. In the traditional synthesis of mesoporous silica nanoparticles involving the use of surfactants, calcination which was necessary to remove excess surfactants, resulted in loss of water-dispersibility of the nanoparticles.32,33 Moreover, Cauda et al. have stated that during calcination, condensation of silanol groups (Si–OH) into silicone (Si–O–Si) linkages occurred leading to the fusion of mesoporous silica nanoparticles.33 To circumvent these issues, we have developed a simple chemical method to fabricate mesoporous HSNPs, that were further surface-modified by functionalization with amine groups, followed by FA and RITC to create RITC-FA-HSNPs endowed with targeting and fluorescent properties respectively.
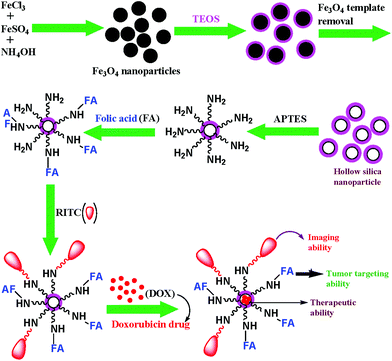 |
| | Scheme 1 Representative schematic illustration for the fabrication of multifunctional designed HSNPs. | |
4.1.1. X-ray diffraction studies.
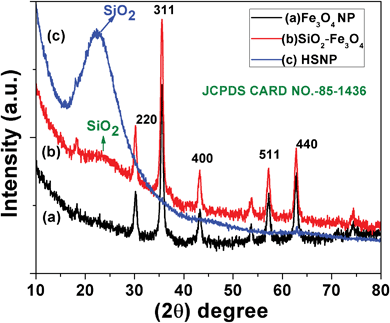
The crystal structure and the crystallite size of the as-prepared nanoparticles were estimated through powder X-ray diffractometry analysis. The XRD patterns of Fe3O4, SiO2–Fe3O4 and HSNPs are shown in Fig. 1. For free iron oxide nanoparticles the peaks are indexed at 18.30°, 30.20°, 35.55°, 43.26°, 57.21° and 62.87°, which correspond to reflection planes of (111), (220), (311), (400), (511) and (440) respectively. The diffraction peaks delineate face-centered lattice and cubic unit cells of nanoparticles. The magnetic nanoparticles are inverse spinel oxide and all peaks match with the usual XRD pattern in accordance with standard data (JCPDS 85-1436). As shown in Fig. 1b, the silica coated magnetic nanoparticles showed all the characteristic peaks of iron oxide nanoparticles. In addition, a broad peak was observed at an angle of 20°–30°, demonstrating the presence of an amorphous silica shell on the surface of the magnetite nanoparticles. The absence of any crystalline planes of iron oxide nanoparticles in Fig. 1c demonstrates that the Fe3O4 nanoparticles were completely removed in the acid etching process. In Fig. 1c for HSNPs, only a single broad peak was observed at 20–30° confirming the formation of amorphous silica material. Hence, the XRD results supported the fabrication of amorphous silica material from crystalline magnetic material in accordance with our proposed scheme which is outlined above (Scheme 1).
4.1.2. FTIR analysis.
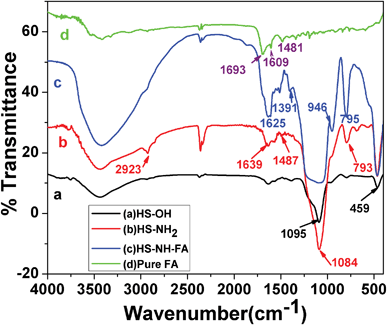
The fabrication of HSNPs using iron oxide nanoparticle as a template, as well as the surface modification of the nanoparticles, was analyzed by FTIR analysis. The FTIR spectra of HSNPs, APTES functionalized HSNPs, pure FA and FA-HSNPs are illustrated in Fig. 2. The absence of a characteristic peak for Fe–O bond vibration at 570–580 cm−1 in Fig. 2a clearly suggests the absence of Fe3O4 nanoparticles in HSNPs. For HSNPs, peaks centered at 1095 cm−1 and 793 cm−1 correspond to asymmetric and symmetric stretching vibrations of the Si–O–Si bond respectively. A band at 459 cm−1 corresponds to Si–O bond vibration. A broad prominent peak at 3400 cm−1 clearly revealed the presence of numerous surface hydroxyl groups on the surface of HSNPs. Further surface modification with APTES was indicated by peaks at 1639 cm−1 and 1487 cm−1, corresponding to N–H bending and C–N stretching vibration of aminated HSNPs. In addition to N–H bond vibration, asymmetric and symmetric C–H bond vibrations were observed at 2923 cm−1 and 2875 cm−1 respectively, indicating the presence of amino-silane moiety on HSNPs. Further, the bonding of FA to the amine moieties of the nanoparticles was evident from Fig. 2c, where vibrations peaks due to the amide bonds were seen to appear at 1625 cm−1 and 1556 cm−1. The peaks observed in the FTIR spectra for free FA molecules were also observed in the spectra for the FA-HSNPs, proving the presence of FA on the HSNPs.
4.1.3. UV-visible absorption study.
Apart from FTIR spectra, UV-Vis spectra were used to confirm the presence of FA conjugation on aminated HSNPs. The UV-Vis spectra for HSNPs, pure FA and FA-HSNP are shown in Fig. 3a. The spectrum for pure FA showed one broad absorption band around 280 nm and one broad shoulder around 361 nm which are ascribed to the π–π* and n–π* transitions in enone moiety of FA, respectively. For FA conjugated HSNPs, the same two peaks of FA are noticeably observed, thus suggesting the successful bonding of FA ligand to HSNPs. Wu et al. also observed two broad absorption peaks at 287 nm and 380 nm for FA conjugated rattle-type magnetic mesoporous silica nanospheres.34
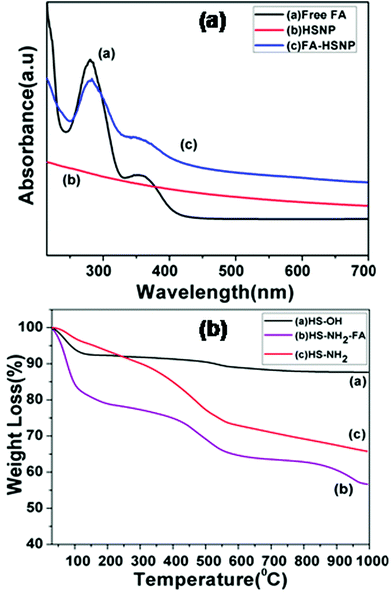 |
| | Fig. 3 (a) UV-Vis spectra of Free FA, HSNPs, FA conjugated HSNPs, (b) TGA curves of HS-OH, HS-NH2 and HS-NH2-FA. | |
4.1.4. TGA measurements.
Step by step surface modification is also evident from thermo-gravimetric analysis (TGA). TGA results of the HSNPs and their surface-functionalized counterparts are presented in Fig. 3b. It was observed that for HSNPs, the decomposition started ∼100–220 °C, possibly due to the evaporation of water and surface hydroxyl groups on HSNPs. The total weight-loss percentage is ∼7–8%. For aminated HSNPs, the initial weight-loss in the range 100–200 °C is contributed to by adsorbed moisture, whereas the major weight loss of ∼30% is accounted by the decomposition of densely aminated silane on HSNPs, thereby demonstrating the presence of amino-silane coupling agent on HSNPs. As for the FA-modified HSNP, the TGA curve showed an initial drastic weight loss of about 20% due to the loss of surface adsorbed water molecules, followed by a major weight loss of ∼40% which could be assigned to loss of surface functionalized APTES and FA molecules.
4.1.5. Nitrogen adsorption–desorption study.
The formation of a mesoporous hollow cavity was confirmed from BET measurements. The nitrogen adsorption–desorption isotherms and the corresponding pore size distribution curves for HSNPs are shown in Fig. 4.
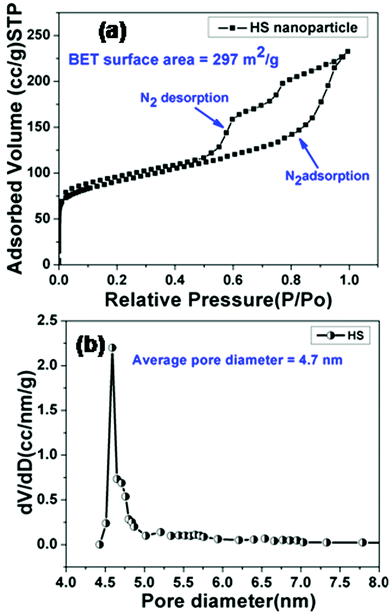 |
| | Fig. 4 (a) Nitrogen adsorption–desorption isotherm curve, (b) BJH pore size distributions of HSNPs. | |
The curve represents a type-IV isotherm pattern, signifying mesoporous behavior of hollow silica nanoparticles. The isotherm curve shows a large hysteresis loop and a leap starts at a relative pressure of 0.5, suggesting the formation of uniform mesopores. The BJH pore size distribution curve shows peaks centered around 4–5 nm and a sharp peak is observed at 4.7 nm. The Brunauer–Emmett–Teller (BET) surface area and Barrett–Joyner–Halanda (BJH) pore volume of the hollow silica nanocarrier are calculated to be 297 m2 g−1 and 0.36 cm3 g−1 respectively. The N2 adsorption–desorption study suggested that the pore diameter was appropriate for adsorption of drug molecules.
4.1.6. Size and surface morphology of nanoparticles.
Besides BET measurement, the formation of HSNPs was examined by HRTEM analysis. The morphology and size of the nanoparticles were obtained from FESEM and HRTEM analysis. TEM, FESEM and selected area diffraction pattern (SAED) of HSNPs are presented in Fig. 5. The HRTEM image of pristine magnetic nanoparticles and their SAED pattern is shown in Fig. 5a and 5b respectively. The free Fe3O4 nanoparticles are spherical and uniform in shape. The corresponding SAED pattern showed a number of well defined crystalline planes of Fe3O4 nanoparticles. The TEM image of silica coated Fe3O4 nanoparticles is shown in Fig. 5c. It was observed that magnetic nanoparticles were coated with a layer of silica. After acid etching, the hard Fe3O4 templates have been successively etched out producing an inner hollow cavity. The HRTEM image consists of dark outer rings and light inner circles with hollow cavity, confirming the formation of hollow silica nanoparticles (presented in Fig. 5d). The HRTEM image of HSNPs showed hollow silica nanoparticles with an average size of 50–70 nm with little agglomeration among particles. One HSNP is shown in the inset of Fig. 5d with higher magnification to demonstrate clearly the presence of an inner hollow core and outer silica shell. From the SAED pattern of HSNP (shown in Fig. 5e), the amorphous nature was clearly observed. The initial crystalline SAED pattern due to crystalline Fe3O4 was diminished, leading to the formation of amorphous HSNPs. The SAED clearly suggested that the acid etching completely removed the inner Fe3O4 magnetic core.
From the FESEM images, it was observed that the HSNPs possess a smooth surface with an average particle size of 70 nm. Similar types of FESEM images were observed for porous silica coated on magnetic nanoparticles as reported by Knezevic et al.35 It is a well known fact that larger sized mesoporous silica materials pose problems for their potential application in drug delivery. In contrast, we have synthesized HSNPs having sizes in the range of 50 nm to 70 nm as an appropriate host material for drug delivery. Both HRTEM and FESEM images clearly revealed that HSNPs are uniform spheres with smooth surfaces and sizes of nanoparticles are found in the range of 50–70 nm. The diameter of inner core cavity was ∼5–7 nm.
4.1.7. Dynamic light scattering and zeta potential.
The hydrodynamic diameter and size distribution of the porous silica nanoparticles and their functionalized counterparts were determined by DLS measurement. The average hydrodynamic diameters (HDs) of HSNPs and the functionalized HSNPs are presented in Table 1. The HD sizes of HSNPs, NH2-HSNPs and FA-HSNPs were observed in the range of 157 ± 15, 242 ± 20, 212 ± 30 nm respectively at physiological pH. From DLS studies, the size distributions of nanoparticles were found to be relatively narrower with polydispersity index (PDI) ∼0.2. Further, zeta potential values of the nanoparticles with different surface functionalization are given in Table 1. Higher zeta potential values for all the nanoparticle suspensions suggested the stable dispersion of the particles in aqueous medium. From Table 1, the zeta potential value for the HSNPs at pH 7.4 was found to be −61.0 mV, the negative zeta values arising mostly from the exposure of the hydroxyl groups on the silica surface. Further, surface functionalization by APTES led to a drop in the zeta potential value to −25.1 mV owing to the introduction of amine groups on surface of the nanoparticles. Again, the attachment of FA molecules to the amine-functionalized nanoparticles increased towards a more negative zeta potential value.
Table 1 Hydrodynamic (HD) size, polydispersity index (PDI) and zeta potential of HSNPs and successively-functionalized HSNPs at physiological pH
| Nanoparticles |
Hydrodynamic diameter (nm) |
PDI |
Zeta potential (mV) at pH 7.4 |
| HSNPs |
157 ± 15 |
0.216 |
−61.0 |
| HS-NH2 |
242 ± 20 |
0.274 |
−25.1 |
| HS-NH-FA |
212 ± 30 |
0.221 |
−50.9 |
4.2. Biological assay
4.2.1. MTT assay.
After proper surface engineering and characterization of the nanoparticles, a number of biological studies were performed. The in vitro cell viability of the FA-HSNPs and DOX-loaded FA-HSNPs was evaluated to determine the selective cancer therapy activity of the drug-loaded nanoparticles by MTT assay.
Fig. 6 represents the cell viability assay of FA-HSNPs and DOX loaded FA-HSNPs on normal cell lines and cancerous cell lines. A cell viability assay revealed that FA-HSNPs caused very low toxicity on both L929 and HeLa cells even at high concentrations (50 μg ml−1), and hence may be safe for in vitro and in vivo applications. In contrast, the DOX-loaded HSNPs exhibited significant cytotoxicity towards HeLa cells even at a lower concentration of 5 μg ml−1. The toxicity is attributed to the synergistic effect of DOX which inhibits the proliferation of HeLa cells. As reflected by the corresponding cell viability assay, about 65% death of HeLa cells was observed at 25 μg ml−1 and ∼78% cell death at 50 μg ml−1. DOX-loaded nanoparticles achieved a synergistic enhancement of apoptosis of HeLa cells. The half maximum inhibition concentrations (IC50) value for the DOX-loaded HSNPs was found to be 13 μg ml−1. In the absence of DOX, a minimum cell viability of 85% was observed on both the cell lines. Moreover, the DOX-loaded nanoparticles induced little toxicity on normal L929 cells which could be attributed to the lack of folate receptors on L929 cells.
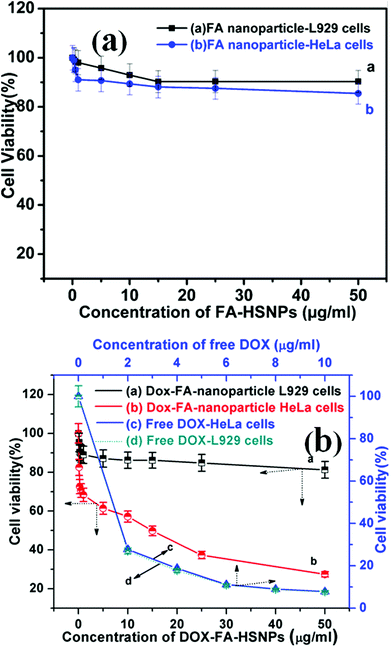 |
| | Fig. 6 (a) In vitro cytotoxicity of FA conjugated HSNPs against L929 cells and HeLa cells, (b) cell viability of DOX loaded FA conjugated HSNPs against L929 cells and HeLa cells, and cell viability of HeLa and L929 cells incubated with various concentration of free DOX. | |
A maximum of 20% cell death was observed for DOX-loaded nanoparticles when incubated with normal cells for 24 h. The difference in cell viability in Fig. 6b originates from the selective conjugation of FA and DOX with HeLa cells. The cytotoxicity of free doxorubicin as a control is presented in Fig. 6b. From the MTT assay, it was found that free DOX caused almost similar cytotoxicity to both L929 and HeLa cells. This also shows the benefit of using DOX-loaded HSNPs which caused low toxicity to the normal L929 cells in contrast to cytotoxic behavior of free DOX on L929 cells.
4.2.2. Intracellular uptake analysis.
Furthermore, to verify the cancer specificity of HSNPs, the cell uptake efficiency of RITC-labeled folate-targeted and non-targeted HSNPs were tested on FR (+) HeLa cells and FR (−) normal L929 cells. The uptake and intracellular distribution of nanoparticles was evaluated by fluorescence study. Both HeLa and L929 cells were incubated with 5 μg ml−1 of water-dispersed RITC-FA-HSNPs for 0 h, 30 min, 1 h, 2 h, and 4 h to check the time-dependent cellular internalization of the nanoparticles. Images were observed under a fluorescence microscope to determine the receptor-mediated endocytosis and cytoplasmic distribution of the nanoparticles. The fluorescence uptake images are presented in Fig. 7. The red fluorescence obtained from the RITC-HSNPs was prominently visualized in the cytoplasm of cells. As compared to the HeLa cells, the uptake efficiency was very much less on normal L929 cells due to lack of folate receptors on normal cells. The fluorescence studies clearly authenticated that the targeting specificity of the folate-conjugated hollow silica nanoparticles was enhanced on FR (+) HeLa cells. Moreover, intracellular uptake of both FA functionalized and non-functionalized RITC-HSNPs were cross-checked on HeLa cells and the images were shown in Fig. 8. As shown in the images, in absence of FA, the cellular internalization of nanoparticles on HeLa cells is lower as compared with FA-conjugated nanoparticles. To check the effect of free FA in the cellular uptake of RITC-FA-HSNPs on HeLa cells, a fluorescence study was performed and the results are shown in Fig. 9. In the absence of free FA in the medium, RITC-FA-HSNPs easily penetrated into HeLa cells and produced bright red fluorescence in the nuclei of HeLa cells (Fig. 9a). Although the presence of excess free FA in the cellular medium decreased the intracellular uptake of nanoparticles inside HeLa cells (Fig. 9b), some extent of intracellular uptake was still observed.
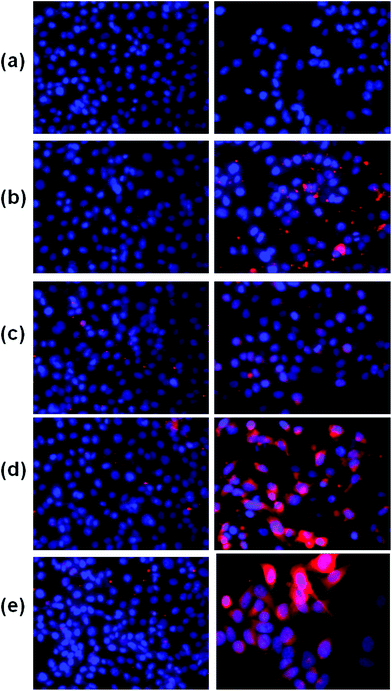 |
| | Fig. 7 Fluorescence microscopy images of L929 cells (left side) and HeLa cells (right side) in the figure. Images acquired after incubating both the cells with 5 μg ml−1 of RITC-FA-HSNPs for (a) 0 min, (b) 30 min, (c) 1 h, (d) 2 h, (e) 4 h incubation. | |
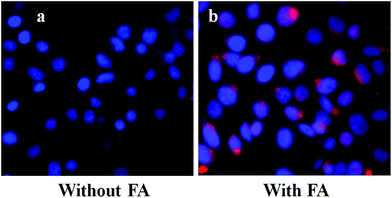 |
| | Fig. 8 Intracellular uptake of HeLa cells incubated with 5 μg ml−1 of (a) RITC-HSNPs and (b) RITC-FA-HSNPs. | |
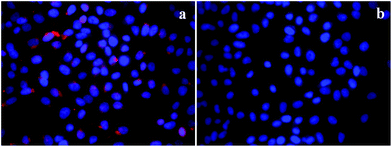 |
| | Fig. 9 Intracellular uptake of HeLa cells incubated with 5 μg ml−1 of RITC-FA-HSNPs (a) in the absence of free FA in the medium and (b) in the presence of excess free FA in the medium. | |
4.2.3. Apoptosis study.
To demonstrate the utility of DOX-loaded HSNPs in targeted cancer therapeutics by inducing cell death and necrosis of cell nuclear morphology, apoptotic studies were performed. Cell nuclei were stained with 4,6-diamidino-2-phenylindole (DAPI), a nuclear staining dye to visualize the morphology of the cell nuclei after treatment with DOX-loaded nanoparticles. On staining with DAPI for 24 h, a blue colour was imparted to the cell nuclei. As evident from the apoptosis results in Fig. 10, there was no visible necrosis of cell nuclei on control treatment. After treatment with DOX-HSNPs, the cells shrank and the morphology of the cell nuclei changed. Apoptotic morphology with condensed and fragmented nuclei was observed and the nuclei fragmentation was gradually enhanced in a dose-dependent manner. It is a well known fact that DOX binds to DNA by intercalation and stimulates a series of biochemical events inducing apoptosis in tumor cells.36 From our apoptotic studies, it was confirmed that DOX-loaded nanoparticles inhibited cell-growth and induced apoptosis. Huge numbers of apoptotic nuclei were noticed when HeLa cells were treated with 25 μg ml−1 of DOX-loaded nanoparticles. In this regard, it can be stated that the DOX-loaded HSNPs could efficiently deliver the drug to the tumor cells, thus preventing the proliferation of HeLa cells effectively.
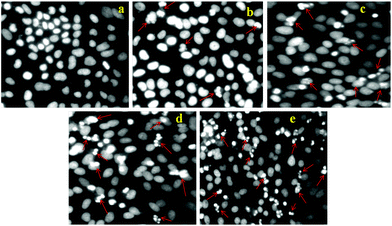 |
| | Fig. 10 Apoptosis study of HeLa cells treated with (a) control (PBS), (b) 5 μg ml−1, (c) 10 μg ml−1, (d) 15 μg ml−1, and (e) 25 μg ml−1 DOX loaded FA-HSNPs. Arrows in the figures indicate apoptotic nuclei. | |
4.2.4. Cell cycle analysis.
Further, to evaluate the cytotoxic efficacy of DOX-HSNPs, apoptosis and cell cycle analysis was carried out using standard propidium iodide (PI) staining followed by a flow cytometry assay. From cell cycle analysis, early apoptosis and the phases of cell death could be investigated. For this purpose, HeLa cells were incubated with different concentration of DOX-loaded nanoparticles. The distribution of HeLa cells in different phases are presented in Fig. 11. From the cell cycle study, it is observed that the maximum distribution of cells occurred in the G0/G1 phase (apoptotic phase) in the control sample, which shifts in the cell cycle phases on treatment with the DOX-loaded nanoparticles. Increasing concentration of DOX-loaded HSNPs showed maximum cell intensification at the G2/M phase, suggesting the cell cycle arrest in the same phase. In the literature, it has already been reported that DOX prevents cancer cell growth by inducing DNA breakage as well as by inducing cell cycle arrest at the G2/M phase.36
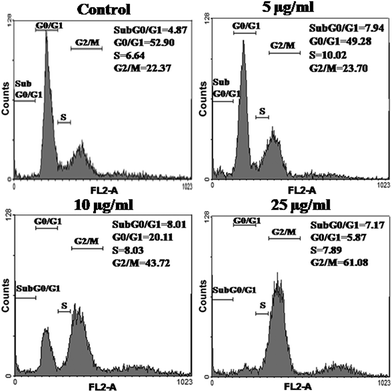 |
| | Fig. 11 Flow-cytometric analysis of cell cycle phase distributions of HeLa cells treated with (a) control (PBS), (b) 5 μg ml−1, (c) 10 μg ml−1 and (d) 25 μg ml−1 of DOX-loaded FA-HSNPs for 24 h. The analysis was performed using the propidium iodide (PI) flow cytometric assay with the aid of FACS Diva software. | |
4.2.5. Drug loading and release behavior.
The loading content and entrapment efficiency of DOX on HSNPs were calculated to be 31.1% and 91.4% respectively. The amount of drug loaded on HSNPs was as high as 31.1%, which is much higher than that given in the literature for the mesoporous silica nanoparticles.37,38 Moreover, the high drug-loading capacity could possibly be due to the high surface-to-volume ratio and the electrostatic interactions between the positively charged DOX molecules and the negatively charged silica nanoparticles.39 The doxorubicin release from the nanoparticles is shown in Fig. 12. The drug release pattern showed an initial fast release rate of the loaded drug followed by cumulative release with time.
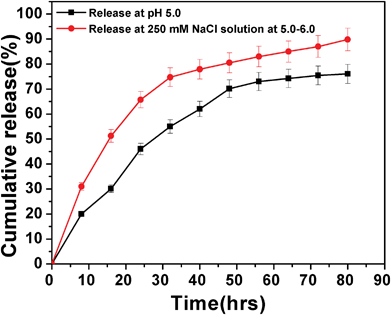 |
| | Fig. 12 DOX release pattern of drug-loaded FA-HSNPs in PBS buffer pH 5.0 and in the presence of 250 mM NaCl in PBS buffer at pH = 5.0–6.0. | |
Nearly 70% of the adsorbed doxorubicin was released from the HSNPs within 60 h at pH 5.0, while the release rate increased in the presence of 250 mM NaCl, where 70% of DOX was released within just 30 h. Since DOX binds with HSNPs through ionic interactions the presence of NaCl weakens the ionic interactions between them facilitating higher amount of DOX release in presence of the salt. From drug release pattern, we observed that the drug was released significantly at endosomal pH (pH = 5.0–6.0) and the drug -loaded HSNPs exhibited elevated cytotoxicity to cancer cells which was evident from the apoptosis and cell cycle results.
5. Summary and conclusion
Multifunctional hollow silica nanoparticles (HSNPs) with cancer-specific targeting properties and imaging capabilities have been prepared using a versatile, inexpensive chemical route. It was shown that HSNPs were taken up specifically by HeLa cancer cells in comparison to normal cells. A drug loading study indicated that these hollow silica nanoparticles encapsulated large amount of the anticancer drug DOX. The loading content and entrapment efficiency reached as high as 31.1% and 91.4% respectively. In vitro biological studies revealed that the DOX-loaded folate-targeted nanoparticles achieved excellent efficacy for simultaneously targeting and destroying cancer cells. They specifically accumulate and release the payloads on HeLa cells through receptor mediated endocytosis, with the drug release profile showing the release of 75% of the loaded drug within 80 h at pH ∼5.0. From all the biological studies, it is envisioned that the multifunctional HSNPs synthesized by us are an excellent candidate that may serve as a vehicle in cancer-specific targeting, imaging and therapeutic application in a single entity.
Acknowledgements
Authors would like to acknowledge DBT, India for providing financial support for this work. Banalata Sahoo thanks the Council of Scientific and Industrial Research (CSIR), New Delhi, for providing a Senior Research Fellowship.
References
- L. Pan, Q. He, J. Liu, Y. Chen, M. Ma, L. Zhang and J. Shi, J. Am. Chem. Soc., 2012, 134, 5722–5725 CrossRef CAS.
- Y. Cheng, J. D. Meyers, A.-M. Broome, M. E. Kenney, J. P. Basilion and C. Burda, J. Am. Chem. Soc., 2011, 133, 2583–2591 CrossRef CAS.
- L. Tang, T. M. Fan, L. B. Borst and J. Cheng, ACS Nano, 2012, 6, 3954–3966 CrossRef CAS.
- C.-H. Tsai, H.-T. Chen, S. M. Althaus, K. Mao, T. Kobayashi, M. Pruski and V. S.-Y. Lin, ACS Catal., 2011, 1, 729–732 CrossRef CAS.
- M. R. Mello, D. Phanon, G. Q. Silveira, P. L. Llewellyn and C. M. Ronconi, Microporous Mesoporous Mater., 2011, 143, 174–179 CrossRef CAS.
- Y. Wang and F. Caruso, Chem. Mater., 2005, 17, 953–961 CrossRef CAS.
- F. Torney, B. G. Trewyn, V. S.-Y. Lin and K. Wang, Nat. Nanotechnol., 2007, 2, 295–300 CrossRef CAS.
- S. B. Hartono, W. Gu, F. Kleitz, J. Liu, L. He, A. P. J. Middelberg, C. Yu, G. Q. M. Lu and A. S. Z. Qiao, ACS Nano, 2012, 6, 2104–2117 CrossRef CAS.
- L. F. Su, L. Miao, S. Tanemura and G. Xu, Sci. Technol. Adv. Mater., 2012, 13, 035003 CrossRef (035006pp).
- W. Zhai, C. He, L. Wu, Y. Zhou, H. Chen, J. Chang and H. Zhang, J. Biomed. Mater. Res., Part B, 2012, 100, 1397–1403 CrossRef.
- T. Shiomi, T. Tsunoda, A. Kawai, F. Mizukamib and K. Sakaguchi, Chem. Commun., 2007, 4404–4406 RSC.
- X. Kang, Z. Cheng, D. Yang, P. Ma, M. Shang, C. Peng, Y. Dai and J. Lin, Adv. Funct. Mater., 2012, 22, 1470–1481 CrossRef CAS.
- P. A. Williamson, P. J. Blowera and M. A. Green, Chem. Commun., 2011, 47, 1568–1570 RSC.
- Z. Luo, K. Cai, Y. Hu, L. Zhao, P. Liu, L. Duan and W. Yang, Angew. Chem., Int. Ed., 2011, 50, 640–643 CrossRef CAS.
- T. Y. Ohulchanskyy, I. Roy, L. N. Goswami, Y. Chen, E. J. Bergey, R. K. Pandey, A. R. Oseroff and P. N. Prasad, Nano Lett., 2007, 7, 2835–2842 CrossRef CAS.
- H.-K. Na, M.-H. Kim, K. Park, S.-R. Ryoo, K. E. Lee, H. Jeon, R. Ryoo, C. Hyeon and D.-H. Min, Small, 2012, 8, 1752–1761 CrossRef CAS.
- C.-Y. Lai, B. G. Trewyn, D. M. Jeftinija, K. Jeftinija, S. Xu, S. Jeftinija and V. S.-Y. Lin, J. Am. Chem. Soc., 2003, 125, 4451–4459 CrossRef CAS.
- C. Kim, S. Kim, W.-K. Oh, M. Choi and J. Jang, Chem.–Eur. J., 2012, 18, 4902–4908 CrossRef CAS.
- Y. Jiao, J. Guo, S. Shen, B. Chang, Y. Zhang, X. Jiang and W. Yang, J. Mater. Chem., 2012, 22, 17636–17643 RSC.
- X. Mei, S. Yang, D. Chen, N. Li, H. Li, Q. Xu, J. Ge and J. Lu, Chem. Commun., 2012, 48, 10010–10012 RSC.
- X. Mei, D. Chen, N. Li, Q. Xu, J. Ge, H. L. B. Yang, Y. Xu and J. Lu, Soft Matter, 2012, 8, 5309–5316 RSC.
- C. Wang, J. Yan, X. Cui and H. Wang, J. Colloid Interface Sci., 2011, 354, 94–99 CrossRef CAS.
- N. Venkatathri, R. Srivastava, D. S. Yun and J. W. Yoo, Microporous Mesoporous Mater., 2008, 112, 147–152 CrossRef CAS.
- S. K. Sahu, S. Maiti, A. Pramanik, S. K. Ghosh and P. Pramanik, Carbohydr. Polym., 2012, 87, 2593–2604 CrossRef CAS.
- M. Das, D. Bandyopadhyay, D. Mishra, S. Datir, P. Dhak, S. Jain, T. K. Maiti, A. Basak and P. Pramanik, Bioconjugate Chem., 2011, 22, 1181–1193 CrossRef CAS.
- M. Das, D. Mishra, P. Dhak, S. Gupta, T. K. Maiti, A. Basak and P. Pramanik, Small, 2009, 5, 2883–2893 CrossRef CAS.
- D. Bhattacharya, M. Das, D. Mishra, I. Banerjee, S. K. Sahu, T. K. Maiti and P. Pramanik, Nanoscale, 2011, 3, 1653–1662 RSC.
- B. Sahoo, S. K. Sahu, D. Bhattacharya, D. Dhara and P. Pramanik, Colloids Surf., B, 2013, 101, 280–289 CrossRef CAS.
- H. N. Li, F. F. Nie, W. Liu, Q. S. Dai, N. Lu, Q. Qi, Z. Y. Li, Q. D. You and Q. L. Guo, Toxicology, 2009, 257, 80–85 CrossRef CAS.
- J. Fried, A. G. Perez and B. D. Clarkson, J. Cell Biol., 1976, 71, 172–181 CrossRef CAS.
- Y. Chen, H. Chen, D. Zeng, Y. Tian, F. Chen, J. Feng and J. Shi, ACS Nano, 2010, 4, 6001–6013 CrossRef CAS.
- J. M. Kim, S. Jun and R. Ryoo, J. Phys. Chem. B, 1999, 103, 6200–6205 CrossRef CAS.
- V. Cauda, C. Argyo, D. G. Piercey and T. Bein, J. Am. Chem. Soc., 2011, 133, 6484–6486 CrossRef CAS.
- H. Wu, G. Liu, S. Zhang, J. Shi, L. Zhang, Y. Chen, F. Chena and H. Chena, J. Mater. Chem., 2011, 21, 3037–3045 RSC.
- N. Z. Knezevic, I. I. Slowing and V. S.-Y. Lin, ChemPlusChem, 2012, 77, 48–55 CrossRef CAS.
- H. Wang, Y. Zhao, Y.-l. H. YanWua, K. Nan, G. Nie and H. Chen, Biomaterials, 2011, 32, 8281–8290 CrossRef CAS.
- C. Li, Z. Hou, Y. Dai, D. Yang, Z. Cheng, P. Ma and J. Lin, Biomater. Sci., 2013, 1, 213–223 RSC.
- Y. Zhu, Y. Fang and S. Kaskel, J. Phys. Chem. C, 2010, 114, 16382–16388 CAS.
- Y. Gao, Y. Chen, X. Ji, X. He, Q. Yin, Z. Zhang, J. Shi and Y. Li, ACS Nano, 2011, 5, 9788–9798 CrossRef CAS.
Footnote |
| † Present Address: Department of Applied Chemistry, Indian School of Mines, Dhanbad 826004, India. |
|
| This journal is © The Royal Society of Chemistry 2013 |
Click here to see how this site uses Cookies. View our privacy policy here.