A pH-sensitive nano drug delivery system of doxorubicin-conjugated amphiphilic polyrotaxane-based block copolymers†
Lan Jiang,
Ze-ming Gao,
Lin Ye,
Ai-ying Zhang and
Zeng-guo Feng*
School of Materials Science and Engineering, Beijing Institute of Technology, Beijing 100081, China. E-mail: sainfeng@bit.edu.cn; Fax: +86-01-68912650; Tel: +86-01-68912650
First published on 19th August 2013
Abstract
A pH-sensitive nano antitumor drug delivery system was prepared by conjugating doxorubicin (DOX) to amphiphilic polyrotaxane (PR)-based block copolymers through a pH-sensitive cis-aconityl moiety. The resulting polymer-drug conjugates were able to self-assemble into polymeric micelles in an aqueous solution with diameters varying from 297 nm to 178 nm after the conjugation as evidenced by DLS measurements. The pH-sensitive cis-aconityl linkage provided a controlled and sustained release of DOX over a period of more than 5 days in an acidic environment mimicking the tumor microenvironment, and a negligible amount of release in an environment with physiological pH. The nanoparticles had lower cytotoxicity than the free drug and can efficiently transfer and release the drug into HeLa cells. With these promising properties, the PR-based block copolymers have the potential to be carriers for the controlled release of antitumor drugs.
Introduction
Polymeric nanoparticles, self-assembled from amphiphilic block copolymers in aqueous solution, are promising carriers or vehicles for safe systemic delivery of antitumor drugs into cancer cells due to their distinct advantages including the improvement of drug solubility, reduced systemic toxicity, and prolonged circulatory time.1–4 These nanoparticles can easily escape from the blood vessel and accumulate in tumor tissue, a phenomenon known as “enhanced permeability and retention (EPR) effect”.5 Based on this EPR effect, they can be passively targeted to tumor sites with modified pharmacokinetics in the body and cellular levels.The development of pH-sensitive polymer-based drug delivery systems for cancer chemotherapy has attracted tremendous interest due to the fact that the microenvironment of tumor tissue is mildly acidic compared to that of healthy tissue.6,7 The pH value of blood is approximately 7.4, while the pH in tumor tissue is often 0.5–1.0 units lower.8 This pH difference could contribute to the extracellular release of antitumor drugs bound to polymers through acid-labile linkers, especially when the polymer conjugates tend to be trapped in the tumor due to the EPR effect. In addition, the pH value in the endocytic compartments of the tumor cells generally ranges from 4.5 to 6.5,9 which could also trigger the release of drug bound to polymers via acid-labile linkage once the drug-polymer conjugates are internalized by the cancer cells. Consequently, an ideal drug-loaded nanoparticle should be stable in the blood and healthy tissue at pH 7.4, but release the free drug specifically in tumor tissues upon being internalized by the cancer cells. To this end, acid-labile moieties such as cis-aconitic acid and hydrazone are widely used in small antitumor drug bioconjugate chemistry.10
Cyclodextrins (CDs) constitute a series of cyclic oligosaccharides composed of 6, 7 and 8 D-glucose units linked by α-1,4 bonds and are named α-, β- and γ-CDs, respectively. The geometry of CDs resembles a hollow truncated cone providing a hydrophobic cavity, and as a result, they can include linear polymers to give rise to novel polypseudorotaxanes (PPRs) and polyrotaxanes (PRs) after being end-capped with bulky stoppers or oligomers. Since CDs are nontoxic and biodegradable, CD-based PRs have been broadly explored not only for the purpose of fundamental supramolecular polymer research,11–13 but also for their use as polymer therapeutics, especially as carriers for controlled drug release due to their unique structural characteristics.14,15 On one hand, there exists a large number of hydroxyl groups located on the CD rings that could be chemically modified, allowing to conjugate bioactive agents to them.16 On the other hand, as the threaded CDs can freely slide and rotate along the included polymer chain, this conformational flexibility is desirable for enhancing the multivalent ligand–receptor interactions and is promising for applications in targeting and drug-mediated drug delivery systems.17,18 Several studies on drug-loaded nanocarriers created from CD-based PRs have been reported in the literature.7,8,19–22 However, most of those releasing strategies are focused on the dissociation of the supramolecular structure via hydrolysis of the terminal bulky groups, which would lead to the drugs being released along with the CD slipping off the polymer main chain. Evidently, the toxicity of the decomposition products (e.g., free CDs) is difficult to avoid, and the systematic investigation of the pH-triggered, targeted release of antitumor drugs from CD-based supramolecular polymers is still limited.
In a previous report, we synthesized certain types of amphiphilic PR-based block copolymers via the in situ atom transfer radical polymerization (ATRP) of poly(ethylene glycol) methyl ether methacrylate (PEGMA) initiated by PPRs formed from a distal 2-bromopropionyl end-capping Pluronic F127 with varying amounts of β-CDs.23 Owing to the hydrophilic nature of the attached PEGMA oligomers, as well as the adaptive flexibility of those entrapped β-CDs along the Pluronic F127 main chain, the copolymers were found to self-assemble into unique core–shell nanoparticles in an aqueous solution, exhibiting their potential as carriers for the controlled release of amphotericin B (AmB).18 Encouraged by this result, a pH-triggered target delivery system of the antitumor agent doxorubicin (DOX) was constructed from one of those amphiphilic PR-based block copolymers in this study. As shown in Scheme 1, DOX was chemically bound to the entrapped β-CDs on the Pluronic F127 chain via an acid-labile cis-aconityl moiety to give the conjugates (PR–DOXs). As expected, these conjugates self-assembled into micelles in aqueous solution and accumulate at the tumor site according to the EPR-mediated passive targeting concept. Once internalized by the tumor cells, where a mildly acidic environment is present, the bound DOX molecules can be released from the conjugates through hydrolysis, leading to a sustained level of DOX within the tumor cells to exert cytotoxic effects.
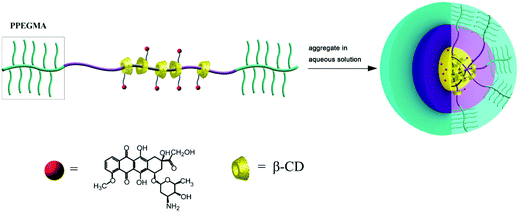 | ||
| Scheme 1 Schematic diagrams of the self-aggregation of the PR-DOX nanoparticles in an aqueous medium. | ||
Experimental section
Materials
N,N′-Carbonyldiimidazole (CDI) and cis-aconitic anhydride were purchased from Alfa Aesar, USA. Doxorubicin·HCl (DOX·HCl) was obtained from HuaFeng United Technology Co., Ltd, Beijing, China. 1-Ethyl-3-(3-dimethylaminopropyl) carbodiimide hydrochloride (EDC·HCl) and N-hydroxysuccinimide (NHS) were available from J&K Scientific Ltd, China. All other solvents and reagents were of analytical grade.Synthesis of PR-based block copolymers
As previously reported,18,23 a distal 2-bromopropionyl terminated Pluronic F127 was prepared by the end-capping reaction of a four-fold molar excess of 2-bromopropionyl bromide to Pluronic F127 in CH2Cl2. The amphiphilic PR-based block copolymers were synthesized via the in situ ATRP of PEGMA initiated by PPRs self-assembled from the resulting distal 2-bromopropionyl end-capping Pluronic F127 and various amounts of β-CDs. For the convenience of expression, the obtained PR-based block copolymers were designated as F-n-m, where F means Pluronic F127, which was used as the polymer main chain, n stands for the feed molar ratio of β-CD to Pluronic F127 and m represents the feed molar ratio of PEGMA to Pluronic F127, respectively.Synthesis of polyrotaxanes having amino pendant groups (PR-EDA)
As a typical example, F-30-20 (200 mg) with a found molar ratio of Pluronic F127 to β-CD to PEGMA equal to 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :21.4:46.5 was dissolved in 4 mL DMF and mixed with CDI (74 mg, 0.462 mmol) dissolved in 2 mL DMF. After stirring for 5 h at 40 °C, ethylene diamine (276 mg, 4.62 mmol) was added dropwise to the mixture, which was continuously stirred at room temperature for 3 h. Finally, the mixture was dialyzed using a dialysis bag (MWCO 3500) for 24 h with the water being changed every 8 hours, and then freeze-dried to obtain the product in 82.5% yield.
:21.4:46.5 was dissolved in 4 mL DMF and mixed with CDI (74 mg, 0.462 mmol) dissolved in 2 mL DMF. After stirring for 5 h at 40 °C, ethylene diamine (276 mg, 4.62 mmol) was added dropwise to the mixture, which was continuously stirred at room temperature for 3 h. Finally, the mixture was dialyzed using a dialysis bag (MWCO 3500) for 24 h with the water being changed every 8 hours, and then freeze-dried to obtain the product in 82.5% yield.
Synthesis of doxorubicin-cis-aconitate (DOX-CA)
The synthesis of doxorubicin-cis-aconitate (DOX-CA) was carried out according to the literature with some modifications.24,25 DOX·HCl (50 mg, 0.086 mmol) was dissolved in 15 mL ice-cold carbonate buffer (0.1 M, pH = 9). cis-aconitic anhydride (50 mg, 0.32 mmol) was added slowly at 0 °C while maintaining a pH of 8.5 by addition of ice-cold NaOH solution (0.5 M). The reaction mixture was stirred at 0 °C for 30 min, and at room temperature for another 30 min. The reaction was cooled on ice and acidified with ice-cold HCl solution (1 M) until precipitation occurred. The precipitates were isolated by centrifugation (5 min at 4000 rpm), and freeze-dried to obtain the product in 38.5% yield.Synthesis of PR-DOX
Doxorubicin-cis-aconitate (28 mg, 0.038 mmol) was dissolved in 12 mL PBS (0.1 M, pH = 7.4), to which EDC·HCl (40 mg, 0.2 mmol) was added. The solution was stirred for 2 min at room temperature, and then NHS (25 mg, 0.12 mmol) was added. The solution was stirred at room temperature for 30 min, after which PR-EDA (120 mg) was added. The resulting mixture was finally stirred for 20 h in the dark. The crude product was dialyzed using a dialysis bag (MWCO 3500) for 24 h with the water being changed every 8 hours, followed by gel purification by column chromatography on Sephadex LH-20 with DMF as the elution solvent to completely remove any unbound DOX. For the convenience of expression, the polymeric conjugates were assigned as F-n-m-DOXp, where p represents the feed molar ratio of DOX to β-CD entrapped on the PR-based triblock copolymer.Preparation of the PR-DOX micelles
The PR-DOX micelles were prepared by directly dissolving PR-DOX in water and allowing it to stand overnight. The solution was then diluted to the desired concentration with the appropriate amount of distilled water.In vitro release of DOX from polymeric micelles
A release study was carried out at 37 °C in a Guo Hua THZ82 incubator shaker (Jiangsu, China). A dialysis bag (MWCO 3500) containing 3 mL PR-DOX micelle solution was placed into a flask containing 60 mL 0.1 M citrate/phosphate buffers at pH = 5 and 7.4, respectively. The flask was shaken at 100 rpm at 37 ± 0.5 °C. At selected time intervals, a 3 mL sample was removed from the dialysis bag for UV-vis analysis and subsequently replaced with the same amount of fresh buffered solution. The DOX concentration was determined based on the absorbance intensity at 480 nm according to the standard line obtained from a series of solutions with different DOX concentrations (Fig. S3 and S4†). The release experiments were conducted in triplicate and the arithmetic means were taken as the results.DOX uptake
The cellular uptake and intracellular release behavior of PR-DOX micelles against HeLa cells were monitored by fluorescence microscopy and confocal laser scanning microscopy (CLSM). HeLa cells were seeded on plates and incubated for 24 h. The media were replaced with fresh medium containing free DOX and PR-DOX micelles with certain DOX concentrations, and incubated in a humidified atmosphere containing 5% CO2 at 37 °C for 1, 2, 8, 24 and 48 h, respectively. After the defined time intervals, the cells were washed 3 times with normal saline solution and immediately observed under green light from the filter block of a fluorescence microscope (Leica DM IL LED, N2.1, Germany) for uptake assessment. The images were acquired at 40× optical zoom. For CLSM (Zeiss LSM 710, Germany) observations, the cells were fixed with 4% paraformaldehyde for 10 min, the cell nuclei were stained with Hoechst 33342, and Dabco was added to prevent the quenching of fluorescence. Finally, the cells were observed by CLSM. Hoechst 33342 and DOX were excited at 405 and 559 nm with emission at 422 and 619 nm, respectively.Evaluation of the in vitro cytotoxicity of PR-DOX
The cytotoxicity of the PR–DOX nanoparticles against HeLa cells (ATCC, USA) was evaluated by using the 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazoliumbromide (MTT) assay. HeLa cells (8 × 104) were seeded in 96-well plates and incubated in DMEM containing 10% FBS in a humidified atmosphere containing 5% CO2 at 37 °C for 24 h. The media were replaced with fresh media containing free DOX and PR-DOX micelles with different DOX concentrations, and incubated for 2, 8, 12, 24, and 72 h, respectively. Thereafter, MTT solution was added. The plates were incubated at 37 °C for another 4 h. The intracellularly metabolized product formazan of MTT was retrieved by addition of DMSO. The plates were read at 550 nm (Biotek synergy2, USA), and the cell viability was calculated.Characterization of the drug-conjugated polyrotaxanes
1H NMR (400 MHz) spectra were recorded on a Bruker ARX-400 spectrometer at room temperature using DMSO-d6 as the solvent and tetramethylsilane (TMS) as the internal standard.UV-vis spectra were recorded on a Hitachi U-2800 spectrophotometer.
Samples for transmission electron microscopy (TEM, Tecnai G2 20 S-TWIN, FEI) were prepared by dropping a few microliters of the aqueous solution (0.5 mg mL−1) of the sample on a copper grid coated with amorphous carbon.
The size and size distribution of the micelles were determined by dynamic light scattering (DLS) (Zetasizer Nano ZS90, England) at 20 °C with angle detection at 90°.
Gel permeation chromatographic (GPC) measurements were carried out at 50 °C on a Waters 2410 instrument using DMF and LiBr (0.05 M) as the eluent mix at a flow rate of 1.0 mL min−1. All the GPC data were calibrated by using polystyrene (PS) standards.
Fourier transform infrared spectroscopy (FTIR) spectra were measured using a Shimadzu IR Prestige-21 FTIR spectrometer at room temperature in the range between 4000 and 500 cm−1, with a resolution of 2 cm−1 and 20 scans. Samples were prepared by dropping their corresponding chloroform solutions on KBr plates and drying them at a high temperature.
Results and discussion
Synthesis and characterization of PR-DOX
In our previous report,23 amphiphilic PR-based block copolymers were synthesized via the in situ ATRP of PEGMA initiated with PPRs made from a distal 2-bromopropionyl end-capped Pluronic F127 (BrP-F127-PBr) (Fig. S1†) with varying amounts of β-CDs at 25 °C. The 2D ROESY NMR and DSC analyses provided solid evidence for the self-assembly of β-CDs with Pluronic F127 producing inclusion complexes (ICs) or PPRs. In this study, the feed molar ratio of BrP-F127-PBr to Cu(I) to PMDETA was kept at 1:2:2.4, and that of PEGMA to BrP-F127-PBr was varied from 10 to 30, but the ratio of the β-CDs was kept at 30. The theoretical/found compositions and yields after thorough purification are summarized in Table 1. About two thirds of the β-CDs were found to be still entrapped on the Pluronic F127 main chain. When the feed molar ratio of PEGMA to BrP-F127-PBr was increased, the degree of polymerization (DP) of PEGMA oligomers attached to two ends of the PPRs was nearly twice the feed one. The results suggest that the in situ ATRP of PEGMA was controllable to some extent in this study.
| Name | Molar composition (F127:β-CD:PEGMA) |
Molecular weight and polydispersity index | Yield (%) | |||
|---|---|---|---|---|---|---|
| Feed ratio | Found ratioa | Mna × 10−3 | Mnb × 10−3 | Mw/Mnb | ||
| a Determined by 1H NMR analysis in DMSO-d6.b Determined by GPC with DMF and LiBr as the eluent mix at 1.0 mL min−1 using PS standards. | ||||||
| F-30-30 | 1:30:30 |
1:23.3:63 |
178 | 447 | 1.07 | 35.6 |
| F-30-20 | 1:30:20 |
1:21.4:46.5 |
139 | 355 | 1.13 | 24.5 |
| F-30-10 | 1:30:10 |
1:15.3:26 |
87.3 | 314 | 1.16 | 17.4 |
| F-30-20–DOX | — | — | 156 | 363 | 1.13 | 73.3 |
Pluronic F127-β-CD PPRs have low water solubility due to hydrogen bonding between the threaded CD's hydroxyl groups, whereas the water solubility of the resulting F-n-m PRs is increased with the DP of the PEGMA oligomers attached to the two ends of the PPRs. However, the water solubility of F-30-10 is still low for the self-assembly into micelles in aqueous solution. Although F-30-30 possesses the highest DP of the PEGMA oligomers, it would be difficult to chemically modify due to the steric hindrance of the brush-like end-capping blocks. As a compromise, F-30-20 was selected as a typical sample to be conjugated with varying amounts of DOX here. The preparation pathway of the PR-DOX conjugates is illustrated in Scheme 2. As each β-CD ring contains 7 glucose units, a large number of hydroxyl groups in the as-prepared PR-based block copolymer are available to be activated and conjugated with DOX. At first, the hydroxyl groups of the β-CDs entrapped on the Pluronic F127 main chain were activated by CDI, followed by reacting them with ethylene diamine to transfer them into ethylene diamine urethane. As compared with the 1H NMR spectrum of the intact F-30-20 (Fig. 1(A)), the signals of both the β-CD and ethylene diamine moieties are visible in the spectrum of PR-EDA (Fig. 1(B)), even though the proton resonance peaks of β-CD are heavily broadened. Based on the integral area ratio of the characteristic peaks at 4.8 ppm (β-CD) to 3.0 ppm (ethylene diamine), about 2.8 molecules of ethylene diamine were grafted onto each β-CD.
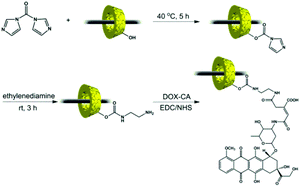 | ||
| Scheme 2 Preparation pathway of the PR-DOX conjugates. | ||
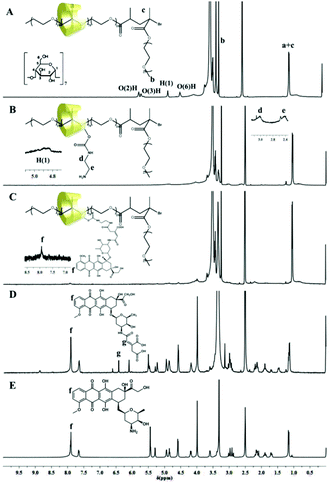 | ||
| Fig. 1 1H NMR spectra of (A) unmodified (intact) F-30-20, (B) F-30-20-EDA, (C) F-30-20-DOX7, (D) DOX-CA and (E) DOX. | ||
DOX-CA was prepared by the reaction of DOX with cis-aconitic anhydride according to the literature.10 Before the reaction, the peaks at about 7.9 ppm in the 1H NMR spectrum of DOX (Fig. 1(E)) are attributed to the protons of the anthracene moiety of DOX. After the reaction, additional peaks at 6.12 and 6.43 ppm appeared, which can be ascribed to the protons of (![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CH–COO–) of the cis- and trans-aconitic anhydride linkages in the DOX-CA (Fig. 1(D)). The TOF-MS spectrum of DOX-CA displays the expected pseudomolecular ions at m/z 700 (M + H), 722 (M + Na) and 738 (M + K) (Fig. S3†), respectively.
CH–COO–) of the cis- and trans-aconitic anhydride linkages in the DOX-CA (Fig. 1(D)). The TOF-MS spectrum of DOX-CA displays the expected pseudomolecular ions at m/z 700 (M + H), 722 (M + Na) and 738 (M + K) (Fig. S3†), respectively.
PR-DOX was prepared by the reaction of the carboxyl groups of DOX-CA with the primary amino groups of ethylene diamine urethane grafted onto the β-CDs in the presence of EDC and NHS in PBS. Fig. 1(C) shows the 1H NMR spectrum of the conjugate F-30-20-DOX7. The peaks of the protons of the anthracene moiety of DOX conjugated to β-CD are clearly visible. As the characteristic peaks of DOX in the 1H NMR are weak due to the high molecular weight of the PR-based block copolymer as shown in Table 1, the drug loading content was alternatively determined by using UV-vis spectroscopy. The results are summarized in Table 2. As can be seen, the drug loading content increased with the feed molar ratio of DOX to β-CD. Meanwhile, more DOX was grafted onto β-CD with decreasing DP of the PEGMA oligomers, likely due to the steric hindrance of the brush-shaped PEGMA oligomers as bulky end-capping stoppers of the PRs. The sample F-30-20-DOX7 with a found molar β-CD to DOX ratio of 1:1.6 was used in the following experiments. The resulting F-30-20, F-30-20-EDA and F-30-20-DOX7 were also characterized by FTIR (Fig. S2†). Compared with the IR spectrum of F-30-20, sharper vibration absorption peaks for amino groups associated with EDA appear at 3430 cm−1 in the IR spectra of F-30-20-EDA and F-30-20-DOX7, clearly indicating that the threaded β-CDs were chemically modified with EDA. The characteristic out-of-plane vibration peaks of the aromatic ring of DOX at 564 and 787 cm−1 were also found in the spectrum of F-30-20-DOX7, which provides direct evidence of DOX being successfully conjugated to the polyrotaxanes.
| Conjugate no. | F-n-m-DOXp | Molar composition (β-CD:DOX) |
DOX (wt%) | |
|---|---|---|---|---|
| Feed ratio | Found ratioa | |||
| a Determined by UV-vis spectroscopy. | ||||
| 1 | F-30-20–DOX1 | 1:1 |
1:0.6 |
3.5 |
| 2 | F-30-20–DOX2 | 1:2 |
1:0.9 |
4.6 |
| 3 | F-30-20–DOX7 | 1:7 |
1:1.6 |
9.3 |
| 4 | F-30-10–DOX7 | 1:7 |
1:2.2 |
23.3 |
GPC measurements of the PR-based block copolymers as well as the resulting PR-DOX conjugates were carried out by using DMF and LiBr (0.05 M) as the eluent mix. LiBr was added to suppress the association of polyrotaxanes and PR-DOX. As shown in Fig. 2(A), the molecular weight of the PR samples was increased with the feed molar ratio of PEGMA to the macroinitiator, and the polydispersity index was in a low range of 1.07–1.16. After the conjugation, the elution time of the PR-DOX sample became shorter and the polydispersity index remained very low (Fig. 2(B)). All the GPC traces exhibited a nearly symmetrical and unimodal peak. This further evidence confirms the preparation of PR-based block copolymers and PR-DOX conjugates in this study. However, the molecular weight determined by GPC was generally not coherent with that identified by 1H NMR analysis. This discrepancy is most likely due to the PS calibration standards used, and DMF is only a marginal solvent for PS.
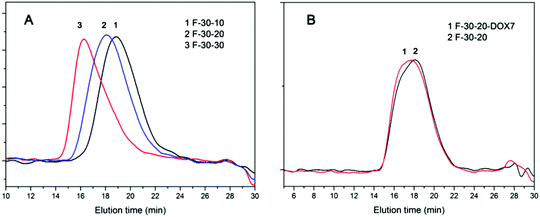 | ||
| Fig. 2 GPC traces of (A) F-30-10, F-30-20 and F-30-30 and (B) F-30-20 and F-30-20-DOX7 with DMF + 0.05 M LiBr as the eluent mix. | ||
Micelle properties of PR-DOX in aqueous solution
As the threaded β-CDs incline to self-aggregate along the included Pluronic F127 chain through neighboring hydrogen bonds, the polyrotaxanes are always characterized by a typical channel-type crystal structure and tend to precipitate from water. However, the PR-based block copolymers in this study appear not to form the channel-type crystal structure because the PPEGMA brushes sterically hinder the β-CDs entrapped on different polymer chains from self-aggregating evenly to a certain extent. Consequently, due to the high flexibility of β-CDs to slide and rotate along the polymer chain, the attachment of PPEGMA endows both the PR-based block copolymers and PR-DOX conjugates with an amphiphilic character leading to their self-assembly into nano-sized micelles in an aqueous solution. Here, the water-soluble PPEGMA chains serve as a hydrophilic shell stabilizing the nanoparticle, and the Pluronic F127 and β-CD moieties constitute the hydrophobic core. Fig. 3(A) and (C) illustrate the morphologies of the micelles formed from the samples F-30-20 and F-30-20-DOX7 in aqueous solutions as evidenced by TEM.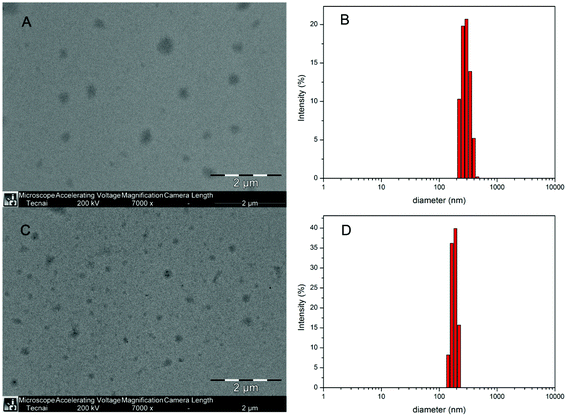 | ||
| Fig. 3 TEM micrographs of the F-30-20 (A) and F-30-20-DOX7 (C) nanoparticles; size and size distribution of F-30-20 (B) and F-30-20-DOX7 (D) determined by DLS. | ||
It can be seen from the TEM images that F-30-20-DOX7 is able to self-assemble into well-defined spherical micelles with regard to the micelles of its precursor F-30-20. The diameter of the observed spheres was about 200 nm for F-30-20 and around 100 nm for F-30-20-DOX7. For comparison, the size and size distribution of the micelles of both the PR-based block copolymer and the PR-DOX conjugate were also determined by DLS measurements, and the data are displayed in Fig. 3(B) and (D). The size of the micelles is about 297 nm for F-30-20 and 178 nm for F-30-20-DOX7, and both of them are larger than those observed by TEM. This may be caused by the shrinkage of the particles on the grids during the drying process prior to TEM observation. In addition, another reason is that the PEO segments could not be observed by TEM without staining, and the particle observed by TEM was only the hydrophobic part of the micelle. After conjugation with DOX, the average diameter of the hydrophobic part of the PR-DOX micelles was significantly decreased, most likely due to the strong π–π stacking between the grafted DOX molecules and the free sliding and rotation of the β-CDs along the Pluronic F127 main chain. The smaller diameter could make it easier for nanoparticles to evade scavenging by the mononuclear phagocyte system (MPS) and attain an appreciable EPR effect at the site of a solid tumor.
Effect of pH on the in vitro DOX release rate
As the pH sensitivity is crucial for a delivery system to release drugs at the target tumor cells (acidic environment), we designed and prepared this pH-responsive PR-based drug delivery system. However, before gel purification was carried out, a large amount of DOX was found to be physically encapsulated into micelles, presumably through π–π stacking and hydrophobic interactions. The drug was quickly released in a neutral medium under these conditions. The physically encapsulate content of DOX was substantially decreased after gel separation was conducted by column chromatography. As a consequence, the release behavior of DOX was evaluated based on the change of absorbance intensity at 480 nm by using UV-vis spectroscopy in two buffered solutions at pH 5.0 and 7.4, respectively, and the results are shown in Fig. 4.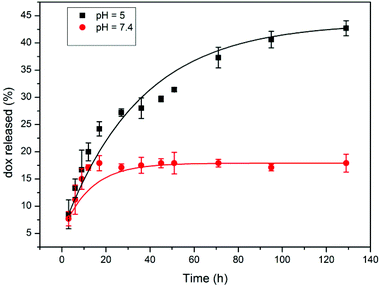 | ||
| Fig. 4 Release profiles of DOX from the F-30-20-DOX7 nanoparticles at different pH values. | ||
In PBS solution at pH 7.4, a typical two-phase release profile was observed, which exhibited an initial burst but a suspended release manner afterward. The amount of released DOX from the F-30-20-DOX7 micelles incubated at pH 7.4 was about 17% of the total drug loaded in the micelles in the first 15 h, and this value was maintained for up to 128 h. However, the micelles incubated at pH 5.0 exhibited a different release behavior with a basically sustained release manner throughout the whole process, and the release of DOX reached 43% of total drug loading after 128 h. This result clearly indicates that the hydrolytically cleavable cis-aconityl linkage connected to DOX and β-CD possesses the desired pH sensitivity, and the conjugated DOX molecules hardly deviate from the delivery system in a normal bloodstream environment (neutral environment). It should be noted that there is a DOX burst release of around 13% at both pHs within the first 5 h, most likely due to some unconjugated DOX still being physically entrapped inside the particles. This means that the gel purification process needs to be further modified for the complete removal of those ungrafted DOX molecules, and on the other hand, the purified PR-DOX conjugates could be used to load another antitumor drug, for example paclitaxel, through physical encapsulation to develop a dual drug loaded system.
Cellular uptake and cytotoxicity
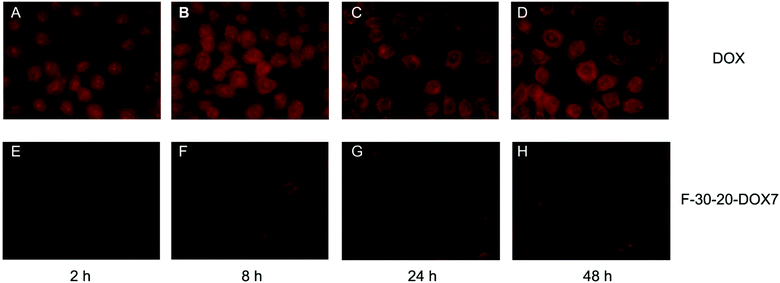 | ||
| Fig. 5 Fluorescence microscopy images of DOX and F-30-20-DOX7 uptake in HeLa cells after 2, 8, 24 and 48 h of incubation (magnification 40×). | ||
To get further insight into the distribution of the released drug within the cells, CLSM was used to examine the internalization processes of free DOX and F-30-20-DOX7 micelles. Cell nuclei were stained in order to determine whether DOX enters the nuclei and to compare the internalization difference between free DOX and PR-DOX conjugate. For most particulate carriers, it is generally assumed that the triggering mechanism of releasing the drugs in the cytoplasm occurs in the endosome.21,28 In contrast, free DOX could quickly diffuse into the cells through the cell membrane and is transported into the nuclei, where it interrupts the DNA replication process as previously reported.29 Thus, although the drug begun to accumulate inside the cells for both free DOX and PR-DOX nanoparticles after 1 h of incubation, the intracellular drug distribution for the PR-DOX conjugate was quite different from that of the free DOX solution (Fig. 6). After 1 h of incubation with the free DOX solution, the fluorescence of DOX in HeLa cells was clearly observed in the cell nuclei, and no obvious fluorescence was detected in the cytoplasm over 24 h (Fig. 6(D–F)), suggesting that DOX indeed diffuses into the cytoplasm through the cell membrane and is transported into the nuclei, where it interrupts the DNA replication process as previously reported.29 However, after 1 h of incubation, there was almost no fluorescence of DOX from PR-DOX in the cell nuclei, and only weak fluorescence of DOX from PR-DOX throughout the cytoplasm (Fig. 6(A)). The results suggest that the micelles are taken up from the extracellular fluid into the cells by endocytosis, and DOX begins to be released from the micelles due to the lower pH value of the endosomal compartments in this period of time. However, after 24 h, more fluorescence was observed in the cytoplasm, and weak fluorescence was also seen in the nuclei. It is clearly shown that internalized PR-DOX was delivered to lysosomes where the slightly acidic conditions permitted more DOX to be released, and a small amount of DOX entered the nuclei (Fig. 6(C)) within 24 h. Therefore, the PR-DOX conjugate is an efficient vehicle to transport DOX into the cytoplasm, and the internalization mechanism of PR-DOX is different from that of free DOX.
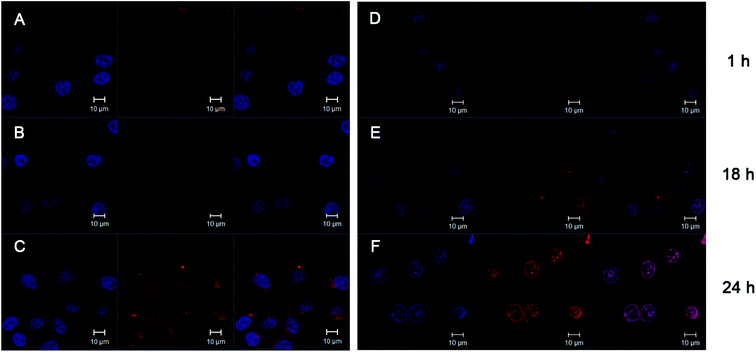 | ||
| Fig. 6 CLSM images of HeLa cells incubated with F-30-20-DOX7 micelles for 1 h (A), 18 h (B) and 24 h (C), and with free DOX for 1 h (D), 18 h (E) and 24 h (F). For each panel, images from left to right show the cells with nuclear staining by Hoechst 33258, with DOX fluorescence and overlays of both images. | ||
Although the results of DOX uptake confirm that the PR-DOX conjugate can transport DOX into cells efficiently, only small amounts of DOX could enter the nuclei as observed by CLSM after 24 h. This is most likely due to the fact that the release had not been completed in one day. As DOX is released from nanoparticles intracellularly and diffuses into the nuclei just as free DOX, it is important to make sure that the micelles disintegrate controllably to allow for more drug to be released from the micelles intracellularly. Therefore, taking advantage of the biodegradable hydrophilic shell and using a more sensitive pH-responsive linker is the subject of our ongoing study.
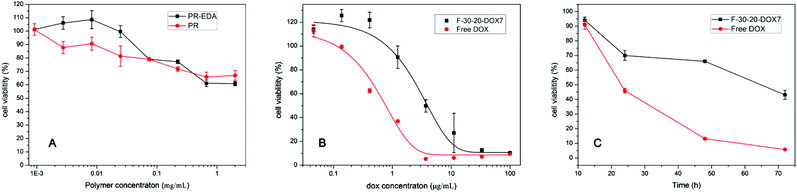 | ||
| Fig. 7 The cytotoxicity of (A) intact F-30-20 and PR-EDA with different polymer concentration, (B) free DOX and PR-DOX7 nanoparticles with different DOX concentrations against HeLa cells (incubation time 72 h), and (C) free DOX and F-30-20-DOX7 nanoparticles with a fixed DOX concentration against HeLa cells at different incubation time. | ||
The cytotoxic effects of free DOX and PR-DOX micelles with different DOX concentrations incubated with HeLa cells for 72 h are depicted in Fig. 7(B). The concentration of free DOX required to inhibit the cell viability by 50% (IC50) is 0.87 μg mL−1. The IC50 of F-30-20-DOX7 is 3.7 μg mL−1, which is four times more than that of free DOX. The relatively low cytotoxicity of PR-DOX correlates well with its drug delivery process, as it is common that the in vitro toxicity of polymer-anticancer drug conjugates is reduced as compared to the free drug due to the long-term drug release process from the nanoparticles.30,31
To further evaluate the in vitro cytotoxicity of the antitumor drug DOX and PR-DOX nanoparticles, the drug concentration was fixed at 10 μg mL−1 to obtain cell viabilities for different incubation times. As shown in Fig. 7(C), free DOX exhibits an instant cytotoxicity as the cell viability drops to 45.9% within the first 24 h and to 13.2% within 48 h, and then decreases very slowly. In contrast, due to the time-consuming drug release from the PR-DOX nanoparticles, the cell viability of F-30-20-DOX7 shows a moderate decrease. After 72 h, it killed 60% of the cells, while free DOX killed the same amount of cells within 24 h, clearly indicating that the PR-DOX indeed exhibits slow release of the drug and low cytotoxicity to HeLa cells, avoiding instant cytotoxicity. Again, the results imply that the PR-DOX nanoparticles are able to perform long-term drug delivery, which could reduce the toxicity to healthy cells in the blood circulation system, and release most of the drug after the nanoparticles accumulate in the tumor tissues as a benefit from the EPR effect.
Conclusion
In this study we prepared particular types of PR-based block copolymers composed of PPR as a central block made from ethylene diamine grafted β-CDs being threaded onto the Pluronic F127 main chain and oligomers formed from the in situ ATRP of PEGMA as outer blocks. The antitumor drug DOX was conjugated to ethylene diamine via acid-sensitive cis-aconityl linkage to give rise to the PR-DOX conjugates. The DOX loading content in the PR-DOX conjugates is in the range of 3.5 to 23.3 wt%. The selected conjugate sample is able to self-assemble into unique spherical nanoparticles in an aqueous solution. There is only a small amount of DOX release at pH = 7.4 in the initial period, indicating that the PR-DOX conjugates would be stable during blood circulation. On the other hand, DOX is released sustainably from the conjugates over a period of more than 5 days at pH = 5.0, demonstrating that DOX could be released in the acidic endocytic compartments once internalized by the tumor cells. The assay for the determination of anticancer activity in vitro revealed that PR-DOX is effective for inhibiting the growth of HeLa cells, while the intact PR-based block copolymers exhibit no significant toxicity. Cell uptake studies indicated that the PR-DOX nanoparticles are internalized by HeLa cells and the grafted DOX is released in the cytoplasm and then enters the cell nuclei, which is different from the localization of free DOX. Overall, the PR-DOX conjugates have a great potential for intracellular drug delivery into tumor cells and achieving highly effective and safe drug therapy.Acknowledgements
The authors acknowledge the support from the Natural Science Foundation of China (no. 20974015), the Doctoral Program Foundation of the Ministry of Education of China (no. 20091101110029) and the NSRIT project (no. 111000737).Notes and references
- I. Brigger, C. Dubernet and P. Couvreur, Adv. Drug Delivery Rev., 2002, 54, 631 CrossRef CAS.
- Y.-C. Wang, Y. Li, T.-M. Sun, M.-H. Xiong, J. Wu, Y.-Y. Yang and J. Wang, Macromol. Rapid Commun., 2010, 31, 1201 CrossRef CAS.
- D. Sutton, N. Nasongkla, E. Blanco and J. M. Gao, Pharm. Res., 2007, 24, 1029 CrossRef CAS.
- Y. Bae, S. Fukushima, A. Harada and K. Kataoka, Angew. Chem., Int. Ed., 2003, 42, 4640 CrossRef CAS.
- M. E. Davis, Z. Chen and D. M. Shin, Nat. Rev. Drug Discovery, 2008, 7, 771 CrossRef CAS.
- G. Pasut and F. M. Veronese, Prog. Polym. Sci., 2007, 32, 933 CrossRef CAS.
- I. F. Tannock and D. Rotin, Cancer Res., 1989, 49, 4373 CAS.
- F. Kratz, U. Beyer and M. T. Schutte, Crit. Rev. Ther. Drug Carrier Syst., 1999, 16, 245 CrossRef CAS.
- X. Q. Yang, J. J. Grailer, S. Pilla, D. A. Steeber and S. Q. Gong, Bioconjugate Chem., 2010, 21, 496 CrossRef CAS.
- J. H. Park, S. Kwon, M. Lee, H. Chung, J. H. Kim, Y. S. Kim, R. W. Park, I. S. Kim, S. B. Seo, I. C. Kwon and S. Y. Jeong, Biomaterials, 2006, 27, 119 CrossRef CAS.
- R. Yang, Y. C. Han, G. Q. Li, D. H. Jiang and H. C. Huang, Biol. Control, 2008, 44, 116 CrossRef.
- A. Harada, J. Li and M. Kamachi, Nature, 1992, 356, 325 CrossRef CAS.
- J. Wang, P. J. Wang, L. Ye, A. Y. Zhang and Z. G. Feng, Polymer, 2011, 52, 5362 CrossRef CAS.
- E. Bilkova, M. Sedlak, A. Imramovsky, P. Charova, P. Knotek and L. Benes, Int. J. Pharm., 2011, 414, 42 CrossRef CAS.
- J. J. Li, F. Zhao and J. Li, Appl. Microbiol. Biotechnol., 2011, 90, 427 CrossRef CAS.
- J. Li and X. J. Loh, Adv. Drug Delivery Rev., 2008, 60, 1000 CrossRef CAS.
- M. Y. Guo, M. Jiang, S. Pispas, W. Yu and C. X. Zhou, Macromolecules, 2008, 41, 9744 CrossRef CAS.
- X. W. Zhang, F. Y. Ke, J. Han, L. Ye, D. H. Liang, A. Y. Zhang and Z. G. Feng, Soft Matter, 2009, 5, 4797 RSC.
- K. Matyjaszewski and J. H. Xia, Chem. Rev., 2001, 101, 2921 CrossRef CAS.
- C. Moon, Y. M. Kwon, W. K. Lee, Y. J. Park and V. C. Yang, J. Controlled Release, 2007, 124, 43 CrossRef CAS.
- C. A. Lackey, O. W. Press, A. S. Hoffman and P. S. Stayton, Bioconjugate Chem., 2002, 13, 996 CrossRef CAS.
- T. W. Wang, C. L. Zhang, X. J. Liang, W. Liang and Y. Wu, J. Pharm. Sci., 2011, 100, 1067 CrossRef CAS.
- X. W. Zhang, X. Q. Zhu, F. Y. Ke, L. Ye, E. Q. Chen, A. Y. Zhang and Z. G. Feng, Polymer, 2009, 50, 4343 CrossRef CAS.
- W. C. Shen and H. J. P. Ryser, Biochem. Biophys. Res. Commun., 1981, 102, 1048 CrossRef CAS.
- J. L. N. Nathalie Lavignac, P. Ferruti and R. Duncan, Macromol. Biosci., 2009, 9, 480 CrossRef.
- H. L. Sun, B. N. Guo, X. Q. Li, R. Cheng, F. H. Meng, H. Y. Liu and Z. Y. Zhong, Biomacromolecules, 2010, 11, 848 CrossRef CAS.
- G. Y. Liu, P. Lv, C. J. Chen, X. F. Hu and J. A. Ji, Macromol. Chem. Phys., 2011, 212, 643 CrossRef CAS.
- C. L. Lo, C. K. Huang, K. M. Lin and G. H. Hsiue, Biomaterials, 2007, 28, 1225 CrossRef CAS.
- K. K. Upadhyay, A. N. Bhatt, A. K. Mishra, B. S. Dwarakanath, S. Jain, C. Schatz, J. F. Le Meins, A. Farooque, G. Chandraiah, A. K. Jain, A. Misra and S. Lecommandoux, Biomaterials, 2010, 31, 2882 CrossRef CAS.
- F. M. Veronese, O. Schiavon, G. Pasut, R. Mendichi, L. Andersson, A. Tsirk, J. Ford, G. F. Wu, S. Kneller, J. Davies and R. Duncan, Bioconjugate Chem., 2005, 16, 775 CrossRef CAS.
- L. J. Wong, M. Kavallaris and V. Bulmus, Polym. Chem., 2011, 2, 385 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: 1H NMR spectra of the ATRP macroinitiator 2-bromopropionyl terminated Pluronic F127 (F-Br), FTIR spectrum of PR and PR-EDA, and standard lines in DMSO and buffered solutions obtained from a series of solutions with different DOX concentrations. See DOI: 10.1039/c3bm60112a |
| This journal is © The Royal Society of Chemistry 2013 |