 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
The composition and end-group functionality of sterically stabilized nanoparticles enhances the effectiveness of co-administered cytotoxins†
Nicole S.
Bryce‡
a,
Binh T. T.
Pham‡
a,
Nicole W. S.
Fong
a,
Nirmesh
Jain
a,
Eh Hau
Pan
a,
Renee M.
Whan
b,
Trevor W.
Hambley
a and
Brian S.
Hawkett
*a
aSchool of Chemistry, University of Sydney, Sydney, NSW 2006, Australia. E-mail: brian.hawkett@sydney.edu.au; Fax: +61 2 9351 8651; Tel: +61 2 9351 6973
bBiomedical Imaging Facility, University of New South Wales, Randwick, NSW 2052, Australia
First published on 13th August 2013
Abstract
Diffusion of active cytotoxic agents throughout an entire solid tumour is a particular challenge to successful drug delivery. Here we show the simple and robust generation of non-toxic, 10–15 nm superparamagnetic iron oxide nanoparticles (SPIONs) that have been sterically stabilized by either 100% anionic or 100% cationic or 100% neutral end-functionalized steric stabilizers or by novel combinations of cationic and neutral end-functionalized polymer. When these nanoparticles were co-administered with various anti-cancer drugs, a significant increase in the diffusion and effectiveness of the cytotoxin in a 3-dimensional model of a solid tumour was shown for specific combinations of surface functionality and cytotoxin. The critical determinant of enhanced cytotoxin diffusion and effectiveness was the end functionality of the steric stabilizers and not the core composition (either iron oxide, silica or gold). We provide evidence that SPIONs stabilized with heterogeneous steric stabilizers enhance nuclear uptake of doxorubicin across multiple cell layers.
Introduction
Chemotherapy, as one of the primary treatment modalities for cancer, requires a balance between the administered dose, efficacy and toxicity of the cytotoxin for successful administration and treatment. Chemotherapeutic drugs often have poor penetration into solid tumours due to the physico-chemical properties of the compound such as high hydrophobicity and charge, pharmacokinetics of individual drugs and the complex tumour microenvironment.1–3 To overcome these problems, there has been considerable effort to design an effective and efficient targeting system for cancer treatment using nanomaterials.4–12Superparamagnetic iron oxide nanoparticles (SPIONs) have been proposed as drug delivery vehicles, however nanoparticle size, toxicity, stabilization, surface charge, functionalization, drug attachment and release of the drug from the nanoparticle are issues that affect their use in medical therapies.13–15 Surface coating of SPIONs with poly(ethylene glycol) (PEG) or dextran prolongs the circulation time of nanoparticles in the bloodstream by shielding the surface charge of the particle.16–18 PEG coating facilitates the decreased uptake of nanoparticles by macrophages, allowing for an increased circulation time of the carrier and increased passive accumulation in the tumour.19–24 However research by Hong et al.25 on nanoparticle–cell interactions showed that although useful in increasing the circulation half-life, the PEG coating may interfere with the interaction of nanoparticles with the endosomal membrane and prevent escape from the endosome. Release of conjugated active compounds from particles is often problematic, resulting in cellular accumulation of the active compound in lysosomes or failure of the active compound to reach the appropriate cellular compartment for optimal cytotoxic activity.26–29Dextran coated nanoparticles have been extensively studied and have FDA approval for use in patients.30 However, the dextran is of high molecular weight and contributes greatly to the overall hydrodynamic size of the particle and as a consequence, lowers the metal content of the particle, which may be disadvantageous to applications such as imaging.
RAFT polymerisation has been shown to be a versatile tool to synthesise polymers of a defined size with a narrow molecular weight distribution.31 Short chain block copolymers have been designed and synthesised by RAFT polymerisation by our group to produce sterically stabilized iron oxide nanoparticles that are stable in water and in physiological solutions with a high salt concentration.32 The short amphiphilic co-polymer molecules are strongly anchored at one end (anchoring block) to the surface of the nanoparticle and the short length of the hydrophilic block (stabilizing block) provides tight control of the overall size of the nanoparticle. The type of anchoring of the stabilising polymer chains to the polymer core depends on the type of core particle (Scheme 1). The stabilisers are anchored to the gold core via the classic gold–thiol bond, whereas anchoring to the silica core particles is via the electrostatic interaction between the quaternary amine and the silica core. Anchorage to the iron oxide core particles is via the co-ordinate/covalent bonds formed between the hydroxyl groups on the surface of the particles and phosphonic acid groups on the anchoring polymer chain. These steric stabilizers also provide SPIONs with surface moieties that can be functionalized for subsequent attachment of drugs or molecules responsible for tumour targeting.32,33
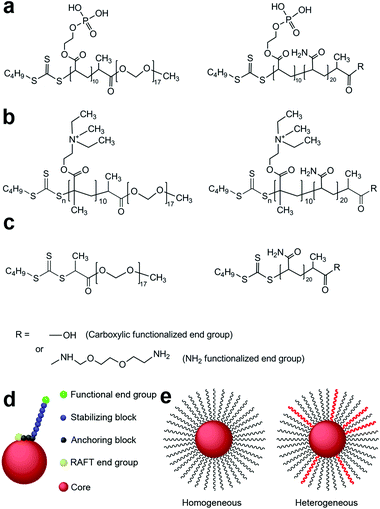 | ||
| Scheme 1 Polymeric steric stabilizers used in this study for (a) iron oxide nanoparticles, (b) silicananoparticles and (c) gold nanoparticles. (d) A diagrammatic representation of a sterically stabilized nanoparticle. (e) Cartoon of homogeneous and heterogeneous polymer stabilized nanoparticles. | ||
We made a fortuitous discovery that co-administration of drugs with our sterically stabilized nanoparticles facilitated the penetration of those drugs across multiple cell layers in a spheroid tumour model and set out to explore the characteristics of the nanoparticles that governed this process. The aim of this study was to determine if the stabilizer end group functionality on nanoparticles influenced the penetration and effectiveness of the co-administered cytotoxin into a model of a solid tumour. We report here a very simple and robust drug delivery system using functionalized sterically stabilized nanoparticles, SPIONs in particular, that significantly enhances drug diffusion and efficacy in 3-dimensional models of avascular colon and ovarian cancer. The nanoparticles were stabilized either with only one type of the steric stabilizer (homogenous coating) or with a heterogeneous mix of the steric stabilizers with neutral and cationic end functionality. Five commonly used chemotherapy drugs of differing molecular size, charge and hydrophobicity were chosen as test compounds to be co-administered with the nanoparticles, rather than physically bound to the nanoparticle. The effectiveness of the nanoparticle/drug co-administration was strongly governed by both the functionalized end group and the composition of the polymeric stabilizers and was unique for each drug.
Materials and methods
Materials
RAFT agents 2-{[(butylsulfanyl)carbonothioyl]sulfanyl}propanoic acid (RAFT-COOH) and methoxy-polyethylene glycol modified 2-{[(butylsulfanyl)carbonothioyl]sulfanyl}propanoic acid (RAFT-MPEG) were kindly provided by Dr Algi Serelis (DuluxGroup). 1,4-Dioxane (Fluka) was distilled under reduced pressure. Monoacryloxyethyl phosphate (MAEP, Aldrich) was passed through an inhibitor removal column (Aldrich). Acrylamide (AAm, Aldrich), 4,4′-azobis(4-cyanovaleric acid) (V-501, Wako), sodium hydroxide (NaOH, Aldrich), N-hydroxysuccinimide (NHS, Aldrich), 1-ethyl-3-(3-dimethylamino-propyl)carbodiimide (EDC, Aldrich) and 2,2′-(ethylenedioxy)bis-(ethylamine) (Aldrich) were used as received. Cisplatin was synthesised as previously described.34Doxorubicin, paclitaxel, 5-fluorouracil and mitoxantrone were used as received (Sigma Aldrich). Milli-Q water was used in all experiments.Synthesis of iron oxide nanoparticles
Maghemite iron oxide cores (γ-Fe2O3) were produced as previously described using the Massart method.32,35 Magnetic iron oxide nanoparticles (SPIONs) were prepared by coating the iron oxide cores with the desired combination of the steric stabilizers.32,33Scheme 1a shows the structures of the polymeric stabilizers used in this study to coat iron oxide cores.Steric stabilization of 10–15 nm maghemite iron oxide nanoparticles in aqueous dispersion
The –COOH end groups on the polymer coatings were then modified to –NH2 end groups using EDC/NHS chemistry.36 Into the above coated nanoparticles (7.8 g), N-hydroxysuccinimide (NHS, 14.4 mg) and then 1-ethyl-3-(3-dimethylamino-propyl)carbodiimide (EDC, 20 mg) were added, mixed by shaking and allowed to react for 2 hours at room temperature. A solution of 2,2′-(ethylenedioxy)bis-(ethylamine) (90 mg in 1 mL of water) was then added to the reaction mixture and allowed to react for a further 3.5 hours. The solution was then dialyzed against excess water with numerous changes, to remove free EDC and the reaction by-products.
All other SPIONs were prepared following the same procedures, using desired amounts of steric stabilizers.
Steric stabilization of sigma ludox AS30 silica particles using 95% poly[2-(dimethylamino)ethyl methacrylate]10-block-poly(ethylene oxide)17 (RAFT-DMAEM10-MPEG17) and 5% poly(2-(dimethylamino)ethyl methacrylate)10-block-poly(acrylamide)20 macro raft agent (RAFT-DMAEM10-AAm20)
16.5 g of the above macro-RAFT solution was diluted with MQ water (17 g) and methyl iodide (0.7 g) added. The mixture was stirred at room temperature for 1 hour before being partially dried using a rotary evaporator. The samples were then placed in the vacuum oven to yield 100% solids.
19.3 g of the above copolymer solution was diluted with MQ water (20 g) and methyl iodide (0.78 g) was added. The mixture was stirred at room temperature for 1 hour be partially dried using a rotary evaporator. The partially dried sample was then placed in the vacuum oven to yield 100% solids.
To the above sterically stabilized silica particles N-hydroxysuccinimide (NHS, 39.4 mg) and then 1-ethyl-3-(3-dimethylamino-propyl)carbodiimide (EDC, 56.2 mg) were added, mixed by shaking and allowed to react for 2 hours at room temperature. 37 mg of 2,2′-(ethylenedioxy)bis-(ethylamine) was then added to the reaction mixture and allowed to react for a further 12 hours. The solution was then dialysed against excess water with numerous changes, to remove free EDC and the reaction by-products.
Steric stabilization of 10–15 nm gold nanoparticles in aqueous dispersion using 95% poly(ethylene oxide)17 (RAFT-MPEG17) and 5% NH2 end functionalized poly(acrylamide)20RAFT agent (RAFT-AAm20)
Into the coated gold nanoparticles (100 mL), N-hydroxysuccinimide (NHS, 4 mg) and then 1-ethyl-3-(3-dimethylamino-propyl)carbodiimide (EDC, 4.1 mg) were added, mixed by shaking and allowed to react for 2 hours at room temperature. A solution of diamine (21 mg of 2,2′-(ethylenedioxy)bis-(ethylamine) in 2 mL of water) was then added to the reaction mixture and allowed to react for a further 3.5 hours. The solution was then dialysed against excess water with numerous changes, to remove free EDC and the reaction by-products.
Particle morphology and size distribution
Nanoparticle morphology was visualized by transmission electron microscopy before and after steric stabilization. One drop of 0.001 wt% dispersion was placed on a carbon coated copper grid and left to dry at RT. Specimens were imaged on a Phillips CM120 Biofilter transmission electron microscope at 120 kV.Particle size distribution was measured by dynamic light scattering using a Malvern Instrument Zetasizer nano series instrument with a detection angle of 173°, 25 °C. Samples (0.01 wt% in deionised water) were filtered through a 0.45 μm membrane prior to measurement.
The composition of coated nanoparticles was determined by Thermal Gravimetry Analysis (TGA, 2950 TA Instruments). The polymer weight composition on the coated SPIONs, silica and gold nanoparticles were 25% (±3%), 60% (±5%) and 45% (±3%) respectively.
Cell culture
DLD-1 human colon carcinoma cells were obtained from ATCC and used within 2 months of resuscitation. Cells were maintained in Advanced DMEM (Invitrogen) and supplemented with 2% FBS and 2 mM Glutamine in a humidified environment at 37 °C and 5% CO2. Spheroids were formed by plating 1.5 × 104 cells onto 0.75% agarose coated 96 well plates and incubated without agitation for 72 h.38 For all co-administration experiments, the drugs and the nanoparticles were mixed together in cell growth medium, incubated for a minimum of 5 minutes at RT, then added to the cells/spheroids. For all confocal microscopy assays, spheroids were dosed with 1 μM doxorubicin or 100 nM mitoxantrone ±10 ppm nanoparticles and incubated for a further 24 hours. All nanoparticles were sterilized by autoclaving at 120 °C, 2 kPa for 20 min in a Tomy high pressure steam sterilizer ES-315 before use in cellular assays.Cytotoxicity assay
1 × 105 cells per mL were seeded into each well of a 96-well plate and incubated under standard culturing conditions. The cells were dosed with a 4-log range of doxorubicin, cisplatin, paclitaxel, mitoxantrone, 5-fluorouracil ±50 ppm nanoparticles. Individual polymers were tested to a maximum concentration of 50 ppm. The cells were then incubated for 72 h before 1.0 mM MTT was added to each well and further incubated for 4 h. The media was removed and 150 μL of DMSO was added to each well. The absorbance of each well was measured at 600 nm after shaking for 1 min in a Victor3V microplate reader (Perkin Elmer). IC50 values were determined as the drug concentration that reduced the absorbance to 50% of that in untreated control wells. Average values from triplicate wells from 3 independent experiments and standard error of the mean are reported.Outgrowth assay
Spheroids were incubated with cytotoxic drugs ±10 ppm nanoparticles for 24 hours. The concentrations of cytotoxic drugs used were 1 μM Dox, 30 nM Mito, 10 μM Cis, 10 μM 5FU and 10 nM Pac. The treated spheroids were transferred to a 24 well plate using a wide bore transfer pipette and the medium replaced with 1 mL of fresh media. The spheroids were then incubated for 48 hours at 37 °C in a 5% CO2 humidified environment, allowing the spheroid to attach to the plate and the cells to grow out from the spheroid onto the surface of the plate. Hoechst 33342 was then added to the wells and incubated for 30 minutes at 37 °C in a 5% CO2 humidified environment. Widefield fluorescence images of Hoechst 33342 stained nuclei were taken of the cells that had grown out from the spheroid by an independent investigator who was blinded to the experimental treatments. To quantitate the outgrowth, the number of nuclei within a 60° angle from the edge of the spheroid was counted. Two regions from each spheroid were measured from a minimum of 6 spheroids. Values were then normalized to the number of cells that had grown out from spheroids treated with doxorubicin alone for comparison. Standard error of the mean was reported and statistical significance determined by the student's t-test.Spectrophotometry
The fluorescence of 1 μM Dox in the cell media and 1 μM Dox in cell media plus 5 or 10 ppm of 90% MPEG/10% NH2nanoparticles was measured on the RF-5301PC Spectro Fluorophotometer (Shimadzu) with an excitation wavelength of 559 nm and slit width of 5.Transmission electron microscopy sample preparation
Spheroids were washed with PBS, fixed with 2.5% glutaraldehyde solution and washed with PBS. Secondary fixation was by 1% osmium tetroxide and the spheroids were washed and dehydrated through an ethanol gradient. The spheroids were then infiltrated with a gradient of Spurr's Resin–100% ethanol, embedded into BEEM capsules and allowed to polymerise overnight in a 60 °C oven. Due to specimen shrinkage from the dehydration, the resin embedded spheroids were approximately 300 μm in diameter (originally ∼600 μm). Ultra thin sections with a nominal thickness of 95 nm were cut through the central region of the spheroid at ∼100 μm from the edge. Sections were expanded using chloroform prior to being transferred onto 100-mesh copper grids for imaging. To enhance the contrast of the samples for imaging, the sections were post-stained using uranyl acetate and lead citrate.Image acquisition
Confocal images were collected on the Olympus FV1000 inverted microscope using an Olympus UPLAPO 10×/0.40 air objective lens and a PLAPO 60×/1.40 water objective lens. A scan rate of 4.0 μs per pixel and Kalman averaging was applied to all images. Samples containing doxorubicin were excited with the 559 nm diode laser and the resulting emission was collected between 580–680 nm. Samples containing mitoxantrone were excited with the 405 nm diode laser and the resulting emission was collected between 580–680 nm.Widefield fluorescence images were collected on an Olympus IX81 microscope using an Olympus UPlanFL N 10× 0.30 Ph1 objective and a Hamamatsu Orca-ER camera using an Olympus MT20 CellR light source and standard filter sets.
Fluorescence lifetime images were collected using a Leica TCS SP5 MP FLIM system. This system had a tunable Mai Tai Deep See multiphoton laser with a repetition rate of 80 mHz (Spectra-Physics) attached to a Leica DMI6000B-CS inverted microscope. Specimens were illuminated with 800 nm and collected in the descanned internal FLIM detectors over the 540–750 nm range using either a HC PL APO 20×/0.70 IMM or a HCX PL APO 63×/1.30 GLYC objective lens. The data was collected with the aid of the B&H SPCM software. Fluorescence lifetimes were determined using time correlated single photon counting (TCSPC) and analyzed with SPCImage software (version 3.1.0.0). The instrument response function was derived from the decay curve of a 0.5% solution of Rose Bengal and the mono-exponential decay curve of a solution of FITC confirmed. 512 × 512 images were collected and 3× binning applied for analysis to ensure greater than 100![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 photons per pixel were analyzed. A minimum of ten 4 second accumulation scans were taken of each sample. For cell free analysis, mono-exponential curves were individually fitted to the entire image to obtain χ2 values closest to 1. Average values from 3 independent experiments were reported. Analysis of fluorescence lifetimes in spheroids required individually fitted bi-exponential curves to regions of interest defined by the outer edge of the spheroid to obtain average χ2 values closest to 1. A total of 10 spheroids selected from 3 independent experiments were analyzed.
000 photons per pixel were analyzed. A minimum of ten 4 second accumulation scans were taken of each sample. For cell free analysis, mono-exponential curves were individually fitted to the entire image to obtain χ2 values closest to 1. Average values from 3 independent experiments were reported. Analysis of fluorescence lifetimes in spheroids required individually fitted bi-exponential curves to regions of interest defined by the outer edge of the spheroid to obtain average χ2 values closest to 1. A total of 10 spheroids selected from 3 independent experiments were analyzed.
Transmission electron microscopy images of the spheroid sections were obtained using a JEOL 1400 TEM at 120 kV. Representative images were taken at the edge and in the center of each of the spheroid sections. Digital images were obtained with a charge coupled device (CCD) camera.
Scanning Electron Microscopy (SEM) and energy dispersive X-ray spectroscopy (EDS) analysis was conducted on the microtomed spheroid sections using a Zeiss ULTRA Plusscanning electron microscope with a Bruker XFlash 4010 EDSdetector, to determine the composition of the nanoparticles. EDS spectra were acquired using an accelerated voltage of 2 kV.
Results and discussion
Nanoparticles penetrate deeply into the middle of spheroids and accumulate within cells
SPIONs with a core size of 10–15 nm were sterically stabilized with short chain block amphiphilic copolymers that differed by the end functionality of the polymer chain (Scheme 1, Table 1, ESI Table 1†). The three different types of block copolymers comprise an anchoring block to the particle surface and a stabilizing block of either; (i) polyethylene oxide with a neutral end functional group (100% MPEG), (ii) polyacrylamide with an anionic end functional group (100% COOH), (iii) polyacrylamide with a cationic end functional group (100% NH2), or (iv) a desired combination of (i) and (iii), for example 90% MPEG/10% NH2 molar ratio of the two stabilizers. The differentially stabilized SPIONs generated for this study are listed in Table 1. Transmission electron micrographs showed the narrow range of SPION sizes before and after steric stabilization (Fig. 1a and b). A representative plot of the average size and polydispersity of the SPIONs is shown in ESI Fig. 1.† | ||
| Fig. 1 Uncoated and sterically stabilized SPIONs. Transmission electron microscope images of (a) uncoated iron oxide cores and (b) sterically stabilized NP3. Scale bars as indicated. | ||
| Abbrev. name | Particle coating (molar % of individual polymer stabilizers) | Core |
|---|---|---|
| NP1 | 100% MPEG(CH3) | Iron oxide |
| NP2 | 98% MPEG 2% NH2 | Iron oxide |
| NP3 | 95% MPEG 5% NH2 | Iron oxide |
| NP4 | 90% MPEG 10% NH2 | Iron oxide |
| NP5 | 80% MPEG 20% NH2 | Iron oxide |
| NP6 | 50% MPEG 50% NH2 | Iron oxide |
| NP7 | 100% NH2 | Iron oxide |
| NP8 | 100% COOH | Iron oxide |
| NP9 | 95% MPEG 5% NH2 | Gold |
| NP10 | 95% MPEG 5% NH2 | Silica |
To investigate the effect of the end functional group of the steric stabilizer on the penetration of the nanoparticle through multiple cell layers, tumour spheroids, made from human DLD-1 colorectal carcinoma cells,39 were dosed with SPIONs and examined by transmission electron microscopy (TEM). As shown in Fig. 2a–h, all of the stabilizer combinations facilitated penetration of the SPIONs into the core of spheroids. We observed no aggregation of nanoparticles and the high magnification images clearly showed that the intracellular SPIONs remained fully dispersed on the individual particle scale. Energy Dispersive X-Ray Spectroscopy (EDS) confirmed that the large electron dense structures contained high concentrations of iron and oxygen, indicative of SPIONs (Fig. 3). In spheroids dosed with NP1, large (up to 5 μm in diameter) vesicular structures containing nanoparticles, were evident throughout the cytoplasm and close to the nucleus (Fig. 2a and e).
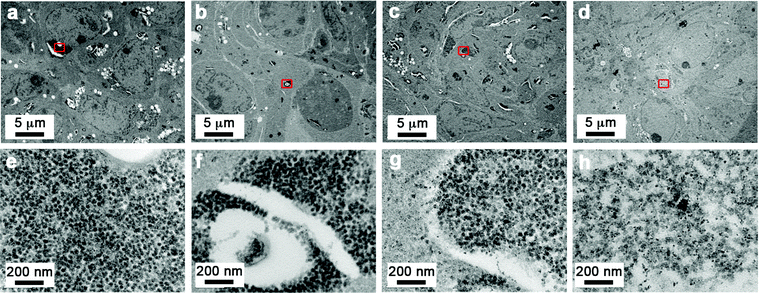 | ||
| Fig. 2 Sterically stabilized SPIONs can penetrate into spheroids and remain evenly dispersed. Transmission electron microscope images of DLD-1 spheroids incubated with SPIONS. Images were taken from the central region of the spheroid from sections that were cut at an approximate depth of 100 μm from the edge of the spheroid. A small region from the centre of the low magnification images (indicated by the red box) is shown in the high magnification images (a and e) NP1, (b and f) NP3, (c and g) NP4, (d and h) NP7. Scale bars as indicated. | ||
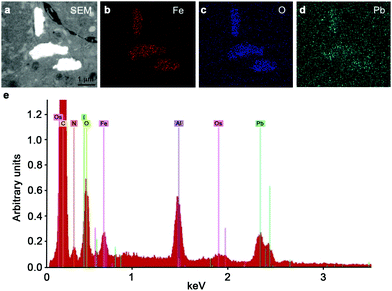 | ||
| Fig. 3 Confirmation of the presence of iron oxide nanoparticles within spheroids energy dispersive X-ray spectroscopy was used to show the presence of an electron dense region (a) consistent with iron (b), oxygen (c) and lead (d) in the sample. (e) Representative EDS elemental spectra showing the iron and oxygen from the NP3 SPIONs present in the sample. | ||
In the presence of NP3 and NP4 (5–10% of NH2 end functionalized steric stabilizers), the size of the SPION containing vesicular structures decreased and localized away from the nucleus (Fig. 2b, c, f and g). For the highly charged NP7 SPIONs, large nanoparticle clusters were less evident than for the neutral NP1 SPIONs, suggestive of a more even distribution of the particles (Fig. 2d and h). The TEM results demonstrated that nanoparticle accumulation and localization within 3D cancer tumour models was significantly influenced by the composition and functionalized end group of the steric stabilizers.
The penetration of anticancer drugs into colon cancer spheroids was significantly enhanced by the presence of SPIONs with specific combinations of steric stabilizers and functionalized end groups.
As all nanoparticles were adept at penetrating the tumour model, we hypothesized that they could be used as carriers of co-administered cytotoxins. The commonly used anticancer drugs, doxorubicin (Dox) and mitoxantrone (Mito) have inherent autofluorescent properties and so were chosen as the basis for this study (ESI Scheme 1†).40,41 Dox has previously been shown to have limited penetration into solid tumours and the diffusion of fluorescent compounds, such as Dox, into spheroids can be easily tracked through fluorescent microscopy.3,41–44 Both Dox and Mito administered individually penetrate no further than 80 μm into spheroids (Fig. 4a and b), however differences in drug penetration were seen when the drugs were co-administered with SPIONs. Upon co-administration of Dox and SPIONs, both the neutral NP1 and the cationic NP7 nanoparticles only slightly enhanced the uptake and penetration of Dox. In contrast, the combination of neutral and positively charged stabilizers on the NP3 nanoparticles resulted in the distribution of Dox throughout the entire spheroid (Fig. 4a). In contrast, when Mito was co-administered with the SPIONs only the 100% cationic end functionalized NP7 SPIONs enhanced the penetration of Mito into the spheroids (Fig. 4b). All other SPIONs tested did not enhance the penetration of either Dox or Mito. Fluorescence spectroscopy showed no significant differences in Dox fluorescence when incubated with the different SPIONS (ESI Fig. 2†), indicating that the nanoparticles were facilitating diffusion of the drugs. As the nanoparticles differed in the proportion of the charged end group of the polymer, we postulate that these results are due to differential interactions between the individual drug and the specific combinations of charges within the microenvironment. The charge of a molecule highly influences its cellular diffusion and cell uptake. Previous studies on the diffusion of a series of platinum based drugs that altered in their charge into the same spheroid model has shown that the more highly charged compounds were able to penetrate further into the spheroid.3,45 Other researchers have utilised the charge of the nanoparticle to influence cell uptake or alteration of the overall charge of the nanoparticle when present in the acidic tumour microenvironment to effect drug release.46–49 This work has manipulated the ratio of charges present at the drug–nanoparticle interface to influence the diffusion of the drug. Future work will determine whether the charge is important for drug association with the nanoparticle or the release of the drug from the nanoparticle. However, our customizable nanoparticles provide great flexibility in determining the optimal charge:drug ratio for in vitro delivery.
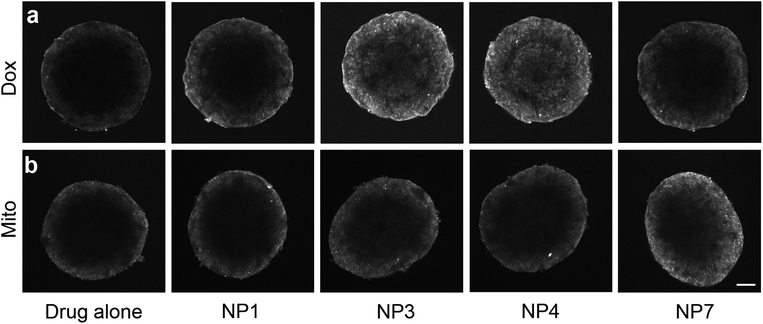 | ||
| Fig. 4 SPIONS influence the diffusion of doxorubicin and mitoxantrone and enhanced diffusion is dependent on stabilizer composition. Single confocal images of (a) doxorubicin or (b) mitoxantrone diffusion into DLD-1 spheroids either alone or with co-administered SPIONs. Scale bar 200 μm. | ||
Enhanced penetration of cytotoxin into the tumour DLD-1 spheroid correlates with increased effectiveness of the cytotoxin
Toxicity assays performed on cells grown in monolayer culture showed that the nanoparticles were not toxic at the highest dose tested (100 ppm) and as such a 72 h IC50 value for the nanoparticles could not be obtained. Similarly, the polymers alone were also found to be non-toxic at the highest dose tested (50 ppm), which is a 4-fold increase in polymer concentration when compared to a dose of nanoparticles at 10 ppm. Conventional measures of cytotoxicity were not suitable for determining the cytotoxicity in 3D due to the large size of the DLD-1 spheroids and to the resistance of the spheroids to either trypsin or collagenase mediated single cell dispersion. To minimize concerns about the diffusion of MTT throughout the entire spheroid, an outgrowth assay was developed to provide a quantitative comparison of cytotoxicity and the effectiveness of the administered cytotoxin. The outgrowth assay requires the placement of the spheroids onto a surface so that the cells can attach and then viable cells migrate away from the main body of the spheroid (ESI Movie 1†). After 48 hours, all of the cells within a 60° angle from one point of the spheroid are counted (Fig. 5a). Dox has previously been shown to inhibit cellular migration on 2-dimensional substrates, while 3-dimensional migration speeds remained unaffected.50 Consequently, the outgrowth experiments were performed in cytotoxin free media after initial treatment.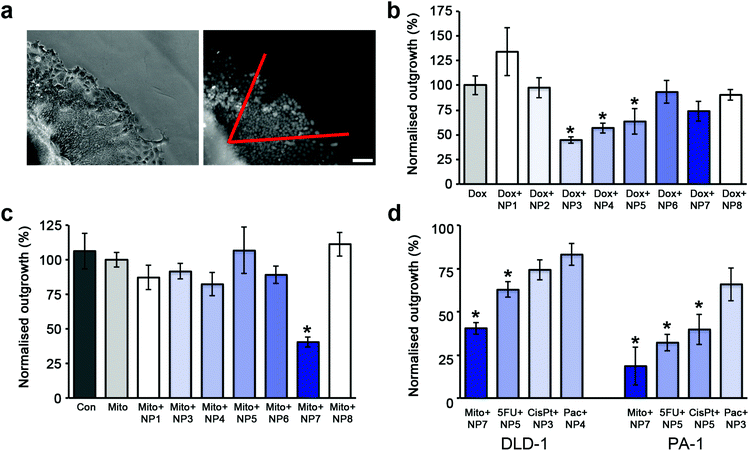 | ||
| Fig. 5 The enhanced effectiveness of cytotoxins when co-administered with SPIONs is dependent on stabilizer composition. (a) Phase contrast image of the edge of a spheroid in the outgrowth assay and the corresponding widefield fluorescent image of Hoechst 33342 stained nuclei. All nuclei within a 60° angle (shown in red) from the edge of the spheroid were counted. Scale bar 50 μm. (b) Plot of cellular outgrowth of DLD-1 spheroids co-administered with Dox and SPIONs normalized to the outgrowth of Dox treated DLD-1 spheroids. The untreated control spheroids (not shown for clarity) had an outgrowth value of 331% ±23 compared to Dox treated spheroids. The shading represents the percentage of NH2 functionalized polymer on the particle. (c) Plot of cellular outgrowth of DLD-1 spheroids co-administered with Mito and SPIONs normalized to the outgrowth of Mito treated DLD-1 spheroids. (d) Plot of cellular outgrowth of the most effective SPION/cytotoxin combination for DLD-1 and PA-1 spheroids. Each combination was normalized to the outgrowth of cytotoxin alone treated spheroids. For graphs (b–d), error bars represent standard error with n = 6. * indicates p < 0.01 compared to cytotoxin alone. | ||
SPIONs alone did not significantly influence the cellular outgrowth from the treated spheroids in comparison to untreated spheroids (ESI Fig. 3†). Cellular outgrowth from the DLD-1 spheroids was reduced by a factor of 6 when Dox was co-administered with the NP3 or NP4 SPIONs (Fig. 5b) compared to untreated spheroids. This was a further 2-fold decrease in the number of outgrown cells compared to spheroids treated with Dox alone. The proportion of NH2 functionality was the critical determinant, as SPIONs coated with less than 5% NH2 and greater than 20% NH2 were ineffective. In contrast, the homogenously coated SPIONs, NP1 and NP7, did not enhance the effectiveness of Dox in the spheroids as shown by the similar numbers of outgrown cells. Taken together with the confocal imaging of Dox distribution, these results reveal that the effectiveness of the drug is directly related to its accumulation and penetration into the spheroid.
In contrast to the treatment with Dox, the strongest inhibition for cellular outgrowth (2.5 fold) was given by Mito co-administered with NP7 SPIONs (Fig. 5c) in comparison with the treatment with Mito alone. All other SPIONs tested did not show significantly enhanced effectiveness of Mito. The outgrowth results for Mito were also in good agreement with the localization of Mito shown by confocal imaging.
We also tested the SPIONs co-administered with Dox or Mito in the PA-1 ovarian cancer cell line. PA-1 cells are extremely sensitive to Dox, and the outgrowth assay was unable to be performed, even using low nanomolar concentrations of Dox (ESI Table 2†). When treated with Mito alone, due to the lower doses used, the cellular outgrowth from the PA-1 spheroid was unaffected, whereas outgrowth was strongly influenced by the co-administration of Mito with all SPIONs tested (ESI Fig. 4a†). Similar to the results seen in the DLD-1 colon cancer cells, the most effective SPIONs/cytotoxin combination was the NP7 particles with Mito, which reduced cellular outgrowth 5-fold over Mito treatment alone (Fig. 5d, ESI Fig. 4a†).
Cellular outgrowth assays were also performed using the non-fluorescent cytotoxins; cisplatin, paclitaxel and 5FU, in both DLD-1 and PA-1 spheroids (Fig. 2d, ESI Fig. 5b–d, 4a–c†). The most effective SPION/cytotoxin combinations varied for each cytotoxin as well as for each cell type. However, it was consistent between cell lines that each specific cytotoxin was most effective with either a homogenous or heterogeneous stabilizer coated SPION (Fig. 5d).
To determine if the core of the nanoparticle had a role in influencing the diffusion of the cytotoxin, nanoparticles were made with either a gold or silica core and stabilized with polymers equivalent to those that coated the NP3 SPIONs (Scheme 1, ESI Fig. 6†). Due to the different surface chemistry of gold and silica compared to iron oxide, changes were made to the anchoring blocks of the amphiphilic co-polymers. However the stabilizing blocks and ratios of end-functionalized groups were consistent between the particles. Both the gold NP9 and silica NP10 particles enhanced doxorubicin penetration into spheroids and inhibited cellular outgrowth when co-administered with Dox similarly to NP3 SPIONs (Fig. 6). This indicated that the increase in the effectiveness and penetration of the cytotoxin was primarily influenced by the stabilizing chains with the core composition and the anchoring block of the amphiphilic co-polymer having no role in this process. These results have implications for the therapeutic use of polymer-stabilized nanoparticles co-administered with cytotoxins. The ratio of charged and uncharged polymeric stabilizers will need to be optimized to enhance the in vivo bio-distribution profile of each cytotoxin.
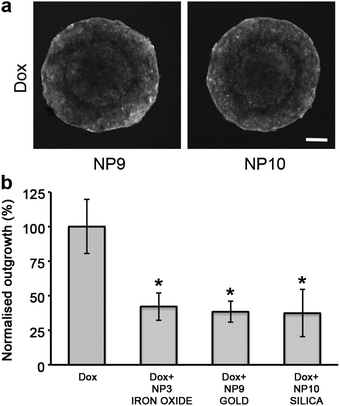 | ||
| Fig. 6 The stabiliser coatings and not the nanoparticle core are responsible for the enhanced diffusion of doxorubicin. (a) Single confocal images of DLD-1 spheroids co-administered with Dox and the SPIONs NP9 (gold) and NP10 (silica). Scale bar 200 μm. (b) Plot of cellular outgrowth of DLD-1 spheroids co-administered with Dox and nanoparticles with different core types: iron oxide, gold and silica. Each nanoparticle core was sterically stabilized with 95% MPEG/5% NH2polymers and the results were normalized to the outgrowth of Dox treated DLD-1 spheroids. The untreated control spheroids (not shown for clarity) had an outgrowth value of 331% ±23 compared to Dox treated spheroids. Error bars represent standard error with n = 6. * indicates p < 0.01 compared to cytotoxin alone. | ||
Nanoparticles act as facilitators for the diffusion of the cytotoxic agent, rather than altering the cytotoxic mechanism of the drug
It was not known if the SPIONs were altering the cytotoxic mechanism of the active molecule or simply facilitating transport of the cytotoxin across multiple cell layers. Cytotoxicity studies showed that the 72 h IC50 value of Dox was unchanged when SPIONs were present (ESI Table 3†), and the SPIONs were not toxic at concentrations 10 fold higher than those used in experimental conditions. Previous work as shown that nuclear localization of Dox is required for cytotoxicity.51TEM imaging of DLD-1 spheroids treated with the co-administration of Dox and NP4 SPIONs showed the lack of SPIONs within the nucleus (Fig. 7a and b). Confocal imaging of DLD-1 cells grown in 2-dimensional culture and dosed with either Dox or Dox co-administered with SPIONs showed no change in the nuclear localization of Dox (ESI Fig. 7†). Fluorescence lifetime microscopy (FLIM) was used to distinguish the different intracellular molecular environments of Dox to determine if Dox had been released from the SPIONs and was free to diffuse into the nucleus. Spectroscopy confirmed that there was little change to the overall fluorescence profile of Dox when nanoparticles were present (Fig. 7c). The Dox fluorescence lifetimes have been determined previously in different cell-free and cellular systems.26,41,52 The values of the fluorescence lifetimes and amplitudes obtained in this study are shown in Table 2. In the cell free system, mono-exponential decay curves were fitted and there was no discernible difference in the fluorescence lifetime (τ) of Dox either alone or in the presence of NP4 SPIONs as shown by the lifetime distribution graphs in Fig. 7d. In the presence of cells, the fluorescence decay curves required a bi-exponential fit to obtain χ2 values closest to 1. Consistent with the cell free system, there was no change in the fluorescent lifetimes τ1 and τ2, nor the amplitude values in spheroids treated with Dox alone or with Dox co-administered with NP4 nanoparticles (Table 2).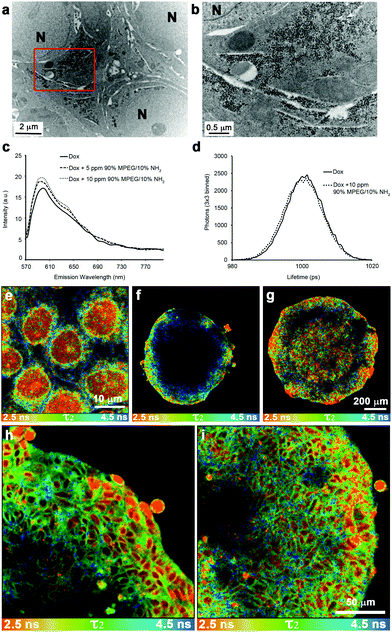 | ||
| Fig. 7 SPIONs stabilized with 10% NH2 functionalized polymer do not localize to the nucleus but enhance doxorubicin uptake into the nucleus of cells deep within the spheroid. (a) Transmission electron micrograph of DLD-1 spheroids co-administered with NP4 SPIONS and Dox. The image was taken from the central region of the spheroid from a section that was cut at an approximate depth of 100 μm from the edge of the fixed and stained spheroid (equivalent to 300 μm depth of an unprocessed spheroid). A higher magnification of the region enclosed by the red box in (a) is shown in panel (b). The nuclei are labelled N and the scale bars are as indicated. (c) Fluorescent spectra of Dox and Dox + NP4 SPIONs. (d) Representative graph of fluorescent lifetime distributions from solutions of Dox and Dox + NP4 SPIONs. Fluorescent lifetime images of doxorubicin treated monolayers and penetration into spheroids with the τ2 lifetime of doxorubicin displayed between 2.5–4.5 ns. The false colour FLIM image of the cell monolayer treated with Dox (e) allows for the cellular localizations of different lifetimes to be easily observed. The red colour indicates nuclear Dox, green indicates cytoplasmic localized Dox and blue indicates membrane bound Dox. (f) FLIM image of Dox treated spheroid. (g) FLIM image of a spheroid co-administered with Dox and NP4. (h) Higher magnification FLIM image of Dox treated spheroid. (i) Higher magnification false colour FLIM image of a spheroid co-administered with Dox and NP4 SPIONs showing increased levels of nuclear Dox (red) further into the spheroid. Scale bars as indicated. | ||
The wider distribution of τ2 values compared to τ1 values in the cellular system allowed false colour images of τ2 values to be generated that easily distinguish between different cellular compartments (Fig. 7e–i). From the cell monolayer image, nuclear (red colour) and membrane localized (blue colour) Dox are both able to be identified (Fig. 7e). In spheroid culture, Dox fluorescence penetrates approximately 80 μm into the spheroid (Fig. 4a). However, from the FLIM data, nuclear localization of Dox (red colour) is only seen in the outer 5–6 layers of cells, a distance of approximately 50 μm, whereas the fluorescence signals at a depth greater than 50 μm have a fluorescent lifetime indicative of membrane localization (Fig. 7f and h). When Dox is co-administered with NP4 nanoparticles, nuclear localization of Dox fluorescence is seen throughout the spheroid (Fig. 7g and i). This indicates that the co-administration of Dox with the NP4 SPIONs increases Dox effectiveness in spheroids by influencing the diffusion of Dox and increasing cellular uptake of Dox at depths greater than 50 μm from the edge of the spheroid. The enhanced nuclear localization of Dox with the NP4 SPIONs is a result of enhanced penetration and cellular uptake as the nanoparticles are localized to discrete vesicular structures in the cytoplasm and not the nucleus (Fig. 2 and 7a,b). The bioavailability of Dox is enhanced by the simple method of co-administration of Dox with the nanoparticles rather than conjugation of Dox to the nanoparticles.
Conclusions
Our studies show that by altering the surface properties of a nanoparticle by fine tuning the functional end groups on the steric stabilizers, it is possible to direct the diffusion of co-administered active molecules throughout a solid tumour model in the presence of serum. Intravenously administered chemotherapy drugs can completely penetrate normal tissue without the aid of SPIONs, however, solid tumours, with their poorly developed blood supply and fast growth rates, have large regions of the tumour that are hypoxic and inaccessible for drug delivery.1 In this situation, co administration of the SPIONs with the chemotherapy drugs offers the potential to provide enhanced penetration of the drugs into solid tumours and thereby provide enhanced outcomes for treatment. As the SPIONs alone are non-toxic and when co-administered with cytotoxins, do not alter the IC50 of the cytotoxin in well oxygenated cells, the SPION/cytotoxin co-administration method may potentially be a suitable clinical treatment option for avascular solid tumours. Additional benefits of the co-administration method compared to other drug loaded or encapsulated nanoparticle delivery systems include the removal of problems such as cytotoxin inactivation when conjugated to the nanoparticle and efficient release of the cytotoxin from the encapsulated nanoparticle. It would be of interest to examine the impact of different charge ratios on the surface of either drug loaded and drug encapsulated nanoparticles to determine if there is a similar enhancement of drug diffusion to that seen with the co-administration of the sterically stabilized nanoparticles and drug. The challenge remains however, to target the SPIONs to the tumour in a manner that will not affect cytotoxin adsorption and release, as we have shown that small changes in surface charge ratios can alter the effectiveness of the SPION/cytotoxin combination. Further studies on the in vivo bio-distribution profile of the SPIONs are required to ensure that the SPIONs do not negatively impact on the bio-distribution of the co-administered cytotoxin. It will be imperative to determine the individual surface properties of sterically stabilized nanoparticles in general and SPIONs in particular to best enhance the effects of each co-administered small molecule in an in vivo environment.Acknowledgements
The authors wish to acknowledge Sirtex Medical Ltd for funding, Dr Algi Serelis of DuluxGroup for RAFT reagents, Assoc. Prof. Ron Clarke for use of the fluorophotometer and discussion. The authors acknowledge the facilities, and the scientific and technical assistance, of the Australian Microscopy & Microanalysis Research Facility at the Australian Centre for Microscopy and Microanalysis, The University of Sydney.Notes and references
- A. I. Minchinton and I. F. Tannock, Drug Penetration in Solid Tumours, Nat. Rev. Cancer, 2006, 6, 583–592, DOI:10.1038/nrc1893.
- N. S. Bryce, J. Z. Zhang, R. M. Whan, N. Yamamoto and T. W. Hambley, Accumulation of an Anthraquinone and its Platinum Complexes in Cancer Cell Spheroids: The Effect of Charge on Drug Distribution in Solid Tumour Models, Chem. Commun., 2009, 2673–2675, 10.1039/b902415h.
- E. K. Rowinsky, L. A. Cazenave and R. C. Donehower, Taxol: A Novel Investigational Antimicrotubule Agent, J. Natl. Cancer Inst., 1990, 82, 1247–1259, DOI:10.1093/jnci/82.15.1247.
- R. K. Jain and T. Stylianopoulos, Delivering Nanomedicine to Solid Tumors, Nat. Rev. Clin. Oncol., 2010, 7, 653–664, DOI:10.1038/nrclinonc.2010.139.
- D. Peer, J. M. Karp, S. Hong, O. C. Farokhzad, R. Margalit and R. Langer, Nanocarriers as an Emerging Platform for Cancer Therapy, Nat. Nanotechnol., 2007, 2, 751–760, DOI:10.1038/nnano.2007.387.
- M. V. Yigit, A. Moore and Z. Medarova, Magnetic Nanoparticles for Cancer Diagnosis and Therapy, Pharm. Res., 2012, 29, 1180–1188, DOI:10.1007/s11095-012-0679-7.
- M. Colombo, S. Carregal-Romero, M. F. Casula, L. Gutierrez, M. P. Morales, I. B. Bohm, J. T. Heverhagen, D. Prosperi and W. J. Parak, Biological Applications of Magnetic Nanoparticles, Chem. Soc. Rev., 2012, 41, 4306–4334, 10.1039/c2cs15337h.
- R. R. Arvizo, S. Bhattacharyya, R. A. Kudgus, K. Giri, R. Bhattacharya and P. Mukherjee, Intrinsic Therapeutic Applications of Noble Metal Nanoparticles: Past, Present and Future, Chem. Soc. Rev., 2012, 41, 2943–2970, 10.1039/c2cs15355f.
- Z. Huang, X. Jiang, D. Guo and N. Gu, Controllable Synthesis and Biomedical Applications of Silver Nanomaterials, J. Nanosci. Nanotechnol., 2011, 11, 9395–9408, DOI:10.1166/jnn.2011.5317.
- N. Kamaly, Z. Xiao, P. M. Valencia, A. F. Radovic-Moreno and O. C. Farokhzad, Targeted Polymeric Therapeutic Nanoparticles: Design, Development and Clinical Translation, Chem. Soc. Rev., 2012, 41, 2971–3010, 10.1039/c2cs15344k.
- T. Panda and K. Deepa, Biosynthesis of Gold Nanoparticles, J. Nanosci. Nanotechnol., 2011, 11, 10279–10294, DOI:10.1166/jnn.2011.5021.
- R. Tietze, S. Lyer, S. Durr and C. Alexiou, Nanoparticles for Cancer Therapy Using Magnetic Forces, Nanomedicine, 2012, 7, 447–457, DOI:10.2217/nnm.12.10.
- T. L. Doane and C. Burda, The Unique Role of Nanoparticles in Nanomedicine: Imaging, Drug Delivery and Therapy, Chem. Soc. Rev., 2012, 41, 2885–2911, 10.1039/c2cs15260f.
- S. P. Egusquiaguirre, M. Igartua, R. M. Hernandez and J. L. Pedraz, Nanoparticle Delivery Systems for Cancer Therapy: Advances in Clinical and Preclinical Research, Clin. Transl. Oncol., 2012, 14, 83–93, DOI:10.1007/s12094-012-0766-6.
- R. Mout, D. F. Moyano, S. Rana and V. M. Rotello, Surface Functionalization of Nanoparticles for Nanomedicine, Chem. Soc. Rev., 2012, 41, 2539–2544, 10.1039/c2cs15294k.
- J. V. Jokerst, T. Lobovkina, R. N. Zare and S. S. Gambhir, Nanoparticle PEGylation for Imaging and Therapy, Nanomedicine, 2011, 6, 715–728, DOI:10.2217/nnm.11.19.
- A. S. Karakoti, S. Das, S. Thevuthasan and S. Seal, PEGylated Inorganic Nanoparticles, Angew. Chem., Int. Ed., 2011, 50, 1980–1994, DOI:10.1002/anie.201002969.
- E. Allard-Vannier, S. Cohen-Jonathan, J. Gautier, K. Herve-Aubert, E. Munnier, M. Souce, P. Legras, C. Passirani and I. Chourpa, Pegylated Magnetic Nanocarriers for Doxorubicin Delivery: A Quantitative Determination of Stealthiness in vitro and in vivo, Eur. J. Pharm. Biopharm., 2012, 81, 498–505, DOI:10.1016/j.ejpb.2012.04.002.
- N. T. Huynh, E. Roger, N. Lautram, J.-P. Benoit and C. Passirani, The Rise and Rise of Stealth Nanocarriers for Cancer Therapy: Passive Versus Active Targeting, Nanomedicine, 2010, 5, 1415–1433, DOI:10.2217/nnm.10.113.
- E. K. U. Larsen, T. Nielsen, T. Wittenborn, L. M. Rydtoft, A. R. Lokanathan, L. Hansen, L. Ostergaard, P. Kingshott, K. A. Howard and F. Besenbacher, et al., Accumulation of Magnetic Iron Oxide Nanoparticles Coated with Variably Sized Polyethylene Glycol in Murine Tumors, Nanoscale, 2012, 4, 2352–2361, 10.1039/c2nr11554a.
- Z. Wang, W.-K. Chui and P. C. Ho, Design of a Multifunctional PLGA Nanoparticulate Drug Delivery System: Evaluation of its Physicochemical Properties and Anticancer Activity to Malignant Cancer Cells, Pharm. Res., 2009, 26, 1162–1171, DOI:10.1007/s11095-010-0308-2.
- L. M. Kaminskas, V. M. McLeod, B. D. Kelly, C. Cullinane, G. Sberna, M. Williamson, B. J. Boyd, D. J. Owen and C. J. H. Porter, Doxorubicin-Conjugated PEGylated Dendrimers Show Similar Tumoricidal Activity but Lower Systemic Toxicity when Compared to PEGylated Liposome and Solution Formulations in Mouse and Rat Tumor Models, Mol. Pharmaceutics, 2012, 9, 422–432, DOI:10.1021/mp200522d.
- H. S. Yoo and T. G. Park, Folate-Receptor-Targeted Delivery of Doxorubicin Nano-Aggregates Stabilized by Doxorubicin-PEG-Folate Conjugate, J. Controlled Release, 2004, 100, 247–256, DOI:10.1016/j.jconrel.2004.08.017.
- K. J. Harrington, C. Lewanski, A. D. Northcote, J. Whittaker, A. M. Peters, R. G. Vile and J. S. W. Stewart, Phase II Study of Pegylated Liposomal Doxorubicin (Caelyx) as Induction Chemotherapy for Patients with Squamous Cell Cancer of the Head and Neck, Eur. J. Cancer, 2001, 37, 2015–2022, DOI:10.1016/S0959-8049(01)00216-7.
- R.-L. Hong, C.-J. Huang, Y.-L. Tseng, V. F. Pang, S.-T. Chen, J.-J. Liu and F.-H. Chang, Direct Comparison of Liposomal Doxorubicin With or Without Polyethylene Glycol Coating in C-26 Tumor-Bearing Mice: Is Surface Coating With Polyethylene Glycol Beneficial?, Clin. Cancer Res., 1999, 5, 3645–3652 CAS.
- X. Dai, Z. Yue, M. E. Eccleston, J. Swartling, N. K. Slater and C. F. Kaminski, Fluorescence Intensity and Lifetime Imaging of Free and Micellar-Encapsulated Doxorubicin in Living Cells, Nanomedicine, 2008, 4, 49–56, DOI:10.1016/j.nano.2007.12.002.
- K. Kaaki, K. Herve-Aubert, M. Chiper, A. Shkilnyy, M. Souce, R. Benoit, A. Paillard, P. Dubois, M. L. Saboungi and I. Chourpa, Magnetic Nanocarriers of Doxorubicin Coated With Poly(ethylene glycol) and Folic Acid: Relation Between Coating Structure, Surface Properties, Colloidal Stability, and Cancer Cell Targeting, Langmuir, 2012, 28, 1496–1505, DOI:10.1021/la2037845.
- Y. Qin, L. Sun, X. Li, Q. Cao, H. Wang, X. Tang and L. Ye, Highly Water-Dispersible TiO2 Nanoparticles for Doxorubicin Delivery: Effect of Loading Mode on Therapeutic Efficacy, J. Mater. Chem., 2011, 21, 18003–18010, 10.1039/C1JM13615A.
- P. Zou, Y. Yu, Y. A. Wang, Y. Zhong, A. Welton, C. Galban, S. Wang and D. Sun, Superparamagnetic Iron Oxide Nanotheranostics for Targeted Cancer Cell Imaging and pH-Dependent Intracellular Drug Release, Mol. Pharmaceutics, 2010, 7, 1974–1984, DOI:10.1021/mp100273t.
- C. Tassa, S. Y. Shaw and R. Weissleder, Dextran-coated iron oxide nanoparticles: a versatile platform for targeted molecular imaging, molecular diagnostics, and therapy, Acc. Chem. Res., 2011, 44, 842–852, DOI:10.1021/ar200084x.
- G. Moad, E. Rizzardo and S. H. Thang, RAFT Polymerization and Some of its Applications, Chem.–Asian J., 2013, 8, 1634–1644 CrossRef CAS.
- N. Jain, Y. Wang, S. K. Jones, B. S. Hawkett and G. G. Warr, Optimized Steric Stabilization of Aqueous Ferrofluids and Magnetic Nanoparticles, Langmuir, 2010, 26, 4465–4472, DOI:10.1021/la903513v.
- N. Jain, X. Zhang, B. S. Hawkett and G. G. Warr, Stable and Water-Tolerant Ionic Liquid Ferrofluids, ACS Appl. Mater. Interfaces, 2011, 3, 662–667, DOI:10.1021/am1012112.
- S. C. Dhara, A rapid method for the synthesis of cis-[Pt(NH3)2Cl2], Indian J. Chem., 1970, 8, 193–194 Search PubMed.
- R. Massart, Preparation of Aqueous Magnetic Liquids in Alkaline and Acidic Media, IEEE Trans. Magn., 1981, MAG-17, 1247–1248, DOI:10.1109/TMAG.1981.1061188.
- C. D. Medley, S. Bamrungsap, W. Tan and J. E. Smith, Aptamer-Conjugated Nanoparticles for Cancer Cell Detection, Anal. Chem., 2011, 83, 727–734, DOI:10.1021/ac102263v.
- G. Frens, Controlled Nucleation for Regulation of Particle-Size in Monodisperse Gold Suspensions, Nat. Phys. Sci., 1973, 241, 20–22, DOI:10.1038/physci241020a0.
- B. J. Kim, T. W. Hambley and N. S. Bryce, Visualising the Hypoxia Selectivity of Cobalt(III) Prodrugs, Chem. Sci., 2011, 2, 2135–2142, 10.1039/C1SC00337B.
- J. Z. Zhang, N. S. Bryce, R. Siegele, E. A. Carter, D. Paterson, M. D. de Jonge, D. L. Howard, C. G. Ryan and T. W. Hambley, The Use of Spectroscopic Imaging and Mapping Techniques in the Characterisation and Study of DLD-1 Cell Spheroid Tumour Models, Integr. Biol., 2012, 4, 1072–1080, 10.1039/c2ib20121f.
- G. An and M. E. Morris, Effects of Single and Multiple Flavonoids on BCRP-Mediated Accumulation, Cytotoxicity and Transport of Mitoxantrone In Vitro, Pharm. Res., 2010, 27, 1296–1308, DOI:10.1007/s11095-010-0108-8.
- O. Hovorka, V. Subr, D. Vetvicka, L. Kovar, J. Strohalm, M. Strohalm, A. Benda, M. Hof, K. Ulbrich and B. Rihova, Spectral Analysis of Doxorubicin Accumulation and the Indirect Quantification of its DNA Intercalation, Eur. J. Pharm. Biopharm., 2010, 76, 514–524, DOI:10.1016/j.ejpb.2010.07.008.
- A. J. Primeau, A. Rendon, D. Hedley, L. Lilge and I. F. Tannock, The Distribution of the Anticancer Drug Doxorubicin in Relation to Blood Vessels in Solid Tumors, Clin. Cancer Res., 2005, 11, 8782–8788, DOI:10.1158/1078-0432.CCR-05-1664.
- P. Mohan and N. Rapoport, Doxorubicin as a Molecular Nanotheranostic Agent: Effect of Doxorubicin Encapsulation in Micelles or Nanoemulsions on the Ultrasound-Mediated Intracellular Delivery and Nuclear Trafficking, Mol. Pharmaceutics, 2010, 7, 1959–1973, DOI:10.1016/j.jconrel.2011.01.022.
- S. J. Wolf, T. Huynh, N. S. Bryce, T. W. Hambley, L. P. Wakelin, B. W. Stewart and D. R. Catchpoole, Intracellular Trafficking as a Determinant of AS-DACA Cytotoxicity in Rhabdomyosarcoma Cells, BMC Cell Biol., 2011, 12, 36, DOI:10.1186/1471-2121-12-36.
- J. Z. Zhang, N. S. Bryce, A. Lanzirotti, C. K. Chen, D. Paterson, M. D. de Jonge, D. L. Howard and T. W. Hambley, Getting to the Core of Platinum Drug Bio-Distributions: The Penetration of Anti-Cancer Platinum Complexes into Spheroid Tumour Models, Metallomics, 2012, 4, 1209–1217 RSC.
- S. Bhattacharjee, D. Ershov, J. Gucht, G. M. Alink, I. M. Rietjens, H. Zuilhof and A. T. Marcelis, Surface Charge-specific Cytotoxicity and Cellular Uptake of Tri-block Copolymer Nanoparticles, Nanotoxicology, 2013, 7, 71–84, DOI:10.3109/17435390.2011.633714.
- S. R. Yoon, H. M. Yang, C. W. Park, S. Lim, B. H. Chung and J. D. Kim, Charge-conversional Poly(amino acids)s Derivatives as a Drug Delivery Carrier in Response to the Tumor Environment, J. Biomed. Mater. Res., Part A, 2012, 100, 2027–2033, DOI:10.1002/jbm.a.34048.
- P. Xu, E. A. Van Kirk, Y. Zhan, W. J. Murdoch, M. Radosz and Y. Shen, Targeted Charge-reversal Nanoparticles for Efficient Anticancer Drug Delivery, Angew. Chem., Int. Ed., 2007, 46, 4999–5002, DOI:10.1002/anie.200605254.
- J. Z. Du, X. J. Du, C. Q. Mao and J. Wang, Tailor-made Dual pH-sensitive Polymer-doxorubicin Nanoparticles for Efficient Anticancer Drug Delivery, J. Am. Chem. Soc., 2011, 133, 17560–17563, DOI:10.1021/ja207150n.
- E. Millerot-Serrurot, M. Guilbert, N. Fourre, W. Witkowski, G. Said, L. Van Gulick, C. Terryn, J. M. Zahm, R. Garnotel and P. Jeannesson, 3D Collagen Type I Matrix Inhibits the Antimigratory Effect of Doxorubicin, Cancer Cell Int., 2010, 10, 26, DOI:10.1186/1475-2867-10-26.
- H. G. Keizer, H. M. Pinedo, G. J. Schuurhuis and H. Joenje, Doxorubicin (Adriamycin): A Critical Review of Free Radical-Dependent Mechanisms of Cytotoxicity, Pharmacol. Ther., 1990, 47, 219–231, DOI:10.1016/0163-7258(90)90088-J.
- G. J. Bakker, V. Andresen, R. M. Hoffman and P. Friedl, Fluorescence Lifetime Microscopy of Tumor Cell Invasion, Drug Delivery, and Cytotoxicity, Methods Enzymol., 2012, 504, 109–125, DOI:10.1016/B978-0-12-391857-4.00005-7.
Footnotes |
| † Electronic supplementary information (ESI) available: 3 tables, 7 figures and a movie are supplied as ESI. See DOI: 10.1039/c3bm60120j |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2013 |