 Open Access Article
Open Access ArticleSize control of laser-fabricated surfactant-free gold nanoparticles with highly diluted electrolytes and their subsequent bioconjugation†
Christoph
Rehbock
,
Vivian
Merk
,
Lisa
Gamrad
,
René
Streubel
and
Stephan
Barcikowski
*
Technical Chemistry I, University of Duisburg-Essen and Center for NanoIntegration Duisburg-Essen CENIDE, Universtitaetsstrasse 7, 45141 Essen, Germany. E-mail: Stephan.Barcikowski@uni-due.de; Fax: +49 201 183 3049; Tel: +49 201 183 3150
First published on 3rd October 2012
Abstract
Size control of laser-fabricated surfactant-free gold nanoparticles is a challenging endeavor. In this work, we show that size control can be achieved by adding ions with low salinity during synthesis. In addition, this approach offers the opportunity to fundamentally study ion interactions with bare nanoparticle surfaces and can help to elucidate the nanoparticle formation mechanism. The studies were carried out in a flow-through reactor and in the presence of NaCl, NaBr and sodium phosphate buffer at minimal ionic strengths. A significant size quenching effect at ionic strengths from 1–50 μM was found, which allowed surfactant-free nanoparticle size control with average diameters of 6–30 nm. This effect went along with low polydispersity and minimal aggregation tendencies and was confirmed by UV-vis spectroscopy, TEM, SEM and analytical disk centrifugation. Our findings indicate that size quenching originates from an anionic electrostatic stabilization depending on the nanoparticle surface area, which may be caused by specific ion adsorption. By subsequent delayed bioconjugation in liquid-flow using bovine serum albumin as a stabilizing agent, nano-bioconjugates with good stability in cell culture media were obtained, which are applicable in toxicology and cell biology.
Introduction
As the application of nanoparticles (NPs) particularly in biology and medicine1 has significantly increased over the last decade, their impact on human health is of growing concern. Even though there are vast amounts of studies providing toxicity data on NPs, the absence of defined standards concerning surface dose, particle properties and cell type significantly limit their comparability.2–4 In order to fabricate suitable NP reference materials low material toxicity as well as controlled particle sizes are required. Gold is generally considered well suited as the bulk material is non-toxic and has been applied in medicine for decades.5–7 Size control of Au NPs can be easily done by chemical synthesis using artificial ligands.8,9 However, up to date these ligands cannot be quantitatively exchanged10 or removed11–13 and they drastically interfere with toxicity assays. This was, e.g. shown for citrate, which had a great influence on viability of alveolar cell lines14 and for the capping agent CTAB, which dramatically affected human skin keratinocytes.15 In this respect NP fabrication by pulsed laser ablation in liquid (PLAL) is a promising alternative method providing totally ligand-free nanoparticles.16 Even though the sizes of these particles can be altered to some extent by varying laser parameters17–19 or including successive laser fragmentation steps,20,21 proper size control during PLAL without organic additives is still difficult and particles tend to exhibit high polydispersity.16 To overcome these limitations, size control can be provided using inorganic salts instead of surfactants during synthesis. For silver NPs, a reduction in the NP size and size distribution in the presence of NaCl as well as a destabilization at higher concentrations is reported by several authors.22–24 A more extensive study reporting on effects of different ions on particle size and stability and giving insight into a potential formation mechanism is provided by Siskova et al.25 Prior work examining the effect of NaCl, KCl, NaOH, HCl and NaNO3 on Au NP formation and particle properties during laser ablation was published by Sylvestre et al.26 They reported a size reduction effect by OH− and Cl−, but only relatively high concentrations >1 mM were examined and the effect of different ionic strengths was not studied. In another study, Amendola and Meneghetti reported a size reduction effect in the presence of different NaCl concentrations allowing size control, even though the formation of small NPs <10 nm was not observed due to NP aggregation.27 Compared to prior works, where PLAL was performed in a batch reactor, we studied the salt effect in a flow-through reactor, where due to different local Au NP concentrations the size quenching occurs at 1000 times lower ionic strengths, so the danger of NP aggregation attributed to high ionic strengths is significantly decreased. Additionally, the effect of phosphate and especially the Cl− homologous halides F− and Br− on Au NP formation during PLAL has not been reported before and may shed light on the specific ion interactions on ligand-free Au NP surfaces.In order to apply Au NPs in biological media, where high ionic strengths are present, additional stabilizing agents are needed subsequent to the salt quenching during PLAL. As artificial ligands cannot be applied due to potential unknown or toxic cross-effects, only substances yet present in the cell culture medium may be used. In this work the serum protein albumin was chosen as a stabilizing agent due to its abundance in many cell culture media and biofluids. The interaction of Au NPs with proteins has been extensively studied for several years.28 It is generally believed that NPs exposed to serum containing media form a stable protein corona,29,30 which provides an enhanced electrostatic and sterical stability.31,32 The conjugation of laser-generated Au NPs with bovine serum albumin (BSA) has been reported elsewhere,33 although conjugation was performed ex situ, which always entails the risk of particle aggregation prior to conjugation. However, the use of in situ conjugation is evenly unbeneficial as proteins may be degraded by post-irradiation,34–36 which can result in degradation products that might have an unpredictable impact on toxicity. Hence, the delayed bioconjugation in liquid-flow, as successfully applied in conjugation experiments with oligonucleotides37 and peptides,38 was used in our study of NP-BSA conjugation.
Here Au NPs are fabricated by PLAL in a flow-through reactor and the different approaches used in this work are summarized in Fig. 1.
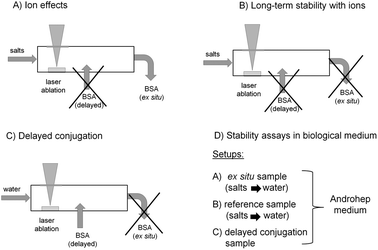 | ||
| Fig. 1 Experimental Au NP synthesis approaches for studying (A) ion effects, (B) long-term stability in the presence of ions, (C) delayed conjugation with BSA and (D) stability in Androhep biological medium. | ||
• The influence of different ions at variable salinities on average particle sizes and polydispersity was studied, while the obtained Au NPs were ex situ stabilized by BSA subsequent to formation (Fig. 1A).
• The long-term stability of the NPs in the presence of ions was checked, while in this case no subsequent stabilization was used (Fig. 1B).
• Using delayed conjugation in liquid-flow, the effect of both BSA concentration and biologically relevant ionic strengths on Au-BSA nanoconjugates was studied (Fig. 1C).
• Finally, the stability of these conjugates in Androhep biological medium was characterized in order to evaluate biological applicability. In this context ligand-free Au NPs as well as NPs stabilized by ex situ and delayed conjugation were tested (Fig. 1D).
Experimental
Material
Sodium chloride, sodium bromide, sodium fluoride, sodium phosphate, Hepes acid, disodium-EDTA, tri-sodiumcitrate-2-hydrate and BSA (≥98%, lyophilized powder) were purchased from Sigma Aldrich. Disodium hydrogen phosphate was obtained from Fluka, sodium dihydrogen phosphate was purchased from Merck and D(+)-glucose monohydrate and sodium hydrogen carbonate were acquired from AppliChem GmbH. All chemicals were used without further purification and dissolved in Milli-Q deionized water. The pH of the used phosphate buffer and the Androhep™ medium was adjusted using 1 M HCl and 1 M NaOH obtained from VWR. Sodium phosphate buffer pH = 7 (NaPP) at 50 mM concentration was made by dissolving 4.250 g of sodium dihydrogenphosphate and 2.711 g of disodium hydrogenphosphate in 1 L of Milli-Q water and was adjusted to pH = 7.Androhep medium was prepared by dissolving 8 g of tri-sodiumcitrate-2-hydrate, 2.4 g of disodium-EDTA, 2.5 g of BSA, 1.2 g of sodium hydrogen carbonate, 9.0 g of Hepes-acid and 26.0 g of D(+)-glucose monohydrate in 1 L of Milli-Q deionized water and adjusted to pH 7.
Methods
Nanoparticle synthesis via laser ablation in liquid was performed using a Nd:YAG ns-laser (Innolas SpitLight DPSS250-100) at λ = 1064 nm with a repetition rate of 100 Hz and a pulse energy of 45 mJ. The raw beam was expanded from 6 mm to 12 mm by an optical telescope and focused via a plano-convex lens (f = 50 mm). The experimental setup is illustrated in Fig. 2. The laser synthesis as well as ex situ and delayed bioconjugation were carried out in a self-designed flow-through chamber made of steel with a total reactor volume of 59 μL. Tubings (d = 3 mm) were connected to the chamber using infusion needles (B Braun). A carrier stream was used to continuously pump a solution through the chamber and a sample stream containing BSA solution was added during delayed conjugation. The streams were propelled by two syringe pumps (ProSense NE 300) each working at a flow rate of 1 mL min−1 resulting in a time delay of Δτ1 = 3 s for delayed conjugation. During ex situ experiments the sample stream was disconnected and the fabricated particles were continuously pumped into a glass vessel with 5 mL of a continuously stirred BSA solution resulting in a delay time of Δτ2 = 125 s. The ablated target was a gold wire (d = 125 μm, Allgemeine Gold) continuously fed via a motion control unit (Faulhaber GmbH, 3242G024BX4 CS 32A 124:1) allowing a continuous mass feed of 147 ± 7 μg min−1. The ablation time was 5 min and considering a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 dilution of the particles by the BSA solution in the sample container during ex situ conjugation or by the sample stream during delayed conjugation leads to a maximum NP concentration of 74 ± 4 μg mL−1.
:1 dilution of the particles by the BSA solution in the sample container during ex situ conjugation or by the sample stream during delayed conjugation leads to a maximum NP concentration of 74 ± 4 μg mL−1.
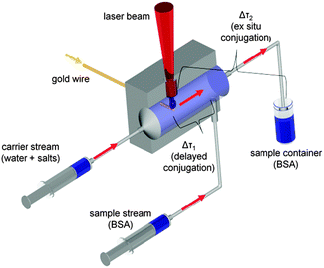 | ||
| Fig. 2 Experimental setup for laser ablation in liquid flow using delayed (Δτ1) and ex situ (Δτ2) conjugation. | ||
UV-vis spectroscopy measurements were carried out using a Thermo Scientific Evolution 201 and a Varian Cary 50 spectrometer in a glass cuvette with 10 mm path length and a volume of 1.5 mL covering a spectral range from λ = 190–900 nm.
Size determination with analytical disk centrifugation (ADC) was conducted with a DC 24000 from CPS instruments at 24000 rpm for 20 min against a saccharose gradient and an external standard (PVC particles at 0.371 μm), using a sample volume of 0.1 mL.
Scanning Electron Microscopy (SEM) measurements were carried out with a Quanta 400 FEG microscope. 5 μL of the sample were pipetted on a polished graphite carrier and dried overnight. Transmission Electron Microscopy (TEM) measurements were performed using a Philips CM 200 FEG on a carbon-coated copper grid. Size determination in both cases was done by averaging over 250–1000 gold NPs.
The mass distribution data obtained from ADC as well as the number distributions deduced from electron microscopy images were fitted with Origin 8.5.1 using a log-normal function. The average particle diameters were identified from the expected values (xc) while the polydispersity index (PDI) was calculated according to DIN ISO 1332139 as variance (σ2) divided by xc2.
Zeta-potential measurements were done with a Malvern Zetasizer “Nano ZS” in a disposable capillary cell with a volume of 750 μL and the pH was determined using a pH-meter PCE-PHD 1 with a suitable electrode.
The influence of NaCl, NaBr, NaF, Na2SO4, Na3PO4 and Na2HPO4/NaH2PO4 buffer (NaPP) at pH = 7 with ionic strengths from 1–2000 μM on nanoparticle properties was determined (Fig. 1A) using UV-vis spectroscopy and ADC as well as SEM, TEM and zeta-potential measurements for representative samples. The respective salt solutions were added to the carrier stream and ex situ stabilization of the obtained nanoparticles was done with 2.5 g L−1 BSA dissolved in 10 mM NaPP, pH = 7 after Δτ2 = 125 s. The concentrations of colloidal gold were determined by UV-vis spectroscopy using a calibration which correlates the ablated mass, gravimetrically determined, to the nanoparticle interband absorption at λ = 380 nm (Fig. S1, ESI†).
Long-term stability measurements were performed with samples fabricated in the presence of 3 μM NaCl, NaBr and NaF as well as with different ionic strengths of NaCl in the carrier stream (Fig. 1B). The unconjugated samples were measured with an UV-vis spectrometer in 20 min intervals for 700 min. For comparison, a BSA-stabilized sample as well as a sample fabricated in pure water were tested. For better comparability the gold concentrations (c), spectroscopically determined, were normalized by the initial concentrations (c0)
Delayed bioconjugation of the Au NPs was done by adding different concentrations of BSA in a range from 5 × 10−4 to 25 g L−1 dissolved in 10 μM and 50 μM NaPP buffer at pH = 7 to the sample stream (Fig. 1C). The carrier stream contained pure Milli-Q water, so the ablation itself was not influenced by ions in that study. The resulting bioconjugates were analyzed by UV-vis spectroscopy.
To evaluate the long-term stability of the bioconjugates in cell culture media a 5 mL sample was fabricated by delayed conjugation using a BSA concentration of 2.5 g L−1 at a buffer concentration of 10 mM. For comparison ligand-free NPs and NPs conjugated ex situ with 2.5 g L−1 of BSA were equally tested (Fig. 1D). The samples were added to 15 mL of Androhep medium and stored in a fridge at 4 °C. Stability was assessed by daily UV-vis spectroscopy measurements over a period of 28 days.
Results and discussion
Results
The averaged nanoparticle concentrations determined by the calibration curve (Fig. S1, ESI†) were 65 ±10 μg mL−1, which is close to the maximum concentration calculated by wire mass feed and volume flow. The variations in the ablated mass can be attributed to fluctuations in the speed of the used wire feeder and irregular flows in the ablation chamber. A significant dependence of the ablated mass on the ionic strength, as it was reported for silver targets by Siskova et al.,25 could not be found in our studies.Further information drawn from UV-vis spectra was the primary-particle-index (PPI). We define the PPI as the ratio between the interband absorption at λ = 380 nm and the scattering signal of aggregates, agglomerates and larger particles at λ = 800 nm to quantify the degree of aggregation of the NPs. A definition of the PPI is given in eqn (1). Accordingly, the PPI is inverse to the agglomeration quotient described earlier.40 Generally, a high PPI is attributed to a low proportion of aggregates and is considered beneficial for stable and less polydisperse colloids. Exemplary spectra illustrating this parameter can be found in Fig. S2 of the ESI.†
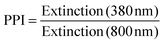 | (1) |
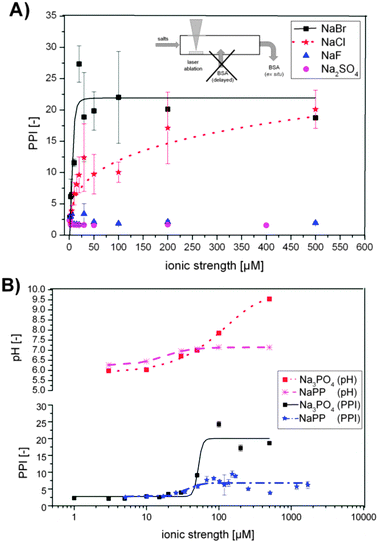 | ||
| Fig. 3 (A) Primary particle index (PPI) as a function of ionic strengths for NaCl, NaBr, NaF and NaSO4 (pH = 6 for all samples). (B) PPI for Na3PO4 and sodium phosphate buffer (NaPP) and respective pH plotted against ionic strengths. | ||
Na2SO4 and NaF show a destabilizing effect on the laser-ablated Au NPs over the entire concentration range, indicated by a purple color of the colloid solution (PPI of 2–4). The Au NPs precipitated within about 15–20 min after synthesis even in the presence of BSA as a stabilizing ligand. The pH = 6 of the electrolytes did not change significantly with concentration.
For NaCl a constant increase of the PPI from 2–20 is evident, while for NaBr the increase is much steeper and already reaches a maximum of PPI = 20 at 30 μM (Fig. 3A). This indicates a decrease in the concentration of aggregates, agglomerates and larger particles, which was confirmed by a color shift of the colloids from purple to deep red and went along with an increased stability. With these ions the pH remained constant at a level of 6 throughout the entire concentration range, which indicates that a specific ion effect is responsible for these trends.
The addition of NaPP and Na3PO4 caused an increase in the PPI with increasing ionic strength as well (Fig. 3B). For both salts a maximum was reached at an ionic strength of 50 μM, which was about 6 for NaPP and about 20 for Na3PO4. These huge differences in PPIs can be attributed to the pH of the carrier solution, which stagnated at pH 7 in the NaPP samples due to buffering while it increased further in the Na3PO4 samples. These findings indicate that higher pH levels lead to decreased aggregation and improved stability of the examined colloids.
The average particle diameters for NaCl, NaBr, Na3PO4 and NaPP determined by ADC as well as SEM data for NaPP are plotted in Fig. 4. Representative distributions from SEM and ADC can be found in Fig. S4 and S5 (ESI†). As all samples containing Na2SO4 and NaF spontaneously aggregated they were excluded from the size determination.
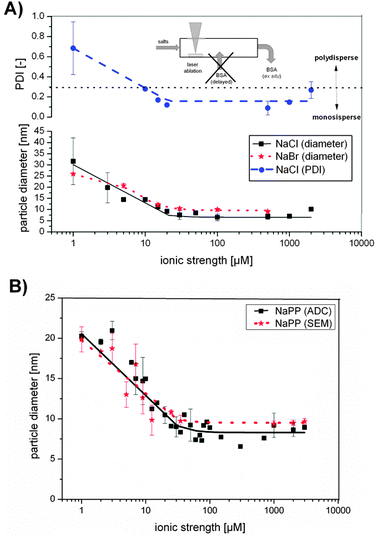 | ||
| Fig. 4 (A) Au NP diameter resulting from size quenching with NaCl or NaBr and PDI with different ionic strength. (B) Au NP diameter resulting from size quenching with sodium phosphate buffer (NaPP) determined by scanning electron microscopy (SEM) and analytical disk centrifugation (ADC) with different ionic strengths. | ||
Au NP sizes in the presence of NaCl and NaBr were significantly decreased with increasing ionic strength following a Hill function. Particle sizes in the case of NaBr decreased from 25 nm to 9 nm, while the PDI remained constant at 0.4 ± 0.06 (Fig. S6, ESI†). Using NaCl as size quencher, the particle size decreased from 30 nm at 3 μM ionic strength to 7 nm at 500 μM ionic strength and while the PDI decreased accordingly from 0.5 to 0.1. So for smaller NPs with average diameter about 7 nm, which were fabricated with ionic strengths >30 μM, monodisperse samples were obtained. However, the standard deviations with low NaCl concentrations were significantly high, indicating low reproducibility, which might be attributed to the impaired stability of the NaCl containing colloids in this concentration regime. For NaPP a similar Hill-fitted decrease of the nanoparticle size with increasing ionic strength was found for data obtained from ADC as well as from SEM, enabling size control in a size range from 20–7 nm. Na3PO4 samples followed a similar trend and can be found in Fig. S7 (ESI†).
Exemplary TEM images and the corresponding size distribution of four samples fabricated in the presence of NaPP (1 μM, 10 μM, 200 μM) and NaCl (100 μM) are presented in Fig. 5. The TEM images clearly confirm the size quenching effect and the determined mean particle sizes derived from ADC and SEM data. Additionally, it is clearly shown that the PDI significantly decreases with increasing ionic strengths as it was found for monodisperse samples (PDI = 0.05)40 for higher ionic strengths.
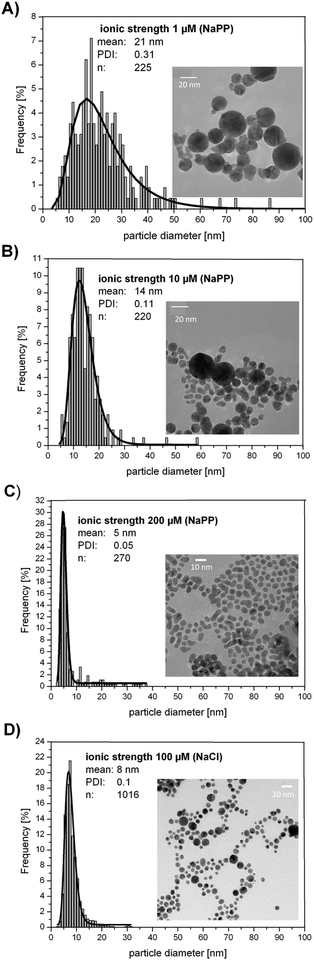 | ||
| Fig. 5 Representative TEM images of Au NPs fabricated in the presence of salts (A) 1 μM NaPP, (B) 10 μM NaPP, (C) 200 μM NaPP, (D) 100 μM NaCl. | ||
So, a significant size quenching effect was found for low ionic strengths (1–50 μM) using NaCl, NaBr and NaPP, which accessorily resulted in a decreased polydispersity at higher ionic strengths. As electrostatic stabilization and surface charges are most likely involved in this process, representative zeta-potential measurements were performed for NaCl in a concentration range from 3 μM–500 μM. The results showed no significant changes with the ionic strength and an average value of −41 ± 4 mV was found, which indicates a good particle stability. However, zeta-potential measurements are based on dynamic light scattering and hence only the surface charges of larger particles are detected. Additionally, smaller entities like gold atoms and clusters remain undetected by this method though they may still have a significant effect on particle stability. Thus, the long-term stability of freshly fabricated Au NPs was examined using UV-vis spectroscopy. The used setup for NP synthesis is illustrated in Fig. 1B and the results from these measurements are shown in Fig. 6. One concentration of NaF, NaCl and NaCl was tested and NaCl was further examined at different concentrations as it is the most relevant salt for applications in cell culture media.
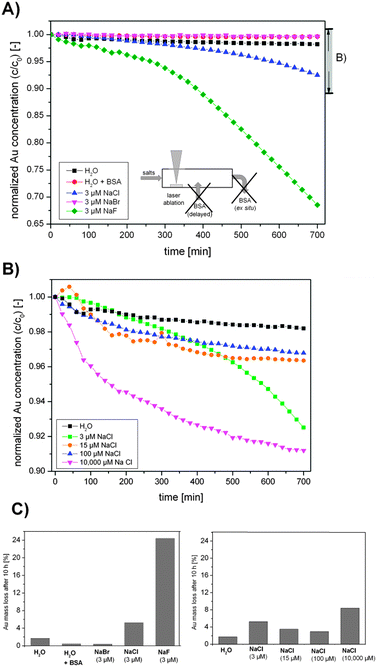 | ||
| Fig. 6 Long-term stability UV-vis measurements of Au NPs fabricated in the presence of the salts (A) NaF, NaCl and NaBr, (B) different concentrations of NaCl and (C) summary of the total Au mass loss after 10 h. Note the different scales in Figures (A) and (B). | ||
The results clearly indicate that particles synthesized in the presence of NaBr possess an improved stability compared to samples fabricated in pure water, which is equal to BSA-stabilized samples. NaCl containing samples showed a decreased stability (Au mass loss in one day = 5%) and for NaF the lowest stability was found (Au mass loss = 24%). The samples synthesized in the presence of different NaCl concentrations all had a decreased stability compared to water. However, low concentrations exhibited a low stability, which first increased up to a concentration of 100 μM (mass loss = 3%), while profoundly higher ionic strengths again decreased stability (mass loss = 8%). These findings clearly indicate that quenching with a biocompatible salt like NaCl decreases stability, though an optimum is reached in a range of 100 μM. However, since the fresh nanoparticles show very high quality in terms of ligand-free surfaces and narrow size distributions, stabilization is envisioned for application of these colloids. As the particles can be successfully stabilized using biocompatible BSA.
The results obtained from the delayed bioconjugation with Δτ1 = 3 s (see Fig. 1C and Fig. 2) of Au NPs synthesized in Milli-Q water with different concentrations of BSA and buffer concentrations of 10 and 50 mM in the sample stream are plotted in Fig. 7. Note that in this case the NaPP was not present during NP synthesis and was not applied for size quenching but was added along with the BSA to simulate salinities found in cell culture media and biological fluids. At low BSA concentrations the PPI remained at a low value, which can be attributed to significant aggregation and impaired stability. An increase in the BSA concentration goes along with higher PPIs until a maximum is reached at the minimal required stabilizing concentration of 0.02 g L−1 at 10 mM NaPP and 0.15 g L−1 at 50 mM NaPP (Fig. 7A). The differences can be attributed to the reduced stability of the colloid at high ionic strengths. At 10 mM NaPP an increase in BSA concentrations did not lead to further aggregation, while at 50 mM NaPP a significantly enhanced aggregation was found (Fig. 7B). Having achieved size-control and stabilization of the Au NPs obtained from PLAL their long-term stability was checked in an exemplary cell culture medium.
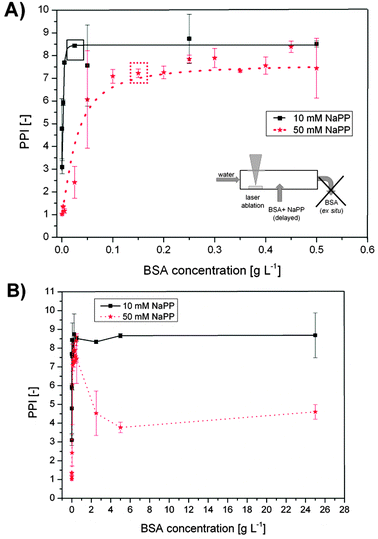 | ||
| Fig. 7 Stabilization of the primary particles obtained from PLA in water and delayed conjugation with BSA at different salinities and concentrations. The primary particle index (PPI) from UV-vis spectroscopy is plotted against BSA concentration NaPP concentrations of 10 mM and 50 mM. (A) Lower BSA concentrations, boxes indicate stabilizing concentrations. (B) Higher BSA concentrations. | ||
The Androhep medium contained citrate, glucose, disodium-EDTA, sodium hydrogen carbonate and Hepes-acid in high concentrations as well as 2.5 g L−1 BSA, which is roughly 17 times higher than the required stabilizing concentration. The reference samples containing unconjugated NPs and ex situ BSA-stabilized particles were not long-term stable in Androhep medium indicated by a considerable amount of black aggregates found after 10 days. In contrast, the samples fabricated by delayed conjugation showed a good long-term stability over a period of at least 28 days, which was confirmed by regular UV-vis measurements. The results are plotted in Fig. 8. The spectra reveal no significant increase in aggregation and no shift in the SPR-band, which may be attributed to a good long-term stability. Variations in the spectra are <5%, do not follow an obvious trend and may be caused by inaccuracies in sample preparation.
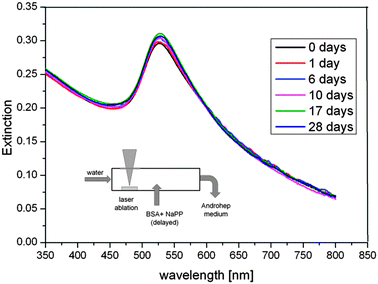 | ||
| Fig. 8 Long-term stability of the surfactant-free BSA-coated Au NPs in Androhep cell culture medium, determined by UV-vis measurements for a period of 28 days. | ||
Discussion
It was found that NPs fabricated by laser ablation in the presence of micromolar aqueous solutes of NaCl, NaBr, NaPP and Na2PO4 showed a considerablely reduced aggregation with increasing ionic strengths. To confirm this, an exemplary sample synthesized at 15 μM NaBr was chosen and the UV-vis spectrum was compared to a simulated spectrum based on a Mie-Gans fitting program previously described by Amendola and Meneghetti.41 The resulting spectra are shown in the ESI† (Fig. S3). The simulation data indicate a fraction of spheres of 100% confirming that no significant aggregation could be found under the used experimental conditions. Additionally, a significant size quenching effect was determined for NaCl, NaBr and NaPP and the polydispersity of the samples notably decreased at higher salt concentrations leading to monodisperse NPs with an average diameter of 5 nm. As the liquid medium is completely surfactant-free during the NP formation process, electrostatic stabilization of the inorganic precursors of the final NPs, namely small NPs and gold clusters, is the most probable cause for the observed effects. BSA is added with a time delay of 125 s and an interference with the NP formation mechanism was not observed. The exemplary zeta-potential measurements with NaCl could not confirm an increased surface charge with increasing ionic strength. However, these values might not be representative because the measurements are based on dynamic light scattering, which significantly overestimates the effect of larger particles and cannot monitor surface charges of small NPs and clusters.It is generally believed that during the laser formation of NPs an ultrafast nucleation process is followed by coalescence and particle growth.42 It is additionally known that nucleation and initial coalescence happen on a very short timescale, so the initial formation of small particles may not be influenced by ions or surfactants.43 This is backed by the fact that regardless of the present ionic strength, the minimum particle diameter never reached values below 5 nm in our present study. An increased surface charge density on the surface of these initial NPs may then stabilize them against further particle growth or coalescence. As the ionic strength decreases, the total surface area which can be stabilized by the present charges is diminished, leading to bigger NPs. Furthermore, a deficit of stabilizing charges may also result in an inhomogeneous distribution of the charges, stabilizing some particles better than others, which can cause uncontrolled growth that leads to a higher polydispersity. The proposed mechanism is illustrated in Fig. 9. It is based on the idea that during the growth of smaller NPs a competition between the present ions and the remaining gold precursors for the NP surface takes place. With increasing ionic strengths the surface of the NPs is totally blocked by the ions and further growth ceases. Additionally, a stabilizing interaction of the ions with the gold clusters may contribute to this growth quenching effect.
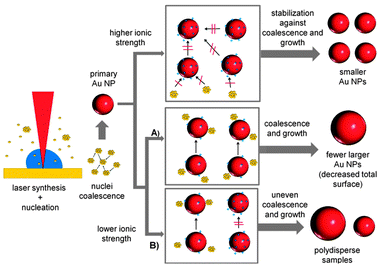 | ||
| Fig. 9 Proposed Au NP growth mechanism routes for PLAL in the presence of ions with low salinity. | ||
To verify the proposed mechanism, the total NP surface area found in a one mL sample is plotted against the amount of present NaCl, NaBr and NaPP in this volume. For NaPP an equivalent dependent on the ionic strength is used. The results found in the concentration regime up to 100 μM are shown in Fig. 10.
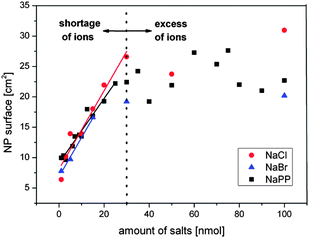 | ||
| Fig. 10 Verification of surface-driven growth quenching mechanism. Resulting NP surface in one mL of colloid after PLAL in saline water plotted against the amount of salts in the respective volume. | ||
These findings clearly show a linear increase of the particle surface with the amount of added salts up to an amount of 30 nmol for NaPP and NaCl and up to 20 nmol for NaBr which nicely correlates with the concentration regime where the size quenching effect takes place and where apparently a shortage of ions is present. From the slopes of the linear regime a respective stabilization amount can be calculated, which was 1.5 nmol cm−2 for NaCl, 1.6 nmol cm−2 for NaBr and 1.9 nmol cm−2 for NaPP, values equivalent to 1800–2290 anions per 8 nm nanoparticle. However, certain deviations from these values may be expected because average diameters were used for the calculations.
Having postulated a suitable mechanism for the salinity-driven size quenching effect itself, it is now necessary to discuss how the surface charge density of the NPs (which is believed to cause this effect) is altered by the ions. In this respect, we believe that a pH effect as well as specific ion interactions may be involved. It is generally assumed that the surface of laser-generated Au NPs is partially oxidized leading to the presence of Au+/Au3+ species. In aqueous solution these positive charges attract water molecules or dissolved O2, which results in the formation of OH/O− species.44,45 Hence, the surface charge is highly pH dependent, which seems to be responsible for the size quenching and stabilization effects attributed to NaPP and Na3PO4 (Fig. 3B). When the concentration of these salts is increased from 3–30 μM the pH increases from 6 to 7. This leads to a deprotonation of the OH groups on the Au NP surface and hence an increased NP surface charge. Accordingly, it goes along with an increase of the PPI in the UV-vis data and the observed size quenching. For higher ionic strengths >30 μM the NaPP buffers the pH at 7, which leads to a stagnation of the PPI while for Na3PO4 the pH continues to increase, which causes even higher surface charges and a corresponding increase of the PPI. This proposed mechanism is illustrated in Fig. 11A.
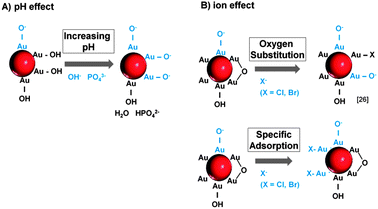 | ||
| Fig. 11 Mechanisms of surface charge generation on ligand-free Au NPs by (A) pH changes and (B) specific ion effects. | ||
For the other examined ions the pH remained constant around 6 over the entire concentration regime. So the ion effect can be studied independently of the pH. Interestingly, samples containing Na2SO4 and NaF showed no stabilizing or size quenching effect and caused immediate Au NP precipitation most likely due to the destabilizing effect of high ionic strengths. For NaCl and NaBr, however, a significant size quenching effect was found (Fig. 4A). As all the used salts contained Na+-ions, it can be assumed that the observed differences in the ion effects are attributed to the anions. We believe that these effects are due to specific interactions of Cl− and Br− with the NP surface. As 3.3–6.6% of the surface atoms on laser-generated particles are oxidized to Au-O44 the increased surface charge might partially result from an exchange of the surface OH groups with Cl− and Br−. This corresponds to findings made on gold nanoparticles obtained from gas phase synthesis46 and to the mechanism proposed by Sylvestre et al.26 for laser-generated NPs.
Our findings indicate that for a successful ion-induced stabilization of the surface of laser-fabricated Au NPs, an amount of at least 1500 pmol cm−2 is required. It is now interesting to compare this value to minimally required NP stabilizing concentrations of organic surfactants reported in the literature. For citrate on Ag NPs a value of 5 pmol cm−2 is found47 and for oligonucleotides on Au NPs a minimum surface coverage of 30 pmol cm−2 is reported.48 Accordingly, much higher ion concentrations are required compared to surfactants. This may result from possible sterical stabilization found in organic ligands and from the fact that the examined organic surfactants carry multiple surface charges that can contribute to NP stabilization. In comparison the amount of free charges on the surface of a laser-fabricated Au NPs is postulated to be 100 pmol cm−2(44), which is 15 times lower than the required amount of ions. This may be attributed to the experimental design used by Muto et al. where the charges were titrated with a bulky organic ligand, which might be able to neutralize more than one surface charge. Another suitable explanation arises from the fact that halides may also directly interact with uncharged gold surface forming monolayers. In our study the calculated footprint for a Cl− ion is 11 nm2, which indicates that no total surface coverage (no monolayer) is reached under stabilizing conditions. The effect of specific ion adsorption on gold surfaces was confirmed using AFM,49 X-ray diffraction50 and STM51 on gold electrodes. The adsorbed Cl− and Br− ions may then transfer their charges to the Au NPs and thus increase the surface charge density, an effect which was already observed for laser-generated Ag NPs in the presence of Br− and l−.52 The reason for this strong affinity of halides with higher ion-diameter (Cl− and Br−) compared to F− and SO42− might result from their larger size and better polarizability, which favors dative and covalent binding to evenly large gold atoms. Furthermore, the solvation of the ions might also play a key role and may be comparable to findings from gold electrodes interacting with different ions.53,54 SO42− and F− are known to be densely hydrated due to their high surface charge, which might hinder their interaction with the NP surface. The differences between Br− and Cl− in the UV-vis spectra, which indicated a much stronger increase of the PPI for Br− (Fig. 3A), may also be attributed to a higher affinity of the Br− to the NP surface caused by differences in solvation, size and polarizability. The proposed mechanism of surface charge generation by ions is illustrated in Fig. 11B.
Our findings basically confirm the size quenching effect in the presence of inorganic salts reported by Amendola and Meneghetti for NaCl.27 However, the ionic strength used in their study to fabricate 6 nm Au NPs was 100 mM, a concentration at which the particles immediately precipitated. In contrast, we found a size quenching effect at salt concentrations 1000 times lower. These differences may arise from higher ablation rates and higher NP concentrations used in their studies. Comparing the two used laser systems the productivity in the study of Amendola and Meneghetti may have been at most 10 times higher, so the huge differences in required ionic strengths for size quenching cannot be solely explained by this. The main differences may be attributed to concentration gradients rather than average concentration levels. Hence, the used batch ablation process compared to our flow-through approach might also play a significant role. When laser ablation is carried out in a liquid batch process there is insufficient mixing because of very high local concentrations close to the ablation site even during magnetic-stirring. This means a considerably higher salt concentration is required to induce the size quenching effect at the point of nanoparticle formation. However, at the same time, the salinity found a few millimetres away from the ablation site may be too high. Taking the ablation zone as a continuous NP source the Au concentration decrease would scale with z3 (z = distance from ablation site), leading to large concentration gradients (Fig. 12A). In a flow-through reactor the fabricated NPs are immediately transported away from the ablation site and diluted by the carrier stream leading to lower local Au NP concentrations and hence fewer total ions are required to induce size quenching (Fig. 12B).
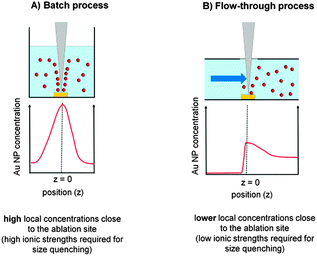 | ||
| Fig. 12 Local NP concentration gradients during batch and liquid flow PLAL. | ||
The results from the long-term stability measurements for NaCl (Fig. 6B) clearly show that the observed size quenching effect by salts does not necessarily increase stability. NP coagulation due to screening out of charges in the electrical double layer has to be considered. The trend showing lower stability for lower concentrations and higher stability for medium concentrations is most likely attributed to the fact that larger particles found in the low concentration regime coagulate faster than small ones. For higher concentrations the destabilizing effects are dominant. Optimal values were obtained at moderate ionic strengths (Fig. 6C). The long-term stability of different ions, however, revealed that Br− seems to be a potent stabilizing agent, while the other tested ions are not (Fig. 6B). This may be caused by a more efficient charge transfer from Br− to the NP surface due to stronger interactions57 though details on the underlying mechanism remain unclear. Even though the Au mass loss at a NaCl concentration of 3 μM after 10 hours is considerable (5%), the decrease in Au mass is negligible for times <1 hours. Hence, stabilization can be done up to one hour after synthesis. This clearly shows that the observed size quenching effect is not limited to fast conjugation systems described in this work but may be used for surfactant-free size control in various other PLAL applications in the future.
BSA conjugation of Au NPs by delayed conjugation in liquid flow clearly revealed a stabilization of the NPs at concentrations of 0.05 g L−1 at 10 mM NaPP and 0.15 g L−1 at 50 mM NaPP (Fig. 7A). At a NP diameter of 10 nm, a sample volume of 10 mL and a colloidal Au concentration of 65 μg mL−1 stabilization can be achieved with 209 BSA/NP (50 mM) and 69 BSA/NP (10 mM). We compared these findings to BSA/NP values found in monolayers on citrate-capped Au NPs and compensated for different particle sizes by only taking the relative surface areas into consideration. Dependent on the used methods, values from 20–40 BSA/NP were found.31,55 So compared to our findings we can assume a multilayer formation on the laser-fabricated NPs, which is typical for Au NP-protein conjugates.56 The increased amount of BSA molecules required for stabilization at higher ionic strengths can be attributed to the impaired stability of the NPs due to screening out of charges in the electrical double layer. This can be compensated by an excess of free BSA molecules, which has already been shown during coagulation experiments.57 However, much higher values of 600 NP/BSA were previously reported during PLAL and ex situ conjugation even though no destabilizing salts were used.33 Note that the residence time of delayed conjugation is far shorter than ex situ conjugation. These findings indicate that protein conjugation by delayed conjugation results in more stable conjugates, possibly due to a tighter interaction with freshly fabricated nanoparticles. This is backed by the long-term stability measurements in Androhep medium (Fig. 8), which indicate an improved stability of the samples generated by delayed conjugation. Additionally, we need to consider the overall negative charge of the BSA, which, besides dative bondings by cysteine residues and sterical stabilization, is most likely responsible for the interaction with the positively charged Au NP surface.31 This leads to an electrostatic repulsion between the BSA molecules, which stabilizes the NP-BSA conjugates. At higher ionic strengths, these surface charges are screened out by the adsorption of counterions.56 This results in a decreased surface charge and hence an impaired interaction with the NP surface and aggregation is favored at higher BSA concentrations due to attractive forces between the BSA molecules. This concept is illustrated in Fig. 13.
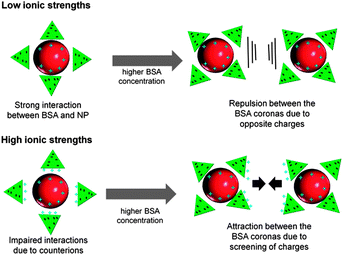 | ||
| Fig. 13 Proposed mechanism showing the influence of ionic strength and protein concentration on the interactions of Au NPs fabricated by PLAL with BSA. | ||
Conclusion
NP fabrication by PLAL offers the opportunity to fabricate totally ligand-free colloids not available by chemical methods. However, size control, polydisperse size distributions and reduced stability in media with high salinities still limit their widespread use. In this work we demonstrated that Au NPs synthesized by PLAL are significantly affected by the presence of Cl−, Br−, PO43− and HPO42−/H2PO4−. Using an advanced flow-through reactor we demonstrated that ionic strengths approximately 100–1000 times lower than those used in other studies triggered a significant surfactant-free size quenching effect. This effect allowed size control in a regime of 6–30 nm and corresponded to a considerable decrease of polydispersity and an increase of the primary-particle-index. This resulted, e.g. in monodisperse NPs with an average size of 6 nm and a polydispersity-index (PDI) of 0.05. Additionally, this approach offered the unique opportunity to study the interaction of ligand-free Au NP surfaces with ions. A suitable mechanism based on electrostatic stabilization by delivery of additional surface charges was proposed and a linear correlation between the stabilized NP surface and the available ions was found. The slopes of these graphs were in a range from 1–2 μmol cm−2 and may be used to calculate the concentrations required for stabilizing a distinct nanoparticle surface by the respective ions. Additionally, a mechanism for surface charge generation based on specific ion adsorption was proposed. Biofunctionalization by delayed conjugation with the protein BSA provided long-term stable conjugates and a significant influence of ionic strengths present during conjugation was demonstrated. Based on this work it is now possible to fabricate stable NP–protein conjugates with controlled particle sizes without the use of artificial surfactants. These species are particularly useful for applications in toxicology and biology where high purity is of paramount importance, for example in reproduction biology.4,7Acknowledgements
We thank the “Deutsche Forschungsgemeinschaft (DFG)” for funding with the priority program 1313, the group of Prof. Epple, Inorganic Chemistry, University of Duisburg Essen, Germany for help with analytical disk centrifuge and SEM measurements and especially Peter Hinze and Dr Ingo Busch from “Physikalisch Technische Bundesanstalt (PTB)” Braunschweig, Germany for support with the TEM measurements.References
- D. A. Giljohann, D. S. Seferos, W. L. Daniel, M. D. Massich, P. C. Patel and C. A. Mirkin, Angew. Chem., Int. Ed., 2010, 49, 3280–3294 CrossRef CAS
.
- S. Sharifi, S. Behzadi, S. Laurent, M. L. Forrest, P. Stroeve and M. Mahmoudi, Chem. Soc. Rev., 2012, 41, 2323–2343 RSC
- Y. Teow, P. V. Asharani, M. P. Hande and S. Valiyaveettil, Chem. Commun., 2011, 47, 7025–7038 RSC
-
U. Taylor, A. Barchanski, W. Garrels, W. Kues, S. Barcikowski and D. Rath, in Nano-Biotechnology for Biomedical and Diagnostic Research, ed. E. Zahavy, A. Ordentlich, S. Yitzhaki and A. Shafferman, Springer Dordrecht, Heidelberg, London, New-York, 2012, ch. 12, pp. 125–133 Search PubMed
- A. E. Finkelstein, D. T. Walz, V. Batista, M. Mizraji, F. Roisman and A. Misher, Ann. Rheum. Dis., 1976, 35, 251–257 CrossRef CAS
- L. Dykman and N. Khlebtsov, Chem. Soc. Rev., 2012, 41, 2256–2282 RSC
- A. Barchanski, U. Taylor, S. Klein, S. Petersen, D. Rath and S. Barcikowski, Reprod. Domest. Anim., 2012, 46, 42–52 CrossRef
- R. Sardar, A. M. Funston, P. Mulvaney and R. W. Murray, Langmuir, 2009, 25, 13840–13851 CrossRef CAS
- B. L. Cushing, V. L. Kolesnichenko and C. J. O'Connor, Chem. Rev., 2004, 104, 3893–3946 CrossRef CAS
- A. Rostek, D. Mahl and M. Epple, J. Nanopart. Res., 2011, 13, 4809–4814 CrossRef CAS
- N. Watanabe and S. Fujihara, Ind. Eng. Chem. Res., 2011, 50, 5611–5617 CrossRef CAS
- S. K. Balasubramanian, L. Yang, L.-Y. L. Yung, C.-N. Ong, W.-Y. Ong and L. E. Yu, Biomaterials, 2010, 31, 9023–9030 CrossRef CAS
- J. A. Lopez-Sanchez, N. Dimitratos, C. Hammond, G. L. Brett, L. Kesavan, S. White, P. Miedziak, R. Tiruvalam, R. L. Jenkins, A. F. Carley, D. Knight, C. J. Kiely and G. J. Hutchings, Nat. Chem., 2011, 3, 551–556 CrossRef CAS
- C. Uboldi, D. Bonacchi, G. Lorenzi, M. I. Hermanns, C. Pohl, G. Baldi, R. E. Unger and C. J. Kirkpatrick, Part. Fibre Toxicol., 2009, 6, 18 CrossRef
- S. Wang, W. Lu, O. Tovmachenko, U. S. Rai, H. Yu and P. C. Ray, Chem. Phys. Lett., 2008, 463, 145–149 CrossRef CAS
- N. G. Semaltianos, Crit. Rev. Solid State Mater. Sci., 2010, 35, 105–124 CrossRef CAS
- W. T. Nichols, T. Sasaki and N. Koshizaki, J. Appl. Phys., 2006, 100, 6 Search PubMed
- A. V. Kabashin and M. Meunier, J. Appl. Phys., 2003, 94, 7941–7943 CrossRef CAS
- T. Tsuji, K. Iryo, N. Watanabe and M. Tsuji, Appl. Surf. Sci., 2002, 202, 80–85 CrossRef CAS
- P. V. Kazakevich, A. V. Simakin, G. A. Shafeev, G. Viau, Y. Soumare and F. Bozon-Verduraz, Appl. Surf. Sci., 2007, 253, 7831–7834 CrossRef CAS
- D. Werner, A. Furube, T. Okamoto and S. Hashimoto, J. Phys. Chem. C, 2011, 115, 8503–8512 CAS
- M. Prochazka, P. Mojzes, J. Stepanek, B. Vlckova and P. Y. Turpin, Anal. Chem., 1997, 69, 5103–5108 CrossRef CAS
- C. H. Bae, S. H. Nam and S. M. Park, Appl. Surf. Sci., 2002, 197, 628–634 CrossRef
- I. Srnova, M. Prochazka, B. Vlckova, J. Stepanek and P. Maly, Langmuir, 1998, 14, 4666–4670 CrossRef CAS
- K. Siskova, B. Vlckova, P. Y. Turpin and C. Fayet, J. Phys. Chem. C, 2008, 112, 4435–4443 CAS
- J. P. Sylvestre, S. Poulin, A. V. Kabashin, E. Sacher, M. Meunier and J. H. T. Luong, J. Phys. Chem. B, 2004, 108, 16864–16869 CrossRef CAS
- V. Amendola and M. Meneghetti, Phys. Chem. Chem. Phys., 2009, 11, 3805–3821 RSC
- M. Mahmoudi, I. Lynch, M. R. Ejtehadi, M. P. Monopoli, F. B. Bombelli and S. Laurent, Chem. Rev., 2011, 111, 5610–5637 CrossRef CAS
- T. Cedervall, I. Lynch, S. Lindman, T. Berggard, E. Thulin, H. Nilsson, K. A. Dawson and S. Linse, Proc. Natl. Acad. Sci. U. S. A., 2007, 104, 2050–2055 CrossRef CAS
- S. H. D. P. Lacerda, J. J. Park, C. Meuse, D. Pristinski, M. L. Becker, A. Karim and J. F. Douglas, ACS Nano, 2010, 4, 365–379 CrossRef
- S. H. Brewer, W. R. Glomm, M. C. Johnson, M. K. Knag and S. Franzen, Langmuir, 2005, 21, 9303–9307 CrossRef CAS
- K. Chen, Y. Xu, S. Rana, O. R. Miranda, P. L. Dubin, V. M. Rotello, L. Sun and X. Guo, Biomacromolecules, 2011, 12, 2552–2561 CrossRef CAS
- V. Amendola and M. Meneghetti, J. Mater. Chem., 2007, 17, 4705–4710 RSC
- Y. Takeda, F. Mafune and T. Kondow, J. Phys. Chem. C, 2009, 113, 5027–5030 CAS
- S. Petersen and S. Barcikowski, Adv. Funct. Mater., 2009, 19, 1167–1172 CrossRef CAS
- S. Petersen, J. Jakobi and S. Barcikowski, Appl. Surf. Sci., 2009, 255, 5435–5438 CrossRef CAS
- C. L. Sajti, S. Petersen, A. Menendez-Manjon and S. Barcikowski, Appl. Phys. A: Mater. Sci. Process., 2010, 101, 259–264 CrossRef
- C. L. Sajti, A. Barchanski, P. Wagener, S. Klein and S. Barcikowski, J. Phys. Chem. C, 2011, 115, 5094–5101 CAS
- Particle size analysis-photon correlation spectroscopy (ISO 13321:1996).
- N. Baersch, J. Jakobi, S. Weiler and S. Barcikowski, Nanotechnology, 2009, 20, 445603–445612 CrossRef
- V. Amendola and M. Meneghetti, J. Phys. Chem. C, 2009, 113, 4277–4285 CAS
- G. W. Yang, Prog. Mater. Sci., 2007, 52, 648–698 CrossRef CAS
- F. Mafune, J. Kohno, Y. Takeda, T. Kondow and H. Sawabe, J. Phys. Chem. B, 2000, 104, 9111–9117 CrossRef CAS
- H. Muto, K. Yamada, K. Miyajima and F. Mafune, J. Phys. Chem. C, 2007, 111, 17221–17226 CAS
- J. P. Sylvestre, A. V. Kabashin, E. Sacher, M. Meunier and J. H. T. Luong, J. Am. Chem. Soc., 2004, 126, 7176–7177 CrossRef CAS
- D. W. Thompson and I. R. Collins, J. Colloid Interface Sci., 1992, 152, 197–204 CrossRef CAS
- P. Wagener, A. Schwenke and S. Barcikowski, Langmuir, 2012, 28, 6132–6140 CrossRef CAS
- S. Petersen and S. Barcikowski, J. Phys. Chem. C, 2009, 113, 19830–19835 CAS
- S. Biggs, P. Mulvaney, C. F. Zukoski and F. Grieser, J. Am. Chem. Soc., 1994, 116, 9150–9157 CrossRef CAS
- O. M. Magnussen, B. M. Ocko, R. R. Adzic and J. X. Wang, Phys. Rev. B: Condens. Matter Mater. Phys., 1995, 51, 5510–5513 CrossRef CAS
- O. M. Magnussen, B. M. Ocko, J. X. Wang and R. R. Adzic, J. Phys. Chem., 1996, 100, 5500–5508 CrossRef CAS
- M. S. Sibbald, G. Chumanov and T. M. Cotton, J. Phys. Chem., 1996, 100, 4672–4678 CrossRef CAS
- D. M. Kolb and C. Franke, Appl.Phys. A: Mater. Sci. Process., 1989, 49, 379–387 CrossRef
- P. Gao and M. J. Weaver, J. Phys. Chem., 1986, 90, 4057–4063 CrossRef CAS
- H. Xie, A. G. Tkachenko, W. R. Glomm, J. A. Ryan, M. K. Brennaman, J. M. Papanikolas, S. Franzen and D. L. Feldheim, Anal. Chem., 2003, 75, 5797–5805 CrossRef CAS
- E. Casals, T. Pfaller, A. Duschl, G. J. Oostingh and V. Puntes, ACS Nano, 2010, 4, 3623–3632 CrossRef CAS
- A. Forough, D. Bahram and Z. A. Farzin, Korean J. Chem. Eng., 2010, 28, 1304–1310 CrossRef
Footnote |
| † Electronic supplementary information (ESI) available: Containing a calibration for determining gold concentrations from UV-vis spectra, data illustrating the primary-particle-index, measured and simulated spectra under optimum synthesis conditions, exemplary size distributions from SEM and analytical disk centrifugation, additional data on particle diameters of gold nanoparticles fabricated in the presence of Na3PO4 and polydispersity index (PDI) data of gold nanoparticles generated in the presence of NaBr. See DOI: 10.1039/c2cp42641b |
| This journal is © the Owner Societies 2013 |