Does the real ReN2 have the MoS2 structure?†
Received
29th August 2012
, Accepted 22nd October 2012
First published on 23rd October 2012
Abstract
Rhenium nitride (ReN2) with the hexagonal MoS2 structure was recently synthesized by metathesis reaction under high pressure. Here the calculated elastic and thermodynamic stabilities and chemical bonding show that the MoS2 phase is unstable based on first-principles calculations. Meanwhile, the MoS2-type ReN2 compound may be stabilized by nitrogen-vacancies from X-ray diffraction and supercell calculations. Structure searches identify a monoclinic C2/m phase for ReN2, which is energetically more stable than previous predictions and MoS2 structure over a wide range of pressures. Above 130 GPa, a tetragonal P4/mbm phase becomes favorable from enthalpy calculations. Both phases have superior mechanical properties, and their syntheses would have important applications fundamentally and technologically.
Introduction
Binary transition metal (TM) nitrides have received increasing interest due to their fascinating properties, such as high thermal and chemical stability, electrical or thermal conductivity, catalytic activity, and superior hardness.1–5 Most of the early TM nitrides (N : TM = 1) belong to the NaCl structure and can be synthesized at ambient conditions, but N-rich nitrides (N : TM > 1) are quite difficult to obtain due to the requirement of a relatively higher nitrogen pressure during synthesis.6 Taking advantage of high-pressure techniques, bulk M3N4 (M = Zr, Hf) with high elastic moduli and hardness has been obtained, opening new avenues for the synthesis of other TM nitrides.7 In 2004, the successful synthesis of platinum nitride (PtN2), which could be used as an alternative to ruby in high pressure experiments due to its structural stability at extremes, stimulated considerable research enthusiasm for 5d TM nitrides.8 Subsequently, osmium and iridium nitrides (OsN2 and IrN2) were synthesized.9,10 These nitrides exhibit intriguing electronic properties and remarkable bulk moduli (B), rendering them as potential hard materials. Recently, Re3N, and Re2N were produced under high pressure high temperature (HPHT) conditions.11 Their bulk moduli (395 GPa for Re3N, 401 GPa for Re2N) are comparable to diamond, making them excellent ultra-incompressible materials, but no experimental evidence supports the existence of other nitrides (e.g. ReN2). Very recently, ReN2 was synthesized by metathesis reaction under high pressure.12 The recovered product at ambient pressure was determined by X-ray Diffraction (XRD) to be ReN2 with the MoS2 structure (space group: P63/mmc, denoted as m-ReN2). The obtained B of this m-ReN2, 173 GPa, is much lower than that of ReB2 (360 GPa) although both structures are so similar to each other. Such a low B value is unusual since the relatively strong covalent bonding between rhenium and nitrogen atoms may enhance the resistance against uniform compression. To address this issue, via first-principles calculations, in this paper we systematically investigated the elastic and thermodynamic stabilities, XRD spectra, and chemical bonding of m-ReN2. The results show that m-ReN2 is unstable. Moreover, calculated XRD and density of states (DOS) hint at the presence of nitrogen-vacancies in m-ReN2. Further structural searching identifies a possible ground state structure and a high-pressure phase for ReN2. Both phases are found to exhibit excellent mechanical properties. The present results could provide a theoretical guidance for the experimental structural redetermination and characterizations of the physical properties of ReN2 for potential technological applications.
Computational method
The structural search for ReN2 is performed using CALYPSO (Crystal structure AnaLYsis by Particle Swarm Optimization) code13 based on a global minimization of free energy surfaces merging ab initio total-energy calculations via PSO technique. The simulation cells for ReN2 are examined at 0 and 50 GPa, within four formula cells (f.u.). Furthermore, the experimentally obtained MoS2-type structure and competing phases based on neighboring and similar compounds, such as OsN2, IrN2, ReB2, WP2 and ReP2, together with previous predictions for ReN2 and WN2 are considered by comparison of their relative stabilities.9,10,14–20
The obtained candidates for ReN2 phase are fully optimized within the framework of density functional theory (DFT) as implemented by the CASTEP package.21 The exchange and correlation functional was treated by the generalized gradient approximation of Perdew–Burke–Ernzerhof (GGA-PBE).22 A 10 × 10 × 10 k-point and cutoff energy of 400 eV were adopted to guarantee high accuracy for both total energy and physical properties calculations. The heat of formation (ΔHf) for candidate ReN2 phases was calculated by the equation of ΔHf = Etotal(ReN2) – (Etotal(Re) + 2Etotal(N)), in which hexagonal Re and a-N2 were adopted for the calculations. Previously synthesized marcasite OsN2, arsenopyrite IrN2, Re2N, and Re3N were also calculated for comparison. Crystal orbital Hamilton populations (COHP) for present nitrides were calculated by the tight-binding linear-muffin-tin orbital calculations with the atomic sphere approximation (TB-LMTO-ASA).23
Results and discussion
The calculated equilibrium lattice parameters for m-ReN2, together with previous experimental results obtained for OsN2, IrN2, Re2N, and Re3N, are presented in Table 1. It can be observed that the lattice parameters are reasonably well reproduced for OsN2, IrN2, Re2N, and Re3N within a maximum error of 1.1%.11,24–28 While for m-ReN2, relatively larger deviations from the experimental lattice parameters are found in both a and c directions (2.1% and 6.2%, respectively).12 Meanwhile, the obtained N–N distance (2.416 Å) in m-ReN2 is much greater than that of the neighboring nitrides (1.438 Å in OsN2 and 1.422 Å in IrN2). Moreover, from calculated elastic constants (Cij), m-ReN2, either unrelaxed or relaxed, is found to be mechanically unstable due to the negative C44 value (−15 and −34 GPa, respectively) according to the Born–Huang criterion for hexagonal structure.29 This conflicting fact between the current calculations and experimental results motivated us to explore its reason and search for a possible ground state structure for ReN2.
Table 1 Calculated equilibrium lattice parameters, a (Å), b (Å), and c (Å), for m-ReN2, OsN2, IrN2, Re2N, and Re3N, with available results for comparison
| |
a
|
b
|
c
|
|
Ref. 12.
Ref. 24.
Ref. 25.
Ref. 26.
Ref. 27.
Ref. 11.
Ref. 28.
|
|
m-ReN2 |
GGA |
2.866 |
|
10.076 |
| Expa |
2.806 |
|
10.747 |
| OsN2 |
GGA |
2.694 |
4.940 |
4.140 |
| GGAb |
2.682 |
4.915 |
4.121 |
| GGAc |
2.70 |
4.93 |
4.13 |
| IrN2 |
GGA |
4.903 |
4.982 |
4.972 |
| GGAd |
4.884 |
4.939 |
4.846 |
| GGAe |
4.876 |
4.926 |
4.917 |
| Re2N |
GGA |
2.854 |
|
9.854 |
| Expf |
2.83 |
|
9.88 |
| GGAg |
2.837 |
|
9.799 |
| Re3N |
GGA |
2.807 |
|
7.167 |
| Expf |
2.78 |
|
7.15 |
| GGAg |
2.8065 |
|
7.112 |
We started the exploration firstly by investigating the effect of nitrogen (N) vacancy on the structural stability of m-ReN2. The XRD spectra of stoichiometric and substoichiometric m-ReN2 are simulated and compared with the obtained pattern in experiment (Fig. 1). Unexpectedly, the obtained diffraction peaks of N-deficient m-ReN2 (ReN1.75 (Re8N14)) match well with the experimental XRD. This substoichiometry might account for its structural instability. Meanwhile, the DOS of this m-ReN2 is calculated to further explore its stability. Other types of impurities or defects, such as oxygen and platinum from capsule material, are also included. The calculated DOS for N-deficient Re8N15 (ReN1.875), Re8N14 (ReN1.75), oxygen-doped Re8N15O (ReN2−xOx, x = 0.125), and platinum-doped Re7PtN16 (Re1−xPtxN2, x = 0.125) are plotted in Fig. 2 based on supercell calculations. All these phases exhibit metallic character because of the nonzero value at the Fermi level (EF). Additionally, the values at Fermi level (N(EF), eV state−1 atom−1) follow a sequence: Re8N14 (0.424) < Re8N15 (0.5076) < Re7PtN16 (0.5834) < Re8N15O (0.7755) < ReN2 (0.8249). Considering the general correlation between stability and the value at the Fermi level in the same metallic system, the presence of vacancies and impurities tend to stabilize the m-ReN2. Specifically, N-deficient m-ReN2 is most stable because of its lowest N(EF) value. Thus, the observed MoS2-structured ReN2 in the previous experiment could be stabilized by the presence of nitrogen vacancies.
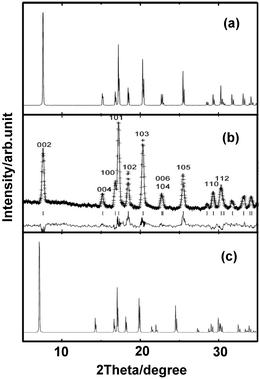 |
| | Fig. 1 Simulated XRD curves for (a) ReN1.75 (Re8N14), (b) experimental result reproduced from ref. 7, and (c) m-ReN2. | |
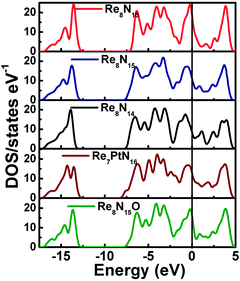 |
| | Fig. 2 Calculated total density of states (DOS) for m-ReN2 (Re8N16), N-deficient ReN1.875 (Re8N15) and ReN1.75 (Re8N14), platinum-doped Re1−xPtxN2 (x = 0.125, Re7PtN16), and oxygen-doped ReN2−xOx (x = 0.125, Re8N15O). | |
We now turn our attention to explore the real structure for ReN2. ΔHf calculations (Fig. 3) uncover a monoclinic structure with space group C2/m for ReN2 (C-ReN2), whose total energy is lower than m-ReN2 and other considered structures. Moreover, the positive ΔHf value suggests that m-ReN2 is thermodynamically instable towards dissociation into its constituent elements, although it may be stabilized kinetically. Also, the possibilities of P42/mnm17 and Pbcn18 phases as the ground state structure are rigorously ruled out in this study. In contrast to m-ReN2, C-ReN2 phase is found to be thermodynamically stable with a negative ΔHf value, −0.148 eV per atom, close to previously obtained experimental values for Re3N (−0.223 eV per atom) and Re2N (−0.073 eV per atom),11 indicating that this nitride could be obtained at moderate pressure. In C-ReN2 phase, shown in Fig. 4, the Re atom is surrounded by seven nearest-neighbor nitrogen atoms with a distorted mono-capped-trigonal-prismatic geometry, different from the six-fold coordination of OsN2 and IrN2 that are composed of MN6 polyhedra. Two types of nitrogen exist: one type forms an N2 dimer located at the center of a trigonal-prism of six Re atoms, similar to OsN2 and IrN2;25,26 the other type forming zigzag chains with Re atoms sitting at the center of a distorted tetrahedron formed by one N and three Re atoms. Furthermore, four denser phases, P4/mbm-, P4/mmm-, Cmc21-, and P21-type structures, are identified as potential high-pressure phases for C-ReN2. The calculated enthalpy–pressure curve with respect to C-ReN2 phase is given in Fig. 5. The C2/m phase is stable up to 130.2 GPa, and then transforms into the P4/mbm structure (P-ReN2). The P4/mmm-type structure suggested in an earlier study19 was found to be unfavorable over a wide range of pressures. Also, P-ReN2 is favored over Re + N2, 1/3Re3N + 5/6N2, and 1/2Re2N + 3/4N2 above 11.5, 17.5, and 19.0 GPa, respectively, which may provide information for the synthesis of this high-pressure phase of ReN2.
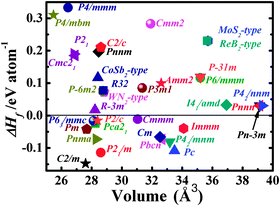 |
| | Fig. 3 The calculated heat of formation (ΔHf) and equilibrium volumes for searched ReN2 phases (Detailed crystal information of the searched phases could be found in the ESI†). | |
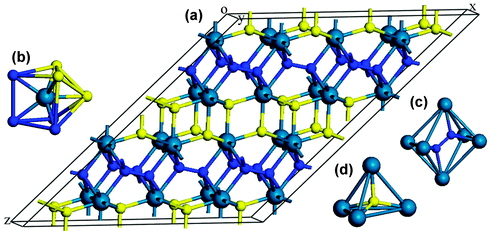 |
| | Fig. 4 Crystal structure of C-ReN2: 3D view (a), coordinative environment of Re atom (b) and two kinds of N atom ((c) and (d)). The predicted C-ReN2 adopts a C2/m symmetry (No. 12), where Re atoms occupy the 4i (0.406, 0.5, 0.231), N atoms occupy 4i (0.009, 0.5, 0.136) and 4i (0.432, 0, 0.530) positions. The structural parameters are optimized to be a = 6.817 Å, b = 2.835 Å, c = 9.363 Å, and β = 142, 40°. | |
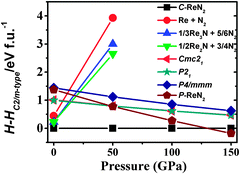 |
| | Fig. 5 Calculated enthalpy differences of possible constituents (Re + N2, 1/3Re3N + 5/6N2, and 1/2Re2N + 3/4N2) and four possible high-pressure phases (Cmc21-, P21-, P4/mmm-type, and P-ReN2) with respect to C-ReN2 phases as a function of pressure at zero temperature. The P-ReN2 is predicted with a P4/mbm symmetry (No. 127), where Re atoms occupy 2b (0, 0, 0.5), N atoms occupy 4g (0.617, 0.117, 0) positions. The optimized structural parameters are a = 4.390 Å and c = 2.644 Å. | |
On the basis of Cij calculations summarized in Table 2, we can check the local stability for C-ReN2 and P-ReN2. C-ReN2 and P-ReN2 exhibit not only mechanical stability, but also greater bulk moduli, against the experimentally obtained m-ReN2. The obtained bulk modulus (B) of C-ReN2 (369 GPa), is slightly greater than those of OsN2 (353 GPa) and IrN2 (333 GPa), but smaller than those of Re-rich nitrides (Re2N (396 GPa) and Re3N (392 GPa)). However, the shear moduli (G) of C-ReN2 and Re-rich nitrides are quite close to each other. P-ReN2 exhibits a quite high G (297 GPa), greater than OsN2 (222 GPa) and IrN2 (205 GPa) by 33.8% and 44.9%, respectively. In view of the greater B and G of P-ReN2 (and C-ReN2), ReN2 could be a hard material for industrial coating applications. Furthermore, P-ReN2 possesses a remarkable C33, 967 GPa, comparable to diamond (1079 GPa),30 revealing the extremely high stiffness along the c-direction. Moreover, the elastic anisotropy, highly correlated with the possibility of the appearance of microcracks and thus significantly affecting the application of a material, is also evaluated based on the universal elastic anisotropy index of AU = 5GV/GR + BV/BR − 6,31 where B and G denote the bulk and shear moduli, and the superscripts V and R represent the Voight and Reuss approximations. The calculations demonstrate that the two predicted ReN2 phases, especially for P-ReN2, have superior elastic isotropic character as indicated by their smaller AU values in comparison with neighboring OsN2 and IrN2.31
Table 2 Calculated single crystal elastic constants Cij (GPa), bulk modulus B (GPa), shear modulus G (GPa), and universal elastic anisotropy (AU) for m-ReN2, OsN2, IrN2, Re2N, and Re3N
| |
C
11
|
C
22
|
C
33
|
C
44
|
C
55
|
C
66
|
C
12
|
C
13
|
C
23
|
B
|
G
|
A
U
|
|
m-ReN2 |
530 |
|
9 |
−34 |
|
|
180 |
15 |
|
87 |
|
|
|
C-ReN2 |
843 |
546 |
652 |
201 |
252 |
210 |
209 |
211 |
256 |
369 |
217 |
0.333 |
|
P-ReN2 |
820 |
|
967 |
258 |
|
252 |
165 |
111 |
|
374 |
297 |
0.183 |
| OsN2 |
772 |
869 |
821 |
167 |
167 |
153 |
72 |
140 |
146 |
353 |
222 |
0.770 |
| IrN2 |
704 |
791 |
754 |
163 |
174 |
141 |
101 |
158 |
118 |
333 |
205 |
0.774 |
| Re2N |
660 |
|
813 |
182 |
|
|
207 |
261 |
|
396 |
210 |
0.101 |
| Re3N |
656 |
|
791 |
194 |
|
|
227 |
247 |
|
392 |
212 |
0.055 |
To further understand the underlying origin of the stability, mechanical properties and chemical bonding, DOS of the ‘isolated’ Re metal and ‘isolated’ N of different positions in the C-ReN2-type structure, and C-ReN2 were calculated as shown in Fig. 6. From Fig. 6(a), a striking feature of DOS is the totally different contributions of different isolated N and Re atoms, the N atom in the N2 dimer (N1) opens a significant gap, whereas the N atom in zigzag Re–N chain (N2) and Re atom strongly localize at the Fermi level. But in real C-ReN2, the presence of direct Re–N and N–N interactions in different coordination environments leads to a profound modification of the electronic structure (Fig. 6(b)). A tremendous shift of both Re and N orbitals to lower energy is discovered due to the orbital mixing. Around −15 eV, the lowest states in energy are dominated by N-2s states. Below the Fermi level, the states are mainly composed of the strong orbital mixing between Re-5d and N-2p states. Assuming the presence of N24− in ReN2,32 Re atom (oxidation state +4) with surplus three d-electrons would locate in the distorted trigonal-prismatic environment. According to crystal field theory, these three d-electrons may split into the fully occupied dz2-orbital at lower energy and be partially occupied with dx2−y2 and dxy orbitals at higher energy, and thus ReN2 is metallic, as expected, consistent with the absence of an energy gap at the Fermi level in the DOS spectrum. Also, this bonding–antibonding splitting further increases the hybridization between Re-5d and N-2p states (Fig. 6(b)) to stabilize the distorted trigonal-prismatic arrangement in C-ReN2. As evidenced by crystal orbital Hamilton populations (COHP) (Fig. 7(a)), a substantial Re–N bonding interaction is observed between −10 and −1.35 eV, and the antibonding state of the Re–N interaction appears from −1.35 eV to the Fermi level. Also, a covalent N–N bonding from the N2 dimer in C-ReN2 is also reflected by COHP. The presence of both Re–N and N–N interactions plays an important role in the exceptional mechanical properties of C-ReN2. With the increase in valence electrons, from Re to Ir, the bonding states of metal and nitrogen gradually filled in the entire occupied region (Fig. 7(b) and (c)), which suggests the increasing stability. But N–N antibonding states start to increase from ReN2 (0.80 eV per bond) to OsN2 (0.87 eV per bond) to IrN2 (0.92 eV per bond), making a negative contribution to B.32 However, for the obtained m-ReN2 by experiment, the N–N antibonding interaction (Fig. 7(d)) is much stronger than the bonding interaction (from integral areas), this negligible N–N interaction in m-ReN2 also supports the aforementioned instability.
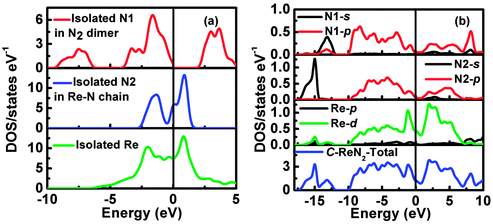 |
| | Fig. 6 Calculated density of states (DOS) of the isolated Rhenium atom and Nirogen atom in different positions in C-ReN2 (a), total and partial DOS of C-ReN2 (b). | |
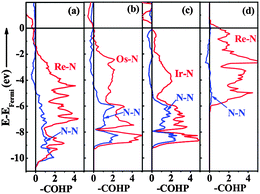 |
| | Fig. 7 Crystal orbital Hamilton Populations (COHP, eV cell−1) of Re–N and N–N interactions in C-ReN2 (a), OsN2 (b), IrN2 (c), and m-ReN2 (d) with the integrated N–N antibonding areas of 0.80, 0.87, 0.92, and 0.39 eV, respectively. | |
Conclusion
From first-principles calculations, we found MoS2-structured ReN2 is unstable as indicated by the negative C44 value and positive heat of formation. Calculated XRD and DOS suggest that nitrogen vacancies may stabilize the MoS2-type ReN2. Furthermore, the structural searching based on CALYPSO code uncovers that a monoclinic C2/m phase for ReN2 is more energetically stable than previous predictions and the experimental MoS2 structure. Also, the C2/m phase will transform to a tetragonal P4/mbm phase above 130 GPa. Furthermore, both monoclinic and tetragonal ReN2 have high bulk moduli and shear moduli, and thus are potential hard materials. The analysis of chemical bonding shows that the combined covalency from N2 dimer and Re–N bonds results in the superior mechanical properties of ReN2. Thus, further synthesis is urgent to understand this nitride.
References
-
L. E. Toth, Transitional Metal Carbides and Nitrides, Academic Press, New York, 1971 Search PubMed.
- S. T. Oyama, J. Solid State Chem., 1992, 96, 442 CrossRef CAS.
- S. T. Oyama, Catal. Today, 1992, 15, 179 CrossRef CAS.
- F. Lévy, P. Hones, P. E. Schmid, R. Sanjinés, M. Diserens and C. Wiemer, Surf. Coat. Technol., 1999, 120, 284 CrossRef.
- A. L. Ivanovskii, Russ. Chem. Rev., 2009, 78, 303 CrossRef CAS.
- A. Friedrich, B. Winkler, E. A. Juarez-Arellano and L. Bayarjargal, Materials, 2011, 4, 1648 CrossRef CAS.
- A. Zerr, G. Miehe and R. Riedel, Nat. Mater., 2003, 2, 185 CrossRef CAS.
- E. Gregoryanz, C. Sanloup, M. Somayazulu, J. Badro, G. Fiquet, H. Mao and R. J. Hemley, Nat. Mater., 2004, 3, 294 CrossRef CAS.
- A. F. Young, C. Sanloup, E. Gregoryanz, S. Scandolo, R. H. Hemley and H. K. Mao, Phys. Rev. Lett., 2006, 96, 155501 CrossRef.
- J. C. Crowhurst, A. F. Goncharov, B. Sadigh, C. L. Evans, P. G. Morrall, J. L. Ferreira and A. J. Nelson, Science, 2006, 311, 1275 CrossRef CAS.
- A. Friedrich, B. Winkler, L. Bayarjargal, W. Morgenroth, E. A. Juarez-Arellano, V. Milman, K. Refson, M. Kunz and K. Chen, Phys. Rev. Lett., 2010, 105, 085504 CrossRef.
- F. Kawamura, H. Yusa and T. Taniguchi, Appl. Phys. Lett., 2012, 100, 251910 CrossRef.
- Y. Wang, J. Lv, L. Zhu and Y. Ma, Phys. Rev. B: Condens. Matter Mater. Phys., 2010, 82, 094116 CrossRef.
- H. Chung, M. B. Weinberger, J. B. Levine, A. Kavner, J. Yang, S. H. Tolbert and R. B. Kaner, Science, 2007, 316, 436 CrossRef CAS.
- F. Hulliger, Nature, 1964, 204, 775 CrossRef CAS.
- R. Rűhl and W. Jeitschko, Inorg. Chem., 1982, 21, 1886 CrossRef.
- E. Zhao and Z. Wu, Comput. Mater. Sci., 2008, 44, 531 CrossRef CAS.
- Y. Li and Z. Zeng, Chem. Phys. Lett., 2009, 474, 93 CrossRef CAS.
- X. Du, Y. Wang and V. Lo, Phys. Lett. A, 2010, 374, 2569 CrossRef CAS.
- H. Wang, Q. Li, Y. Li, Y. Xu, T. Cui, A. R. Oganov and Y. Ma, Phys. Rev. B: Condens. Matter Mater. Phys., 2009, 79, 132109 CrossRef.
- M. D. Segall, P. L. D. Lindan, M. J. Probert, C. J. Pickard, P. J. Hasnip, S. J. Clark and M. C. Payne, J. Phys.: Condens. Matter, 2002, 14, 2717 CrossRef CAS.
- J. Perdew, K. Burke and M. Ernzerhof, Phys. Rev. Lett., 1996, 77, 3865 CrossRef CAS.
- R. Dronskowski and P. E. Blőchl, J. Phys. Chem., 1993, 97, 8617 CrossRef CAS.
- Z. Chen, X. Guo, Z. Liu, M. Ma, Q. Jing, G. Li, X. Zhang, L. X. Li, Q. Wang, Y. Tian and R. Liu, Phys. Rev. B: Condens. Matter Mater. Phys., 2007, 75, 054103 CrossRef.
- J. A. Montoya, A. D. Hernandez, C. Sanloup, E. Gregoryanz and S. Scandolo, Appl. Phys. Lett., 2007, 90, 011909 CrossRef.
- Y. Wang, M. Arai, T. Sasaki and C. Fan, Phys. Rev. B: Condens. Matter Mater. Phys., 2007, 75, 104110 CrossRef.
- Z. Wu, E. Zhao, H. Xiang, X. Hao, X. Liu and J. Meng, Phys. Rev. B: Condens. Matter Mater. Phys., 2007, 76, 054115 CrossRef.
- R. Zhang, Z. Lin, H. Mao and Y. Zhao, Phys. Rev. B: Condens. Matter Mater. Phys., 2011, 83, 060101 CrossRef.
-
M. Born and K. Huang, Dynamical Theory of Crystal Lattice, Oxford University Press, Oxford, 1954 Search PubMed.
- H. T. Mcskimin and P. Andreatch, J. Appl. Phys., 1972, 43, 985 CrossRef CAS.
- M. Segall, R. Shah, C. Pickard and M. Payne, Phys. Rev. B: Condens. Matter, 1996, 54, 16317 CrossRef CAS.
- W. Michael and R. Dronskowski, J. Am. Chem. Soc., 2010, 132, 2421 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c2cp43010j |
|
| This journal is © the Owner Societies 2013 |
Click here to see how this site uses Cookies. View our privacy policy here.