
Highly correlated ab initio study of the low frequency modes of propane and various monosubstituted isotopologues containing D and 13C†
Abstract
With the purpose of providing some clues that could encourage the spectral recordings of 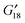 , respectively) is established for the classification of the levels and torsional splittings. The rotational constants are determined with CCSD(T)/CBS (A0 = 29263.46 MHz, B0 = 8454.10 MHz and C0 = 7466.64 MHz) using a non-relativistic procedure. Fundamental anharmonic frequencies corresponding to the high and medium amplitude modes are computed for all the isotopologues. The adjusted parameters are accurate enough to be employed in further spectral analysis.
, respectively) is established for the classification of the levels and torsional splittings. The rotational constants are determined with CCSD(T)/CBS (A0 = 29263.46 MHz, B0 = 8454.10 MHz and C0 = 7466.64 MHz) using a non-relativistic procedure. Fundamental anharmonic frequencies corresponding to the high and medium amplitude modes are computed for all the isotopologues. The adjusted parameters are accurate enough to be employed in further spectral analysis.

- This article is part of the themed collection: Spectroscopy and dynamics of medium-sized molecules and clusters
 Please wait while we load your content...
Please wait while we load your content...

