Accelerated electron transport from photosystem I to redox partners by covalently linked ferredoxin†
Gal
Wittenberg‡
a,
William
Sheffler
b,
Dana
Darchi
a,
David
Baker
b and
Dror
Noy
*ac
aPlant Sciences Department, Weizmann Institute of Science, Rehovot 7610001, Israel
bDepartment of Biochemistry, University of Washington, Seattle, WA 98195, USA
cMigal-Galilee Research Institute, South Industrial Zone, Kiryat Shmona 1101602, Israel. E-mail: drorn@migal.org.il
First published on 1st October 2013
Abstract
Photosystem I is a highly efficient and potent light-induced reductase that is considered to be an appealing target for integration into hybrid solar fuel production systems. However, rapid transport of multiple electrons from the reducing end of photosystem I to downstream processes in vivo is limited by the diffusion of its native redox partner ferredoxin that is a single electron carrier. Here, we describe the design and construction of a faster electron transfer interface based on anchoring ferredoxin to the reducing end of photosystem I thereby confining the diffusion space of ferredoxin to the near vicinity of its photosystem I binding and reduction site. This was achieved by fusing ferredoxin to the PsaE subunit of photosystem I by a flexible peptide linker and reconstituting PSI in vitro with the new fusion protein. A computational algorithm was developed in order to determine the optimal linker length that will confine ferredoxin to the vicinity of photosystem I's reducing end without restricting the formation of electron transfer complexes. According to the calculation, we reconstituted photosystem I with three fusion proteins comprising PsaE and ferredoxin separated by linkers of different lengths, namely 14, 19, and 25 amino acids, and tested their effect on electron transfer rates from photosystem I to downstream processes. Indeed, we found a significant enhancement of light dependent NADPH synthesis using photosystems containing the PsaE-ferredoxin fusion proteins, equivalent to a ten-fold increase in soluble ferredoxin concentration. We propose that such a system could be used for other ferredoxin dependent redox reactions, such as the enzymatic production of hydrogen, a promising alternative fuel. As the system is comprised entirely of natural amino acids and biological cofactors, it could be integrated into the energy conversion apparatus of photosynthetic organisms by genetic engineering.
Introduction
The efficient coupling of light-driven charge separation to elaborate redox catalytic processes in photosynthesis inspires the development of synthetic processes for solar to chemical energy conversion known as “solar fuel” production.1,2 Yet, the natural process that produces sugars still remains the only sustainable solar energy conversion process. An appealing strategy towards the production of more economically and technologically viable solar fuels is to construct hybrid systems that integrate various natural and optionally artificial components in a new non-biological context.3 In this way we can benefit from many redox enzymes that catalyze the production of potential fuel molecules, but are not necessarily parts of the photosynthetic metabolic pathways. Photosystem I (PSI) that uses light to drive the reduction of NADP+ to NADPH in oxygenic photosynthesis is a promising potential target for use as a photosensitizer in hybrid solar fuel production systems.4 This is due to its high quantum efficiency, long-lived charge separated state, and the very negative redox potential at its reducing end that provides sufficient driving force for proton reduction to molecular hydrogen, a promising fuel, at neutral pH.5The key to successful integration of PSI with redox catalysts is providing an interface for efficient transfer of multiple electrons from PSI to the catalytic site. The natural system relies on the small soluble iron–sulfur cluster protein ferredoxin (PetF – after its encoding gene) to carry one electron at a time from the reducing end of PSI to the active site of ferredoxin dependent NADP+ reductase (FNR) that requires two electrons for the reduction of one NADP+ molecule to NADPH. Several attempts were made to bypass this diffusion-limited process and divert electron flow directly to artificial redox catalysts or to the catalytic sites of hydrogen producing enzymes. These include depositing Pt nanoparticles onto the reducing end of PSI,6,7 anchoring nickel–iron hydrogenases to PSI's reducing end next to the PetF docking site,8–10 and wiring the reducing end of PSI directly to the H-cluster catalytic site of di-iron hydrogenase by organic molecular wires.11 Yacoby et al. implemented a somewhat different strategy by fusing PetF using a flexible linker to HydA, an algal di-iron hydrogenase that is a native redox partner of PetF.12
In addition to FNR and HydA, ferredoxins are involved in various metabolic reductive processes and thereby natively interact with many different redox catalytic enzymes.13 In this work, we wish to capitalize on PetF's capacity to serve as an electron carrier from PSI to many redox enzyme targets, and use it as a multipurpose electron transfer interface to PSI, while circumventing limitations caused by diffusion. For this purpose, we anchored PetF to the near vicinity of its docking site to PSI by a short flexible peptide linker that was designed to minimize the diffusion space of PetF without interfering with the formation of PSI–PetF electron transfer complex. The actual construction of the new interface capitalized on the inherent modularity of PSI's reducing end, the so called stromal ridge that is comprised of PsaC, PsaD, and PsaE protein subunits. PsaC harbors the two [4Fe–4S] clusters, FA and FB, that make up the reducing terminal of PSI's electron transport chain while PsaD and PsaE secure it onto the PSI core and provide a specific docking site for PetF.
We designed and prepared recombinant fusion proteins whereby PetF is linked to the C-terminal of PsaE by flexible peptide linkers of variable lengths selected by a computational algorithm. By reconstituting these PsaE::PetF fusion proteins with PsaC, PsaD, and PSI that were chemically stripped off its stromal ridge14,15 we assembled PSI with a new stromal ridge that maintains a covalently linked PetF. We tested the charge recombination kinetics of the electron transport chain in the new system, and found the electron transport chain to be extended toward the anchored PetF. Furthermore, assays of light induced NADPH production by FNR, a native redox partner of PetF, revealed substantial acceleration of NADPH synthesis rates when PetF was anchored PSI with respect to PSI with soluble PetF at equimolar concentrations. The successful application of a computational algorithm for our fusion protein design is further emphasized by a recent report of a 2.3-fold inhibition of NADPH production in a similar system that was comprised of PsaE::PetF fusion proteins with shorter linker lengths.16
Materials and methods
Cyanobacteria growth conditions
The thermophilic cyanobacteria Synechococcus elongatus Nägeli f. thermalis Geitler strain Kovrov 1972 (courtesy of Dr David Kaftan, University České Budějovice, Czech Republic) were grown in BG11 medium17 at 43 °C for four weeks in one liter cultures, under low light conditions (≈25 μmol photons per s m−2). Six liters of culture were used to inoculate a 25 liter fermentor, in which the bacteria were grown for an additional two weeks at 55 °C under similar light conditions.Protein expression and purification
All of the recombinant proteins used in this study were heterologously expressed in Escherichia coli BL21 cells. E. coli cultures for protein expressions were grown in LB medium at 37 °C. PsaC was expressed using the pET36C plasmid, purified and assembled with iron–sulfur clusters according to published procedures.18 PsaE was cloned into a pET28 expression vector in frame to an N terminal maltose binding protein (MBP), a His6 tag and a TEV protease digestion site. Following expression, MBP:His6:TEV:PsaE was purified on a Ni affinity column (His-Trap HP, GE Healthcare Life Sciences). PsaE was then cleaved from the fusion protein using TEV protease, and was separated from MBP:His6 as the flow through of a second separation on the Ni column. PetF and PsaE::PetF fusion constructs were cloned into a pET28 vector modified to contain an in frame “Profinity” tag (Bio-Rad). For the expression of PetF and all PsaE::PetF variants, the culture was supplemented with 1 ml per liter benzyl-alcohol 20 minutes prior to the induction of expression.19 During induction with 10 μM isopropyl β-D-1-thiogalactopyranoside (IPTG) the culture was supplemented with 56 mg per liter FeSO4. Following expression, PetF and PsaE::PetF fusion proteins were purified using a Profinity eXact column (Bio-Rad) according to the manufacturer's guidelines. Recombinant protein purity was verified by SDS-PAGE and protein staining using Instant Blue (Expedeon). Nostoc CytC6 cloned in frame to an N terminal His6 tag in the expression vector pET15b was expressed according to Bialek et al.21 in cells containing the plasmid pEC86, which encodes for cytochrome C maturases (courtesy of Prof. Dr Linda Thöny-Meyer, EMPA, St Gallen, Switzerland),20 and purified on a Ni affinity column. FNR was produced by amplifying the petH gene of Synechocystiss PCC 6803 genomic DNA while omitting 225 bp coding for the N-terminal phycobilisome linker analog,22 cloning into the same pET28 vector as used for PsaE, expressing, and purifying the protein the same way as PsaE. The plasmids pET36C, and pET15b containing CytC6 were kindly provided by Prof. John Golbeck (Pennsylvania State University, USA). Cofactor assembly into PsaC, PetF, CytC6 and FNR was assessed by recording the sample's absorption spectrum using a Jasco 7200 spectrophotometer, and comparing the unique visible absorption bands of each cofactor to the protein's UV absorption band.Photosystem I isolation and stromal ridge stripping
All procedures involving PSI were performed under green light, or darkness when possible. PSI trimers were extracted from S. elongatus as described in ref. 23. PSI stromal ridge proteins were removed to produce a PSI core as in Parrett et al.,15 with modifications that were required in order to adapt the protocol to the thermostable PSI of S. elongatus. Thus, stromal ridge removal was carried out by exposing PSI complexes to 7 M urea at 42 °C in PSI reaction buffer (50 mM Tris pH 8.3, 10 mM NaCl, 10 mM MgCl2, 0.05% triton-X, 15% glycerol). The removal was monitored by following the decay kinetics of the P700+˙ primary donor cation radical of PSI, as described below. The decay is dominated by charge recombination, and when this occurred within the timescale indicating recombination from the FX iron sulfur cluster of the PSI core, and not the iron sulfur clusters of PsaC, FA and FB4,15 the reaction was stopped by diluting it 50-fold with PSI reaction buffer without urea and cooling on ice. The PSI core was then separated from the soluble stromal ridge subunits by ultrafiltration using an Amicon stirred cell with a membrane cutoff of 100![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 Da (Amicon-Millipore), concentrated to 1 mg chl ml−1, and stored at −80 °C.
000 Da (Amicon-Millipore), concentrated to 1 mg chl ml−1, and stored at −80 °C.
Photosystem I stromal ridge reconstitution
50 μg chlorophyll per ml PSI core was incubated with 6 μM PsaD, 12 μM PsaC and 6 μM PsaE or PsaE::PetF fusion protein in PSI reaction buffer overnight in darkness under anaerobic conditions. Then, the reaction was diluted 20-fold in the same buffer and aerated. Unbound protein subunits were removed by ultrafiltration, as mentioned above, under aerobic conditions. Reconstituted PSI was concentrated by ultrafiltration to about 1 mg chlorophyll per ml and stored at −80 °C.Linker length prediction for PsaE::PetF fusion proteins
Computational docking of PetF onto PSI was performed using a variation of the RosettaDock methodology24 with the additional constraint that the electron transfer centers are separated by less than 15 Å.Photosystem I charge recombination kinetics and NADP+ reduction
Charge recombination kinetics of 50 μg chlorophyll per ml PSI (or PSI core) were determined in PSI reaction buffer containing 5 mM ascorbic acid (Sigma) as a sacrificial electron donor and 10 μM 2,6-dichlorophenolindophenol (DCPIP, Sigma). Measurements were done using the fast kinetics P700 mode of a DUAL-PAM 100 (Heinz Walz GmbH). Exponential curve fitting and the analysis of the kinetic components of charge recombination were done using IGOR Pro 6 (WaveMetrics Inc.). NADP+ reduction was measured in PSI reaction buffer containing 10 μg chlorophyll per ml reconstituted PSI (0.12 μM), 1.2 μM FNR, 1.2 μM CytC6, 100 μM NADP+ (Sigma), 5 mM ascorbic acid, 10 μM DCPIP, and PetF to the desired final concentration. NADP+ reduction was determined for 1 minute in the dark and 3 minutes under illumination by 150 μmol photons per s m−2 using the NADPH fluorescence module of the DUAL-PAM 100 (Heinz Walz GmbH).37 The rate of NADPH fluorescence increase was calculated from the slope of the line fitted to the time dependent NADPH fluorescence rise during illumination.Results and discussion
PsaE::PetF fusion protein design
We chose to fuse PetF to the PSI subunit PsaE by a flexible peptide linker. An optimal linker should keep PetF as close as possible to the donor side of PSI while providing enough flexibility and length to allow PetF and PSI to form an electron transfer complex. The molecular structure of this complex is yet unknown,25 but extensive biochemical and spectroscopic studies provided a good approximation of the docking site of PetF on PSI.26–31 We used this information with the individual structures of PetF and PSI to generate a set of possible docked configurations in which the electron donor FB, the terminal redox cofactor of PSI's electron transport chain, and the electron acceptor [2Fe–2S] cluster of PetF are within the electron transfer distance. The maximum distance threshold was set to 15 Å which was shown to be the maximum distance for physiologically relevant electron tunneling.38 This resulted in a diverse ensemble of 2737 possible electron transfer complexes that finely covered the space of possible configurations (Fig. 1A). Then, Monte Carlo optimization was performed within Rosetta32 to determine the shortest possible linker length compatible with each complex. For simplicity, the linker was comprised of flexible GGSGS repeats.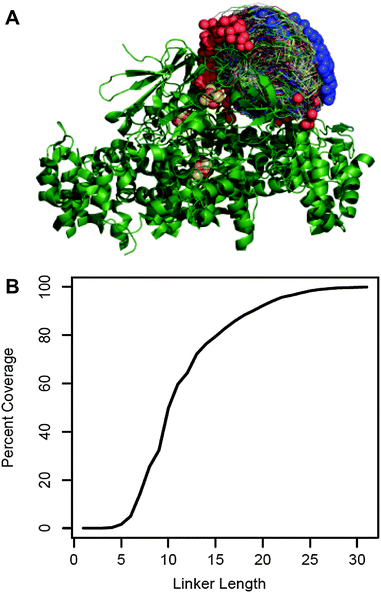 | ||
| Fig. 1 Linker length prediction for fusion proteins. (A) 2737 possible docked configurations in which the electron transfer centers are separated by less than 15 Å (allowing electron transfer) were generated by a variation of the RosettaDock algorithm using the crystal structures of the cyanobacterial PSI (PDB reference 1JB0) and green algal PetF (PDB reference: 1AWD). PSI is shown in green ribbon representation with the iron sulfur co-factors in brown and yellow. The PetF conformations are shown in trace representations with blue and red spheres marking their N and C terminus, respectively. (B) The fraction of docked configurations compatible with a given linker length out of the total docked configuration (percent coverage) plotted as a function of linker length. Compatibility was determined by calculating the shortest possible linker for each configuration using a Monte Carlo optimization performed within Rosetta. The configuration was deemed compatible with all the linker lengths greater than or equal to this minimal length. | ||
Fig. 1B shows the fraction of possible electron transfer complexes that is covered by linkers up to 35 residues long. Assuming that the ensemble of virtual complexes contains at least one complex that reflects the actual binding configuration, higher coverage corresponds to a higher likelihood that a given linker length will allow the native electron transfer complex to form. Thus, we chose to express and characterize three different PsaE::PetF fusion proteins with linker lengths of 14, 19, and 25 amino acids (AA), corresponding to 60%, 80% and 95% coverage, respectively.
Preparation of PsaE::PetF fusion proteins
Fusion proteins with three different linker lengths (Fig. 2A) were heterologously expressed in Escherichia coli. Recombinant proteins were purified by affinity chromatography, and were found by SDS-PAGE to be of high purity. The UV-vis absorption spectra of the fusion proteins (Fig. 2B) were typical of plant-type ferredoxin and featured a protein absorption band at 278 nm, and a broad band with peaks at 320 and 420 nm that is typical of the [2Fe–2S] cluster chromophore.33 The ratio between the peak absorption at 420 nm and 278 nm was found to be ∼0.3 for all three fusion proteins, and 0.58 for PetF. The latter is very close to the expected ratio of 0.56 for fully assembled PetF.33 The lower ratio in the fusion proteins is due to the contribution of the PsaE domain to the absorption band at 278. The calculated molar extinction coefficients at 278 nm of PetF and PsaE are 7000 M−1 cm−1 and 12600 M−1 cm−1, respectively. The expected ratio for fully assembled PsaE::PetF fusion is therefore 0.56 × 7000/(12600 + 7000) = 0.2. The slightly higher observed value most likely reflects the deviation of the theoretical calculated extinction coefficients from the actual values.
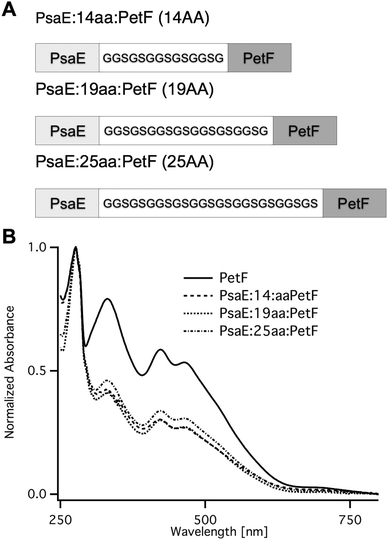 | ||
| Fig. 2 PsaE::PetF fusion protein expression and purification. (A) Sequences of the PsaE::PetF fusion proteins with three linker lengths, 14AA, 19AA and 25AA, corresponding to coverage of 60%, 80% and 95% of the possible docked configurations, respectively. (B) UV-vis absorbance spectroscopy of purified PetF and PsaE::PetF fusion proteins normalized to the absorbance at 278 nm. | ||
Charge recombination kinetics in PSI-PsaE::PetF complexes
Following published procedures,11,14 the stromal ridge of PSI was removed by chemical treatment leaving an intact FX-containing PSI core. Then either PsaE (as a control) or one of the three PsaE::PetF fusion proteins was used together with PsaC and PsaD for reconstituting the stromal ridge onto the PSI core. In order to avoid variations in PSI activity due to the chemical treatment the same batch of PSI cores preparation was used in all the reconstituted samples. The reconstituted photosystems were separated from any unbound subunits by ultrafiltration, the charge recombination kinetics through their reaction center's electron transport chain was assayed by monitoring the decay of the P700+˙ primary donor after excitation by a ten microsecond light flash, and the recorded profiles were fitted to multi-exponential decay curves. A typical fit and a comparison of decay curves from different samples are shown in Fig. 3. All the kinetic traces featured a slow decay phase in the range of 1 s in addition to one or two fast decay phases on the millisecond time scale. The slow decay reflects the slow reduction of P700+˙ by the soluble external donor that is ascorbate-reduced DCPIP, whereas the shorter phases reflect the kinetics of charge recombination across the electron transport chain.28,34,35 Since the concentration of external donors was the same in all the samples we assumed the slow decay phase to have the same time constant in all the samples. Thus, we applied a global fitting routine, whereby all the kinetic traces were fitted with a common long lifetime, and different short decay lifetimes. The fit resulted in a common lifetime of 1.34 ± 0.05 s; the short decay lifetimes and the relative amplitudes are listed in Table 1. In samples of PSI core stripped off its stromal ridge, the fast decay was dominated by a 3 ms phase that is typical of charge recombination from the FX iron–sulfur cluster. The decay of native PSI samples, as well as the PSI core reconstituted with native PsaE was dominated by a 45 ms decay phase typical of the charge recombination lifetime between P700+˙ and [FA/FB]−, the reduced iron–sulfur clusters of PsaC. Charge recombination slowed down two-fold to 90 ms in PSI reconstituted with either PsaE:14aa:PetF, PsaE:19aa:PetF, or PsaE::25aa::PetF. The slower recombination time and the overall decay profiles of the PSI core reconstituted with the PsaE::PetF fusion proteins were unlike those observed for a system comprising the PSI core reconstituted with native PsaE, and soluble PetF. In the latter, an increase of PetF concentrations led to an increase in the relative amplitude of the slow decay phase, and a decrease in the decay time of the faster phase to about 20 ms (Fig. 3B).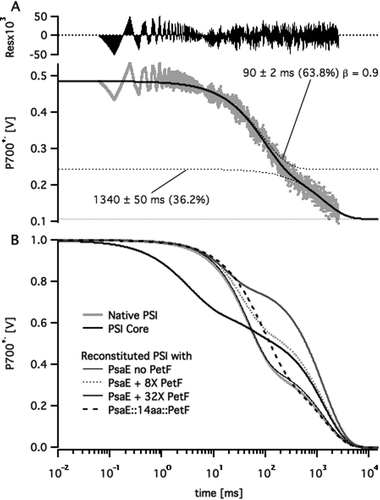 | ||
| Fig. 3 Multi-exponential curve fitting to P700+˙ decay profiles. (A) P700+˙ decay profile of PSI reconstituted with PsaE::14aa::PetF following a saturating pulse of white light (grey dots) fitted to a two stretched exponential decay curve (solid black line). Dotted lines represent the fitted curve and its individual decay components. Labels mark the lifetime, relative amplitude and stretch factor (β) for each component. (B) Fitted decay curves of native, core, and reconstituted PSI samples with different PetF concentrations. Curves were normalized to a P700+˙ signal amplitude of one at t = 0. | ||
| Fast decay | Slow decayb | ||
|---|---|---|---|
| Lifetime (ms) | Relative amplitude (%) | Relative amplitude (%) | |
| a Determined by fitting P700+˙ decay profiles following a saturating pulse of white light to a multi exponential decay curve (see Fig. S1, ESI). b The lifetime of the slow decay phase was common to all curves and was determined by a global fitting routine to be 1.34 ± 0.05 s. c An additional decay phase of 45 ± 9 ms accounting for 11.7% of the amplitude was observed in the PSI core (see Fig. S1, ESI). | |||
| Native PSI | 47.6 ± 0.3 | 61.9 | 38.1 |
| PSI corec | 3.1 ± 0.2 | 32.9 | 55.4 |
| PsaE | 45.6 ± 0.4 | 59.5 | 40.5 |
| PsaE:14aa:PetF | 90 ± 2 | 63.8 | 36.2 |
| PsaE:19aa:PetF | 90 ± 2 | 65.9 | 34.1 |
| PsaE:25aa:PetF | 87 ± 5 | 73.2 | 26.8 |
| [PetF]/[PSI] = 8 | 34.9 ± 0.4 | 40.8 | 59.2 |
| [PetF]/[PSI] = 16 | 23.8 ± 0.4 | 30.7 | 69.3 |
| [PetF]/[PSI] = 32 | 17.1 ± 0.4 | 21.2 | 78.8 |
The observed kinetic profiles may be explained by considering the typical charge recombination times of PSI's iron–sulfur clusters,35 and the well characterized mechanism of electron transfer from PSI to PetF.29 In the absence of PetF, dissolved oxygen acts as a terminal electron acceptor from the reducing end of PSI; in this case P700+˙ is reduced slowly by an external soluble donor, giving rise to the slow decay phase. Alternatively, P700+˙ reduction by charge recombination from reduced iron–sulfur clusters FX−, FA−, and FB− has typical lifetimes of 1–2 ms, 20 ms, and 65 ms, respectively. PetF is rapidly reduced by FB− within microseconds,27,28 and the dissociation rate constant, koff, of the PSI complex with reduced PetF was found to be 3200 ± 500 s−1.36 Reduced PetF is reoxidized by dissolved oxygen with a lifetime of 30–100 ms.27 Thus, increasing the concentration of PetF decreases the relative contribution of charge recombination to P700+˙ reduction kinetics by effective abstraction of electrons from the electron transport chain. This explains the observed increase in relative amplitude of the slow decay phase. The observed decrease in apparent lifetime of the short decay phase may also be explained by charge recombination from FA− becoming more predominant with respect to FB− as PetF concentration is increased. In the case of PsaE::PetF fusion proteins, docking PetF to PSI is likely to decrease koff of the PSI–PetF electron transfer complex such that charge recombination from reduced PetF to oxidized FB competes with dissociation. Moal and Lagoutte16 recently validated this assumption by showing that a mutation that destabilizes the PSI–PetF complex recovers NADPH production activity that was inhibited by connecting Petf to PsaE with a 10-residues linker. Stabilizing the PetF–PSI complex is expected to slow down charge recombination to P700+˙ on one hand, and decrease the relative contribution of reduction by external donor, as observed for all the samples of PSI reconstituted with PsaE::PetF fusion proteins.
NADP+ reduction by PsaE::PetF reconstituted photosystem I
In order to study how covalently bound PetF affects light dependent electron transfer to downstream acceptors from PSI, we reconstructed in vitro the native electron transport chain from individual protein components. Light dependent NADP+ reduction in a ternary system comprising PSI reconstituted with PsaE::PetF fusion proteins, cytochrome C6 (CytC6), and ferredoxin dependent NADP+ reductase (FNR) was compared to a quaternary system in which the first component was PSI reconstituted with native PsaE, and different concentrations of soluble PetF (Fig. 4A). In the dark no NADP+ reduction was observed for any of the samples (data not shown). Upon illumination of the quaternary system, a NADPH fluorescence signal emerged at a constant rate, indicating the reduction of NADP+ to NADPH. While the in vivo ratio of PSI and PetF was found to be in the range of 1:1, doubling PetF concentration in vitro has been previously shown to increase NADPH reduction rates by 50%.16 In accordance with these findings, the addition of increased amounts of PetF, up to 40-fold the molar concentration of PSI resulted in accelerated light dependent NADP+ reduction in our system (Fig. 4B). In PSI reconstituted with the PsaE:14AA:PetF, and PsaE:19AA:PetF fusion proteins, NADP+ reduction rates were comparable to the rate observed with soluble PetF at a ten-fold molar excess with respect to PSI reconstituted with native PsaE. The rate observed for PSI reconstituted with PsaE:25AA:PetF was somewhat slower than the other two constructs but significantly faster than that observed for a five-fold molar ratio of soluble PetF to PSI reconstituted with native PsaE. Since no PetF was added to PSI reconstituted with all three fusion proteins, the PetF:PSI molar ratio is 1:1. Thus, the confinement of PetF to the near vicinity of its reduction site by docking onto the stromal ridge has the equivalent effect of a ten-fold increase in the concentration of soluble PetF with respect to native PSI. Moal and Lagoutte reported that in a similar system the fusion of PseE and PetF with similarly flexible but shorter linkers of 5, 7 and 10 amino-acids decreased the rate of NADP+ reduction compared to a native PSI by 2.3-fold.16 This is in good agreement with our calculations showing that these short linkers allow only a small percent of the possible docked configurations to be formed (Fig. 1B). Yet, Moal and Lagoutte demonstrated that the decreased NADPH synthesis rates are due to strong association of PetF with PSI, which is brought about by the short linker. This was not considered explicitly in our calculations but the substantial rate acceleration observed for the 14-residues linker suggests that the constraint of having PetF associated too strongly with PSI is lifted at a linker length between 11 and 13 amino acids.
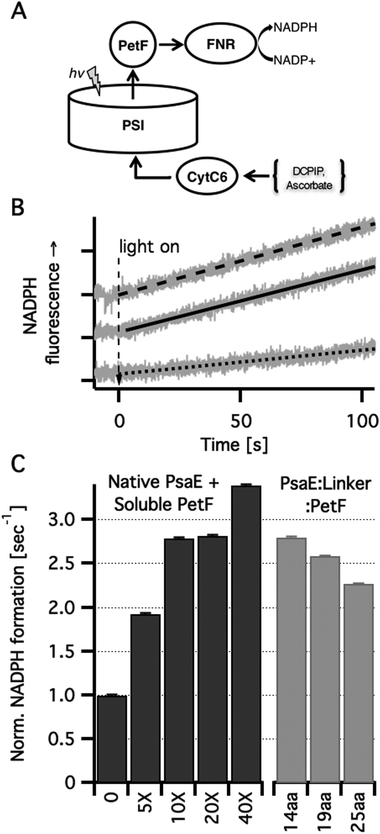 | ||
| Fig. 4 In vitro light dependent NADP+ reduction. (A) Scheme of the electron transport chain in solution. Photoexcited PSI was used to reduce PetF that carried the electrons to FNR where they were used for NADP+ reduction to NADPH. Ascorbic acid was used as a sacrificial electron donor by reducing (via DCPIP) CytC6 that transferred the electrons to the oxidizing end of PSI. (B) NADPH formation rates were measured in vitro by monitoring the linear increase in NADPH fluorescence. Typical traces (grey lines) and their linear fits are shown for PSI with no PetF (dotted line), PSI with ten-fold PetF molar excess (dashed line) and PSI reconstituted with PsaE:19aa:PetF (solid line). (C) NADPH formation rates normalized by the rate for PSI reconstituted with native PsaE without PetF. Molar excess of soluble PetF to PSI are indicated. No PetF was added to the PSI complexes reconstituted with PsaE::PetF fusion proteins. | ||
Conclusions
Efficient electron transfer from PSI via PetF to downstream acceptors such as FNR depends on two possibly contradicting factors. First, the redox centers of PSI and PetF must be in close proximity,38 which would require freedom to allow the correct docking configuration to occur. Then, PetF must be kept in close proximity to the donor side of PSI, limiting its diffusion and effectively decreasing the koff of the complex. Using a computational approach we were able to design linker lengths that would allow both to occur. By using well-established methods for chemical stripping and reconstitution of PSI's stromal ridge subunits, we were able to construct PSI variants in which PetF is covalently anchored to the stromal ridge by three flexible linkers of variable lengths. Complexes containing a 14aa and a 19aa linker, corresponding to 60% and 80% of the allowed docked configurations, were found to increase electron transfer rates to the native redox partner FNR. A longer linker showed less of an increase in electron transfer rates, suggesting diffusion in this system might be a limiting factor. Taken together with previous results16 our results support the use of a computational approach when designing de novo proteins for efficient electron transfer. This has important implications for using the photosynthetic apparatus for driving a variety of reductive chemical processes for the production of alternative solar fuels, particularly in view of recent demonstration of surpassing natural photosynthetic electron transfer throughput in hybrid systems that combine artificial and natural components.11 The advantage of the system presented in this work is that it is comprised entirely of natural amino acids and biological cofactors, which may enable straightforward integration into the energy conversion apparatus of photosynthetic organisms by genetic engineering. However, the effect of introducing a PsaE::PetF fusion protein into photosynthetic organisms remains to be determined.Acknowledgements
This work was funded by a grant number I/85 096 from the Volkswagen foundation. We thank Prof. John Golbeck, Penn State University, State College PA, USA for sharing with us his PSI stromal ridge reconstitution methods, and Anat Shperberg, Weizmann Institute of Science for the preparation of FNR and help with PsaC preparations, and growing cyanobacteria.Notes and references
- R. E. Blankenship, D. M. Tiede, J. Barber, G. W. Brudvig, G. Fleming, M. Ghirardi, M. R. Gunner, W. Junge, D. M. Kramer, A. Melis, T. A. Moore, C. C. Moser, D. G. Nocera, A. J. Nozik, D. R. Ort, W. W. Parson, R. C. Prince and R. T. Sayre, Science, 2011, 332, 805–809 CrossRef CAS PubMed.
- T. W. Woolerton, S. Sheard, Y. S. Chaudhary and F. A. Armstrong, Energy Environ. Sci., 2012, 5, 7470–7490 CAS.
- E. Reisner, Eur. J. Inorg. Chem., 2011, 1005–1016 CrossRef CAS.
- C. E. Lubner, R. Grimme, D. A. Bryant and J. H. Golbeck, Biochemistry, 2010, 49, 404–414 CrossRef CAS PubMed.
- M. L. Ghirardi, A. Dubini, J. Yu and P.-C. Maness, Chem. Soc. Rev., 2009, 38, 52–61 RSC.
- I. J. Iwuchukwu, M. Vaughn, N. Myers, H. O'Neill, P. Frymier and B. D. Bruce, Nat. Nanotechnol., 2010, 5, 73–79 CrossRef CAS PubMed.
- J. F. Millsaps, B. D. Bruce, J. W. Lee and E. Greenbaum, Photochem. Photobiol., 2001, 73, 630–635 CrossRef CAS.
- M. Ihara, H. Nakamoto, T. Kamachi, I. Okura and M. Maeda, Photochem. Photobiol., 2006, 82, 1677–1685 CAS.
- M. Ihara, H. Nishihara, K. S. Yoon, O. Lenz, B. Friedrich, H. Nakamoto, K. Kojima, D. Honma, T. Kamachi and I. Okura, Photochem. Photobiol., 2006, 82, 676–682 CrossRef CAS PubMed.
- H. Krassen, A. Schwarze, B. Friedrich, K. Ataka, O. Lenz and J. Heberle, ACS Nano, 2009, 3, 4055–4061 CrossRef CAS PubMed.
- C. E. Lubner, A. M. Applegate, P. Knörzer, A. Ganago, D. A. Bryant, T. Happe and J. H. Golbeck, Proc. Natl. Acad. Sci. U. S. A., 2011, 108, 20988–20991 CrossRef CAS PubMed.
- I. Yacoby, S. Pochekailov, H. Toporik, M. L. Ghirardi, P. W. King and S. Zhang, Proc. Natl. Acad. Sci. U. S. A., 2011, 108, 9396–9401 CrossRef CAS PubMed.
- A. M. Terauchi, S.-F. Lu, M. Zaffagnini, S. Tappa, M. Hirasawa, J. N. Tripathy, D. B. Knaff, P. J. Farmer, S. P. D. Lemaire, T. Hase and S. S. Merchant, J. Biol. Chem., 2009, 284, 25867–25878 CrossRef CAS PubMed.
- J. H. Golbeck, T. Mehari, K. Parrett and I. Ikegami, FEBS Lett., 1988, 240, 9–14 CrossRef CAS.
- K. G. Parrett, T. Mehari, P. G. Warren and J. H. Golbeck, Biochim. Biophys. Acta, 1989, 973, 324–332 CrossRef CAS.
- G. Moal and B. Lagoutte, Biochim. Biophys. Acta, Bioenerg., 2012, 1817, 1635–1645 CrossRef CAS PubMed.
- R. Y. Stanier, R. Kunisawa, M. Mandel and G. Cohen-Bazire, Bacteriol. Rev., 1971, 35, 171–205 CAS.
- T. Mehari, K. G. Parrett, P. V. Warren and J. H. Golbeck, Biochim. Biophys. Acta, 1991, 1056, 139–148 CrossRef CAS.
- A. de Marco, L. Vigh, S. Diamant and P. Goloubinoff, Cell Stress Chaperones, 2005, 10, 329–339 CrossRef CAS PubMed.
- E. Arslan, H. Schulz, R. Zufferey, P. Kunzler and L. Thöny-Meyer, Biochem. Biophys. Res. Commun., 1998, 251, 744–747 CrossRef CAS PubMed.
- W. Bialek, M. Nelson, K. Tamiola, T. Kallas and A. Szczepaniak, Biochemistry, 2008, 47, 5515–5522 CrossRef CAS PubMed.
- J. J. van Thor, R. Jeanjean, M. Havaux, K. A. Sjollema, F. Joset, K. J. Hellingwerf and H. C. P. Matthijs, Biochim. Biophys. Acta, Bioenerg., 2000, 1457, 129–144 CrossRef CAS.
- N. Li, P. V. Warren, J. H. Golbeck, G. Frank, H. Zuber and D. A. Bryant, Biochim. Biophys. Acta, 1991, 1059, 215–225 CrossRef CAS.
- J. J. Gray, S. Moughon, C. Wang, O. Schueler-Furman, B. Kuhlman, C. A. Rohl and D. Baker, J. Mol. Biol., 2003, 331, 281–299 CrossRef CAS.
- P. Fromme and I. Grotjohann, in Bioenergetic Processes of Cyanobacteria, ed. G. Renger, G. A. Peschek and C. Obinger, Springer, Netherlands, 2011, pp. 285–335 Search PubMed.
- P. Barth, I. Guillouard, P. Setif and B. Lagoutte, J. Biol. Chem., 2000, 275, 7030–7036 CrossRef CAS PubMed.
- P. Q. Setif and H. Bottin, Biochemistry, 1994, 33, 8495–8504 CrossRef CAS.
- P. Q. Setif and H. Bottin, Biochemistry, 1995, 34, 9059–9070 CrossRef CAS.
- P. Setif, in Photosystem I: The Light-Driven Plastocyanin: Ferredoxin Oxidoreductase, ed. J. H. Golbeck, Springer, Netherlands, 2006, pp. 439–454 Search PubMed.
- P. Setif, N. Harris, B. Lagoutte, S. Dotson and S. R. Weinberger, J. Am. Chem. Soc., 2010, 132, 10620–10622 CrossRef CAS PubMed.
- A. Amunts and N. Nelson, Structure, 2009, 17, 637–650 CrossRef CAS PubMed.
- C. A. Rohl, C. E. Strauss, K. M. Misura and D. Baker, Methods Enzymol., 2004, 383, 66–93 CAS.
- W. J. Rogers, M. Hodges, P. Decottignies, J. M. Schmitter, P. Gadal and J. P. Jacquot, FEBS Lett., 1992, 310, 240–245 CrossRef CAS.
- I. R. Vassiliev, Y. S. Jung, M. D. Mamedov, A. Y. Semenov and J. H. Golbeck, Biophys. J., 1997, 72, 301–315 CrossRef CAS.
- I. R. Vassiliev, M. L. Antonkine and J. H. Golbeck, Biochim. Biophys. Acta, Bioenerg., 2001, 1507, 139–160 CrossRef CAS.
- V. Fourmond, B. Lagoutte, P. Setif, W. Leibl and C. Demaille, J. Am. Chem. Soc., 2007, 129, 9201–9209 CrossRef CAS PubMed.
- U. Schreiber and C. Klughammer, PAM application notes, 2009, 2, 1–13 Search PubMed.
- C. C. Page, C. C. Moser, X. Chen and P. L. Dutton, Nature, 1999, 402, 47–52 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c3cp53264j |
| ‡ Current address: Max-Planck-Institut für Molekulare Pflanzenphysiologie, Am Mühlenberg 1, 14476, Potsdam-Golm, Germany. E-mail: wittenberg@mpimp-golm.mpg.de |
| This journal is © the Owner Societies 2013 |