Photoorganocatalysis. What for?
Davide
Ravelli
,
Maurizio
Fagnoni
and
Angelo
Albini
*
PhotoGreen Lab, Department of Chemistry, University of Pavia, V.Le Taramelli 12, 27100 Pavia, Italy. E-mail: angelo.albini@unipv.it
First published on 18th September 2012
Abstract
In the reactions reviewed, an organic molecule under irradiation catalyzes a chemical reaction. The activation is based either on hydrogen or on electron transfer. Commonly used photoorganocatalysts are aromatic ketones, quinones, heterocycles, dyes; intermediates formed are radicals, radical ions and ions from precursors such as alkanes, alkenes, amines, ethers etc. Oxidation (mainly oxygenation) and reduction processes are obtained along with the α-functionalization of amines and ketones, conjugate additions, cycloadditions etc. The key characteristic of the method is the smooth generation of highly reactive intermediates under mild conditions.
 Maurizio Fagnoni, Davide Ravelli and Angelo Albini | Davide Ravelli was born in 1984 and obtained his PhD from the University of Pavia in 2012 with a thesis on tetrabutylammonium decatungstate photocatalyzed C–C bond forming reactions (Prof. A. Albini as the supervisor). He is currently a post-doc student at the same University, pursuing the study on photocatalytic processes and evaluating the adoption of ionic liquids as electrolytes in lithium batteries. His research interests focus on radical mediated synthetic procedures and on the rationalization of organic reaction mechanisms by means of computational tools. |
Maurizio Fagnoni graduated in Pavia in 1992 under the guidance of Prof. Angelo Albini. He is currently an Associate Professor of the Department of Chemistry at the University of Pavia. He is interested in the generation of reactive intermediates e.g. radicals, cations and radical ions by photochemical means. Particular attention has been given to eco-sustainable synthesis by using photocatalysis and photogenerated phenyl cations. In 2011 he was a recipient of the CINMPIS prize for “Innovation in organic synthesis” and of the “Exploration of new research frontiers Award” from Cariplo Foundation. |
Angelo Albini completed his studies in Chemistry in 1972, did postdoctoral work at the Max-Planck Institute for Radiation Chemistry in Muelheim, Germany (1973–74) and joined the Faculty at Pavia in 1975, where he is currently a Professor of Organic Chemistry, after a period at the University of Torino and stays at the Universities of Western Ontario (Canada, 1976–77) and Odense (Denmark, 1983). He is active in the field of organic photochemistry, organic synthesis via radicals and ions, photoinitiated reactions, mild synthetic procedures in the frame of the increasing interest for sustainable/green chemistry and applied photochemistry. |
1. Introduction
Transition-metal catalysis has become one of the most important tools in organic synthesis due to the variety of reactions accessible.1 Some drawbacks remain, however, such as the high cost and toxicity of the catalyst employed and in some cases the problems related to the disposal of the catalyst at the end of the reaction. In the last few years, a metal-free approach emerged that makes use of organic compounds as catalysts and has been dubbed as “organocatalysis”. The work in the field has rapidly progressed with application in reactions such as nucleophilic substitutions, Michael additions, cycloadditions and aldol reactions. The asymmetric version of such processes has been likewise successfully developed.2 Organocatalysts can be divided into two main classes depending on the interaction, covalent or non-covalent (H-bonding, proton transfer, ion pair formation) with the organic substrate within the catalytic cycle.2In the former case, an organocatalyst (OC, see Scheme 1) reacts with an organic molecule (the first reagent, R1) to form a stable organic compound or an intermediate (OC–RA). At this stage, the activation induced by OC makes possible the attack of the second reagent (R2) to form a second adduct (OC–RB) that releases the end product P with the concomitant regeneration of OC.
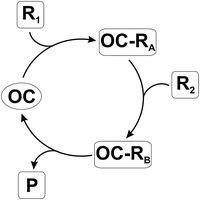 | ||
| Scheme 1 | ||
Most of the OCs used are (chiral) secondary amines and R1 are electrophiles, typically aldehydes, ketones or α,β-unsaturated carbonyls. The OC promotes the generation of either an iminium ion (Scheme 2) or an enamine (Scheme 3). This causes, respectively, an increase in the electrophilicity of the starting carbonyl derivative – promoting nucleophiles addition or cycloaddition reactions – or the Umpolung of the starting carbonyl – leading to electrophilic addition onto the enamine intermediate.2 Similarly, N-heterocyclic carbenes (NHCs), another class of widely used OCs, promote the Umpolung of an organic moiety (mainly aldehydes or Michael acceptors) for C–C bond formation reactions.3 When chiral, the OC may make the process stereoselective.2
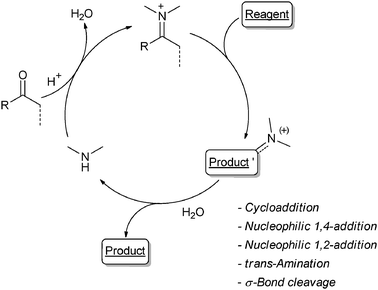 | ||
| Scheme 2 | ||
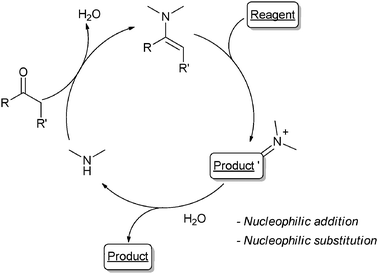 | ||
| Scheme 3 | ||
Photocatalysis, where an electronically excited species PC* acts as the catalyst, has been gaining interest and various organic transformations under such conditions have been reported recently, in most cases involving C–C bond formation (via activation of C–H bonds) or oxidations.4 In many cases the catalyst is a transition metal complex,4,5 as is e.g. the case of the use of coloured Ru and Ir complexes for the now rapidly developing visible light photocatalysis.5 However, reactions where organic molecules play this role are known, indeed some of them are among the longest known photochemical reactions, and are also of interest for some peculiarities, which prompted activity in the field.4a,6–7 As an example, their role in the photocatalyzed oxidation of pollutants has been recently reviewed.8
A photocatalytic cycle is depicted in Scheme 4. The excited catalyst activates a reagent R1 that is transformed into RA through a chemical reaction. The deactivated photocatalyst (PCD) is then regenerated through a reverse process by an intermediate RB, arising through a (series of) reaction(s) from RA. PC re-enters the cycle and the final product P is obtained (Scheme 4).
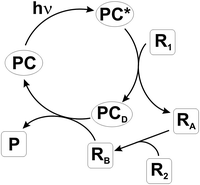 | ||
| Scheme 4 | ||
The present review refers to the case where the photocatalytic cycle involves only organic species, although this does not necessarily mean that a metal containing species does not participate in the follow-up steps.
Physical energy transfer (sensitization) is excluded from the definition,9 as is the formation of a ground state complex,10 and thus of a different species with a different absorption spectrum,10,11 as well as the photochemical formation of a catalyst4a or of a thermally active reagent.12 A representative example of the last case is shown in Scheme 5, where a cyclohexanone derivative (1) is converted into the corresponding gem-dihydroperoxide (2) by visible light irradiation in i-PrOH in the presence of oxygen and a catalytic amount of anthraquinone (3). Here the role of 3 is that of photogenerating the oxidant, be it the hydroxy hydroperoxide 4 or hydrogen peroxide from it (Scheme 5).12
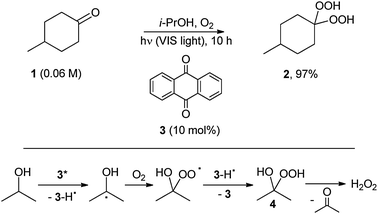 | ||
| Scheme 5 | ||
Likewise, a bio-inspired system was recently reported, where a dye (Rose Bengal, 7, 0.1 mol%) was involved in the reduction of various alkyl chlorides including the toxic 1,1-bis(4-chlorophenyl)-2,2,2-trichloroethane (DDT) via a visible-light-driven process (Scheme 6).13 This does not fit with the above definition because, although 7 absorbs light, the active species is the photogenerated reduced form of vitamin B12 derivative (8), a ground state compound present in small amounts (1 mol%).13 Chloroamide 5 was efficiently dechlorinated to 6 in a satisfactory yield by irradiation with a 200 W tungsten lamp under these conditions (Scheme 6).
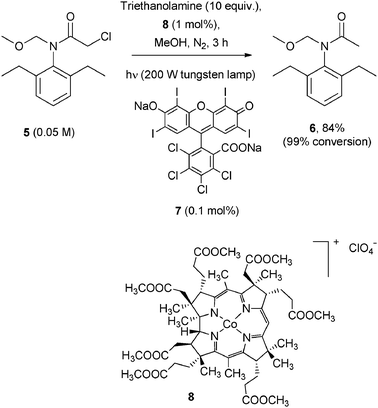 | ||
| Scheme 6 | ||
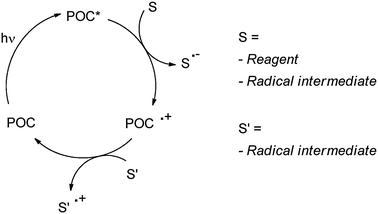
As mentioned above, photocatalytic reactions necessarily involve the activation of (one of) the reagent(s) (that is incorporated into the end product) by excited photoorganocatalysts (POC*, or by other short-lived intermediates directly generated from them, such as radical ions POC˙+ or POC˙−) and thus must be a fast process. Adopting the above conditions coupled with the requirement that the catalyst is regenerated in practice limits the viable modes of action of a photocatalyst to hydrogen atom transfer (HAT, Scheme 7) or electron transfer (ET, Schemes 8 and 9) as illustrated in previous reviews.4
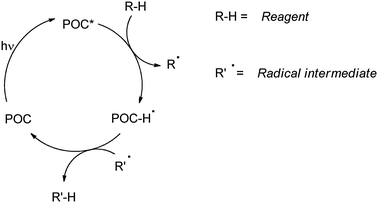 | ||
| Scheme 7 | ||
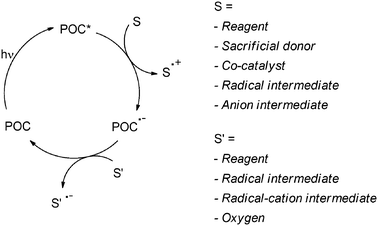 | ||
| Scheme 8 | ||
 | ||
| Scheme 9 | ||
In the HAT mechanism, homolytic hydrogen abstraction from a C–H bond (e.g. in ethers, aldehydes, alcohols and alkanes) leads to the formation of a nucleophilic carbon-centered radical prone to react with Michael acceptors in conjugate addition reactions.4a The reaction is successful when the POC-H˙ species is able to back-donate a hydrogen atom to a radical intermediate formed in the reaction (Scheme 7). Known POCs acting in this way are limited to aromatic ketones and quinones.4a,14
In the ET mechanism either an oxidative or a reductive activation is involved (see Schemes 8 and 9). The situation depicted in Scheme 8 is quite common. In that case, electron transfer from reagent S generates the radical cation S˙+ that undergoes the chemistry. The POC is regenerated, often by the oxygen mediated oxidation of POC˙− from the first step (Scheme 8). In some instances, however, other donors are initially oxidized, such as a sacrificial donor (usually a tertiary amine), a co-catalyst (again another organic molecule) or radical or anionic intermediates in turn generated in the process. In a limited number of cases the reaction starts with the reduction of a reagent by photogenerated POC˙−. Moreover, the regeneration of the POC can be accomplished by an ET reaction between POC˙− and radicals or radical cations (Scheme 8). On the other hand, the capability of POC* to reduce a reagent is less commonly exploited (Scheme 9).
In contrast to the HAT mechanism, there is a wide variety of POCs able to behave as oxidants or reductants when in the excited state. These include acridinium salts, (thia)pyrylium salts,15 diazapyrenium salts, riboflavin tetraacetate, aromatic esters and nitriles, substituted benzophenones,14 polycyclic aromatics (e.g. phenanthrene) and more recently dyes.7 In a couple of cases the POC mimics the action of titanium dioxide as a microelectrode, namely the semiconductor mesoporous carbon nitride polymer (a heterogeneous POC),16 and the 9-mesityl-10-methylacridinium ion.
Table 1 gathers the key electrochemical parameters for the ground and excited states of some organic photoredox catalysts in comparison with those of Ru(bpy)32+, the most widely used metal complex in visible light photocatalysis, along with their absorption. It is apparent that POCs are excellent one electron oxidants in the excited states (E*red ranging between 1.0 and 3.0 V vs. SCE). In contrast, transition metal photocatalysts usually undergo an initial reduction or oxidation via the excited state (e.g. Ru2+* to either Ru+ or Ru3+) and this leads to the species actually involved in the catalytic cycle, either a good reductant (Eredca. −1.4 V vs. SCE for Ru+) or a modest oxidant (Eoxca. 1.3 V vs. SCE for Ru3+).5b
| Photocatalysts (PC) | E red (PC/PC˙−) | E ox (PC˙+/PC) | E*red (PC*/PC˙−) | E*ox (PC˙+/PC*) | Absorptionb (nm) |
|---|---|---|---|---|---|
| a Redox potentials are reported in V vs. SCE; bpy = 2,2′-bipyridine. b Absorption maxima or the convenient absorption range for preparative irradiations. | |||||
| Rose Bengal (7) | −0.78 | 1.09 | 0.99 | −0.68 | 559 |
| 10-Methylacridinium ion (19) | −0.43 | 2.32 | 320–470 | ||
| Eosin Y (23) | –1.14 | 0.72 | 1.18 | –1.60 | 539 |
| 9,10-Dicyanoanthracene (46) | –0.89 | 1.97 | 330–440 | ||
| Benzophenone (62) | –1.68 | 1.55 | 320–380 | ||
| Phenanthrene (81) | –2.49 | 1.83 | 1.10 | –1.76 | 300–350 |
| Ru(bpy)32+ | –1.33 | 1.29 | 0.84 | –0.86 | 450 |
Most POCs exhibit some activity even when exposed to solar light, since most of them absorb strongly in the UV-A region and a significant fraction is coloured (Table 1).
The aim of the present review is to highlight the emerging trends in the application of a photoorganocatalyst in organic synthesis. For most of the reported reactions, the amount of the organocatalyst used tends to be larger than that of the metal-based catalyst. The present discussion will be limited to reactions taking place with a <20% molar proportion of the photocatalyst, excluding reactions where the additive is used in a stoichiometric amount, although some of these are closely analogous to the catalytic proper reactions.
2. Applications of photoorganocatalysts (POC) in synthesis
Known reactions that answer the above definition by using POC are limited in number, but quite varied. In this section these have been classified into oxidations, reductions, alkylations and other C–C bond forming reactions and arylations. The collection is not exhaustive and is biased toward the most recent applications. Further examples can be found in previously published reviews.4a,b,62.1. Oxidations
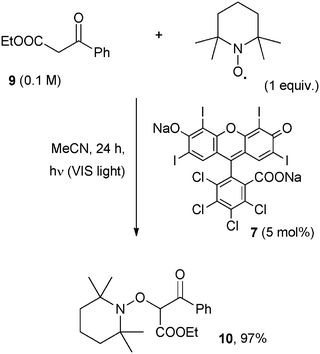
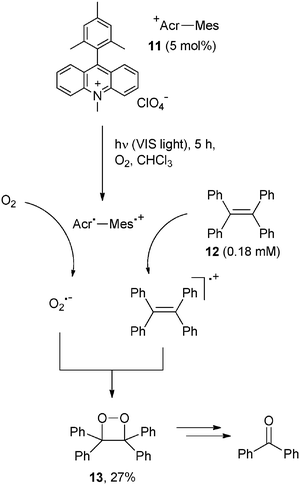
In contrast, oxygen was required in the photocatalytic oxidation of anthracenes and olefins, where visible light (Xenon lamp) was likewise effective.18 In this case a charge-separation dyad was used as a photocatalyst, such as the 9-mesityl-10-methylacridinium ion (11, Acr+–Mes, 5 mol%) developed by Fukuzumi et al. Accordingly, irradiation of an oxygen saturated solution of 9,10-dimethylanthracene or tetraphenylethylene resulted in the formation of dimethylepidioxyanthracene and tetraphenylethylene dioxetane, respectively. The reaction was initiated by the photoexcitation of Acr+–Mes that caused an intramolecular electron transfer reaction to form Acr˙–Mes˙+ that in turn was able to oxidize the organic substrate (to the corresponding radical cation) as well as to reduce oxygen to O2˙−. The coupling between the two last species gave rise to a dioxetane.18Scheme 11 illustrates the typical mode of action of Acr+–Mes in the photooxidation of tetraphenylethylene (12) where the corresponding dioxetane 13 was isolated in 27% yield.19 The oxidation potential of the dioxetane is quite low (ca. 1.56 V vs. SCE) and accordingly, further oxidation can occur upon prolonged irradiation and benzophenone is ultimately formed.18,19
 | ||
| Scheme 10 | ||
 | ||
| Scheme 11 | ||
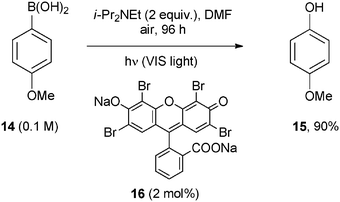
The direct aerobic oxidative hydroxylation of arylboronic acids to phenols is another interesting application of a dye photocatalyzed formation of a C–O bond by using air as the oxidant.20 The process was usually carried out by using Ru or Ir based complexes, but in a single case (see Scheme 12) Acid Red 87 (Eosin Y) disodium salt (16) was successfully adopted. The reduced photocatalyst (formed by a photoinduced ET reaction with an amine such as i-Pr2NEt) was able to reduce oxygen to the superoxide radical anion that in turn added to the boron atom of the arylboronic acids (e.g.14). This caused a series of rearrangements that upon final hydrolysis gave the corresponding phenol (15) in an excellent yield (Scheme 12).20
 | ||
| Scheme 12 | ||
Acridinium salts (19) are effective POCs also in the Baeyer–Villiger oxidation of cyclobutanones (17) to γ-lactones (18) that occurs from modest to good yields in the presence of 1.2 equiv. hydrogen peroxide (Scheme 13).21
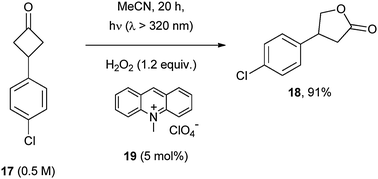 | ||
| Scheme 13 | ||
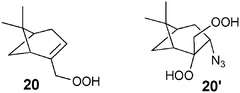
Two dyes (Rhodamine B and Rhodamine 6G, ca. 0.1 mol%) were used for the azidohydroperoxidation of myrtenyl hydroperoxide (20) in the presence of the azide anion and oxygen. The photoinduced oxidation of the azide anion generated an azidyl radical prone to be added to the double bond of 20. Trapping of the resulting radical by oxygen followed by a further ET step led to the azido bis-hydroperoxide 20′.22 Despite the modest yields (not exceeding 13%), the reaction was notable for the concomitant addition of the N3 and OOH groups onto the double bond of 20.
A case of C–P bond formation by photocatalysis was recently reported and involved the reaction of a number of N-aryl tetrahydroisoquinolines (21) in the presence of an excess of diethyl phosphite, resulting in an oxidative synthesis of α-amino phosphonates (22, Scheme 14).23 Green LEDs were used as the light source and Eosin Y (23, 2 mol%) as POC, regenerated by the air dissolved. In common with other photocatalytic reactions of the easily oxidized tetrahydroisoquinolines, the mechanism involves oxidation by POC* and H transfer from the radical cation to O2˙−, in this case followed by trapping of the resulting iminium ion by the phosphorus nucleophile (yield of the phosphonate exceeding 80%, Scheme 14).23
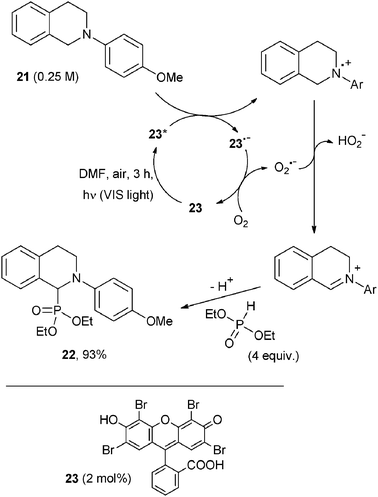 | ||
| Scheme 14 | ||
Very recently, substituted N-aryl tetrahydroisoquinolines were used as the starting materials for the preparation of isoquino[2,1-a]pyrimidines via an intramolecular photocatalyzed formation of a C–N bond again by using Eosin Y disodium salt (16, 0.5 mol%) as POC.24 The strategy was in this case based on the intramolecular addition of a pendant N-tosyl moiety onto the iminium ion formed. Thus, 4-methyl-N-(2-(7-methyl-3,4-dihydroisoquinolin-2(1H)-yl)benzyl)benzenesulfonamide (24) was functionalized under mild conditions to give 3-methyl-5-tosyl-5,6,12,13-tetrahydro-4bH-isoquinolino[2,1-a]quinazoline (25) in 85% yield upon 25 h irradiation by a 36 W fluorescent light (Scheme 15).24
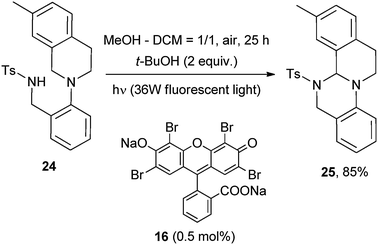 | ||
| Scheme 15 | ||
![[double bond, length as m-dash]](https://www.rsc.org/images/entities/b_char_e001.gif) O bonds.
Oxidation processes forming X
O bonds.
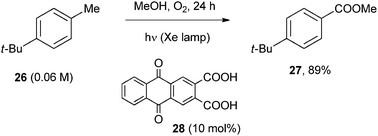
Oxidation processes forming X![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O bonds (X = C, S) have also been reported, and the largest part involved the conversion of methyl aromatics and of benzyl alcohols to the corresponding carbonyl or carboxyl derivatives. Again, molecular oxygen mostly is the oxygen source and the activation involves either hydrogen or electron transfer. With anthraquinone derivatives the first mechanism operates. Thus, excited anthraquinone-2,3-dicarboxylic acid (28) abstracted a benzylic hydrogen in 4-t-butyltoluene (26) and the resulting benzyl radical was trapped by oxygen. In methanol the corresponding hemiacetal was formed and a second hydrogen abstraction/oxygen addition sequence afforded the methyl ester 27 (Scheme 16).25 The reaction was effective even with polymethylated benzenes, provided that a larger amount of POC was used. Under these conditions, p-xylene was oxidized to dimethyl terephthalate in 46% yield.25
O bonds (X = C, S) have also been reported, and the largest part involved the conversion of methyl aromatics and of benzyl alcohols to the corresponding carbonyl or carboxyl derivatives. Again, molecular oxygen mostly is the oxygen source and the activation involves either hydrogen or electron transfer. With anthraquinone derivatives the first mechanism operates. Thus, excited anthraquinone-2,3-dicarboxylic acid (28) abstracted a benzylic hydrogen in 4-t-butyltoluene (26) and the resulting benzyl radical was trapped by oxygen. In methanol the corresponding hemiacetal was formed and a second hydrogen abstraction/oxygen addition sequence afforded the methyl ester 27 (Scheme 16).25 The reaction was effective even with polymethylated benzenes, provided that a larger amount of POC was used. Under these conditions, p-xylene was oxidized to dimethyl terephthalate in 46% yield.25
 | ||
| Scheme 16 | ||
Moreover, when the reaction was carried out in ethyl acetate and by using 2-chloroanthraquinone (8 mol%) as the POC, the corresponding benzoic acids were formed.26 In this aerobic oxidation, improved yields were obtained in the presence of both bases (potassium carbonate) and acids (trifluoroacetic acid, TFA).
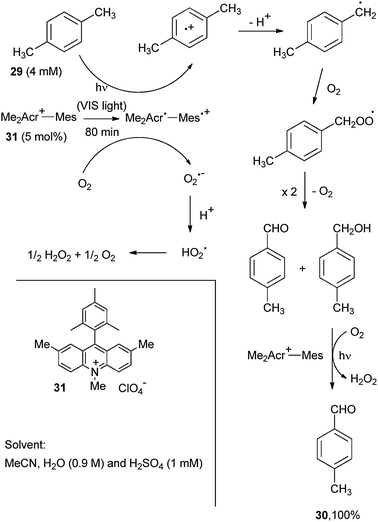
An example of the ET mechanism is the clean and chemoselective oxidation of methyl aromatics (e.g. p-xylene, 29) by using the 9-mesityl-2,7,10-trimethylacridinium ion (31, Me2Acr+-Mes, 5 mol%) as the photocatalyst.27 ET oxidation followed by deprotonation yielded benzyl radicals that could add oxygen leading to a mixture of p-tolualdehyde and p-methyl benzyl alcohol (Scheme 17). The oxidation conditions allowed the further oxidation of the alcohol to the aldehyde 30, but the high oxidation potential of the latter prevented any further oxidation by the Mes˙+ moiety. As shown in Scheme 17 an equivalent amount of hydrogen peroxide was likewise liberated in the reaction. Another example is the oxidation of β-methylnaphthalene to β-naphthaldehyde with 9-mesityl-10-methylacridinium perchlorate as the catalyst and again oxygen as the oxidant, albeit a non-negligible amount of 2-methyl-1,4-naphthoquinone was also formed. Notice that this catalyst acts as an oxygen sensitizer with less easily oxidized substrates, such as alkenes.28
 | ||
| Scheme 17 | ||
The oxidation of primary and secondary benzyl alcohols to aromatic aldehydes and, respectively, ketones was likewise efficient with acridinium salts as POCs (λ > 310 nm).29,30 Yields were good (>80%), independently of the presence of ring substituents. Various benzylic oxidations could be carried out by having recourse to riboflavin tetraacetate (10 mol%) that is another convenient POC for the benzylic oxidation of alkylbenzenes, as well as of N-acylbenzylamide to the corresponding imide (83% yield in aqueous acetonitrile, 30 min irradiation).31
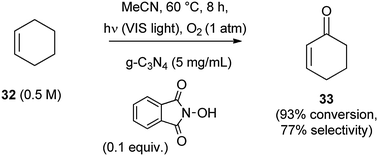
As hinted in the introduction, heterogeneous POCs are also available. Thus, mesoporous carbon nitride (mpg-C3N4)16,32 is a solid-state semiconductor based on a polymeric melon (a slightly disordered precursor of graphitic carbon nitride having a band gap of 2.7 eV, which corresponds to an optical wavelength of 460 nm). This has some similarities with TiO2, but absorbs in the visible region. The oxidation of benzyl alcohols with mpg-C3N4 (50 mg per mmol of alcohol) required the use of a stainless steel autoclave.32 The selectivity was high, but the conversion not complete. Graphitic carbon nitride (g-C3N4)16 in conjunction with N-hydroxyphthalimide acted as a metal-free photocatalytic system for the allylic oxidation of various alkenes,33 as shown in Scheme 18 for the conversion of cyclohexene (32) into cyclohexenone (33). The actual C–H activating agent was the corresponding nitroxyl radical.
 | ||
| Scheme 18 | ||
The same protocol was applied to the oxidation of styrene to benzaldehyde, of ethyl benzene to acetophenone and of cyclohexane to cyclohexanone (in the last case, however, the conversion was limited to ca. 6%).33
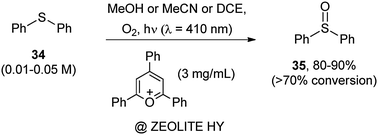
The formation of SO bonds by oxidation of sulfides can be treated here. The use of 2,4,6-triphenylpyrylium salts15 encapsulated within zeolite Y as an inert support34 (Scheme 19) was found effective for the chemoselective oxidation of diphenyl sulfide 34, whether in medium polar and in polar protic solvents. The encapsulation avoided the degradation of the photocatalyst generally observed in solution. In most cases the process gave mixtures of oxidized derivatives namely disulfides, sulfinic and sulfenic esters as well as sulfonic acids. However, in the case of aromatic sulfides the formation of the corresponding sulfoxides 35 was predominant and in some cases exclusive (see Scheme 19).34
 | ||
| Scheme 19 | ||
Carbon nitride (mpg-C3N4) has been likewise used for the photocatalytic selective oxidation of sulfides to sulfoxides with O2 as the oxidant at room temperature.35
Resin-supported Eosin Y was used as the POC in the reduction of 4-nitrophenol (36) to 4-aminophenol (37) under visible light. The reaction gave good yields and occurred in the presence of NaBH4 that reduced the excited state of the dye (Scheme 20). An important advantage of the method is that the catalyst can be reused at least three times with no significant loss in the rate constant of the reaction.38
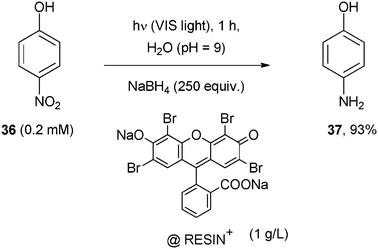 | ||
| Scheme 20 | ||
2.3. Formation of C–C bonds
As an example, the one electron oxidation of N-methylpyrrolidine by excited 4,4′-dimethylaminobenzophenone (38) or 4,4′-dimethoxybenzophenone (39, Scheme 21) and selective deprotonation from a ring α-position gave a nucleophilic radical that was trapped by conjugate addition onto electron-poor olefins.39 Photoaddition onto methyl acrylate, acrylonitrile and dimethyl maleate was obtained in good yields by using a 10 mol% amount of 39.39
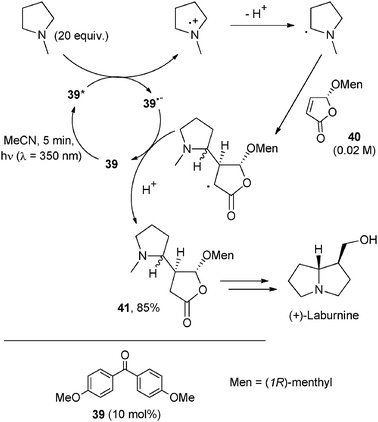 | ||
| Scheme 21 | ||
Interestingly, when using a chiral olefin such as (5R)-5-menthyloxy-2[5H]-furanone (40) the addition took place with facial diastereoselectivity (≥90%).40 The adduct formed (41, 85% yield)39 was later elaborated in a few steps for the preparation of pyrrolizidine and indolizidine alkaloids including (−)-isoretronecanol and (+)-laburnine (Scheme 21).41
The addition of dithiocarbamate to the solution appeared to improve efficiency and selectivity of some of these reactions. In the extreme case the alkylation of N,N-dimethylacrylamide took place only in the presence of such an additive.42
A variation of this process led to the tetrahydroquinoline derivative 42 in a good yield and with excellent stereoselectivity by addition of the α-amino radical generated from N,N-dimethylaniline to furanone 40 (bearing a chiral menthyloxy group) and by cyclization (see Scheme 22).43 The reaction was photocatalyzed by Michler's ketone (38) at λ = 350 nm.
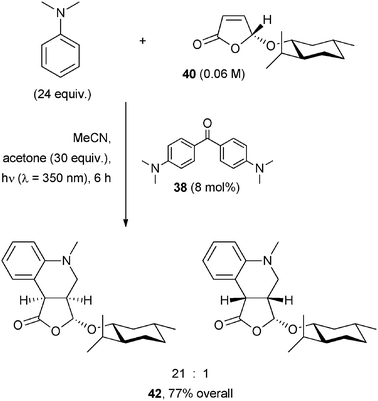 | ||
| Scheme 22 | ||
An intramolecular version of the reaction was developed by Bach and coworkers, where a catalytic enantioselective reaction was performed. Here the POC (a functionalized benzophenone) was tethered to a chiral complexing agent (44) able to embed the substrate 43 through hydrogen bonding.44 The irradiation of the complex generated a α-amino radical prone to react intramolecularly in a chiral environment. As a result, the chiral pyrrolizidine 45 was formed in a good yield with a satisfactory ee (Scheme 23).44
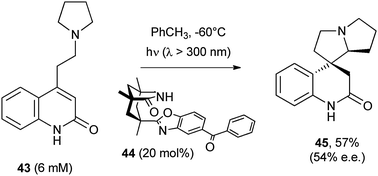 | ||
| Scheme 23 | ||
On the other hand, using Eosin Y for the radical (from methyldiphenylamine) alkylation of diethyl ethylidenemalonate resulted in a poor yield (9%), compared with 90% when an Ir complex was chosen.45
α-Amino radicals can be likewise generated from α-silyl amines (48). The trimethylsilyl (TMS) group lowers the oxidation potential of the substrate, thus making a cyanoarene such as 9,10-dicyanoanthracene (46) able to cause photoinduced ET46 in the presence of a co-catalyst (biphenyl, 47, see Scheme 24). Desilylation of the radical cation (in a protic medium), intramolecular cyclization onto a double bond, reduction by 46˙− and protonation gave carbamate 49 in a decent yield and in high diastereomeric excess. Related cyclizations were used for inducing structural changes in oligopeptides by introducing the structural motif present in compound 48 into the peptide chain.
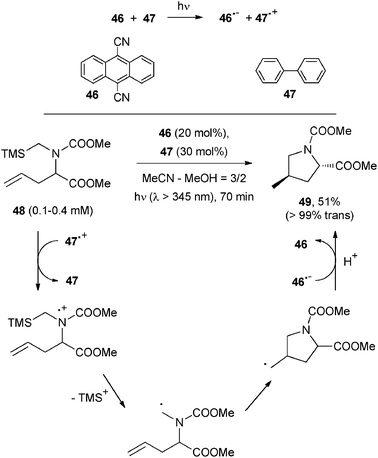 | ||
| Scheme 24 | ||
An alternative way consisted of the homolytic hydrogen abstraction from the amine radical cation previously formed to generate an electrophilic iminium ion that could then be trapped by carbon nucleophiles. Oxygen was usually required as a hydrogen atom scavenger. In a pioneering work, the Santamaria group photocyanated a series of alkaloids by using the visible light absorbing POC N,N′-dimethyl-2,7-diazapyrenium-bis-(tetrafluoroborate) (52, DAP2+, 2 BF4–, 1 mol%) and Me3SiCN as the cyanide source.47 Under these simple conditions, atropine 50 gave α-aminonitrile 51 in 90% yield. The success of the reaction was in part attributed to the stability of the radical cation generated by monoelectronic reduction of the diazapyrenium salt (Scheme 25).
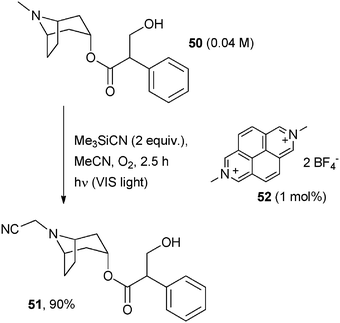 | ||
| Scheme 25 | ||
Rose Bengal photocatalyzed the cyanation of the N-acetyl-2,16-dihydro derivative of the alkaloid tabersonine (33% yield, 2 h irradiation)48 as well as that of N-aryl-tetrahydroisoquinolines (5 mol% catalyst, in combination with graphene oxide, 53).49 The mild conditions required allowed for the C–H functionalisation of these tertiary amines by using Me3SiCN as the nucleophile. Using 53 improved the rate and the efficiency of the reaction. The process was likewise applied for the trifluoromethylation of the isoquinoline derivative by using TMSCF3 as the nucleophile.49 The reaction was considered ‘green’ because of the use of a cheap organic dye as POC and air as the oxidant.
Cyanoarenes (e.g. 1,4-dicyanonaphthalene, 56) are again suitable for the regioselective generation of iminium ions by oxidation of unsymmetrical tertiary amines. In this case the cation could be formed by oxidation of the α-amino radical by the ground state cyanoarene. Substituted pyrrolidines (e.g.54) were then purposely employed for the complete regioselective synthesis of tetrahydro-1,3-oxazines such as 55 (Scheme 26).50
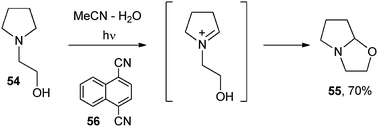 | ||
| Scheme 26 | ||
N-aryl-tetrahydroisoquinolines underwent smooth dehydrogenative coupling reaction in the presence of dyes (Rose Bengal and Eosin Y gave the best performance), thus offering a facile metal-free way of building sp3–sp3 C–C bonds.23,51,52 The α-functionalisation of tertiary amines via Henry and Mannich reactions was thus obtained. As an example, irradiation with green light LEDs of a mixture of the isoquinoline and nitromethane (or another nitroalkane) gave the corresponding Henry coupled product in a high yield (at least with benzylamines),51 which was further improved when solar light was used. Henry adducts from N-aryl tetrahydroisoquinolines were likewise reported by using Eosin Y bis-(tetrabutylammonium salt) (2 mol%) as the POC.52 In this work, further insights into the mechanism were obtained and the role of superoxide radical anion (O2˙−) was documented52 (see further Scheme 14).
The dehydrogenative-Mannich reaction catalysed by Rose Bengal 7 was likewise reported as illustrated in Scheme 27. Here the nucleophile was an enamine generated in situ from acetone (the solvent of the reaction) and an organocatalyst (pyrrolidine), promoted by TFA.51 Under these conditions, the substituted isoquinoline 57 smoothly gave aminoketone 58 after 20 h irradiation in almost quantitative yield (Scheme 27). When using cyclohexanone, substituting (L)-proline for pyrrolidine improved the yield and imparted a good diastereoselectivity.51
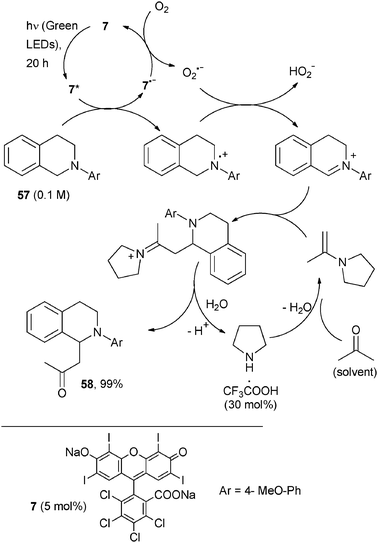 | ||
| Scheme 27 | ||
The oxidative coupling of tetrahydroisoquinolines with dialkyl malonates (used as the solvent) was similarly obtained by irradiation at 530 nm in the presence of 2 mol% Eosin Y. The products were isolated in excellent yields since the excess of dialkyl malonate was easily eliminated by Kugelrohr distillation.23
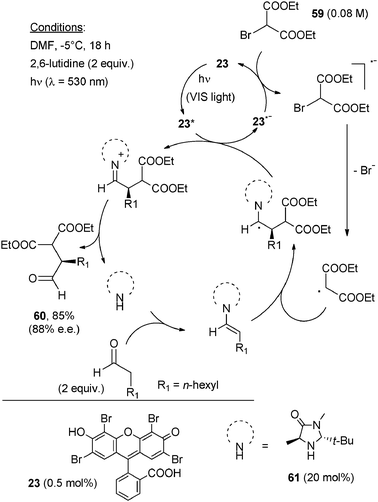 | ||
| Scheme 28 | ||
A slight modification of the reaction described in Scheme 28 was recently reported and made use of Rose Bengal (0.5 mol%) and of the same imidazolidinone organocatalyst, along with LiCl (10 mol%) and 2,6-lutidine (2 equiv.) as additives.53
The second approach exploits the ET activation of organoselenium compounds to build carbocation equivalents. Thus, oxidation of an alkyl phenyl selenide by excited 1,4-dicyanonaphthalene (10 mol%), addition to an enol silyl ether and PhSe radical loss gave a α-functionalized ketone in up to 80% yield. The reaction was completed in 8 to 10 hours in MeCN – H2O (4/1) and equilibration with dissolved air was sufficient for reoxidizing the photocatalyst.54
 | ||
| Scheme 29 | ||
1,3-Dioxolanes were studied in more detail for the introduction of a masked formyl (or acyl) group. In this case, benzophenone had to be used in a stoichiometric amount, but anthraquinone 3 worked at a lower concentration, as seen in the 3 photocatalyzed addition of 1,3-dioxolane onto phenyl vinyl ketone (65) to give the monoprotected 1,4-ketoaldehyde 66 in 65% yield (Scheme 30).56
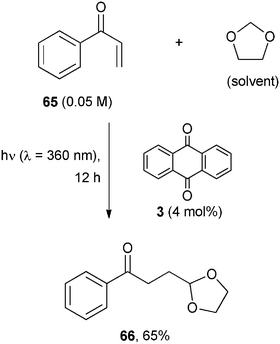 | ||
| Scheme 30 | ||
When using chiral enones such as 1,3-dioxin-4-one derivatives, the alkylation (benzophenone, 15 mol% as the POC) occurred from the a-side in both inter- and intramolecular reactions, leading to highly diastereoselective processes. As one may expect, the most efficient reaction occurred with the unsubstituted 1,3-dioxin-4-one.57
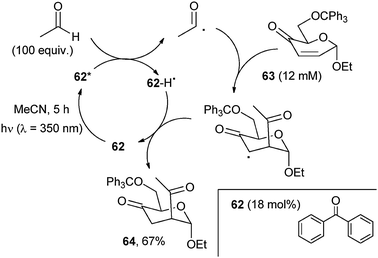
This mild reaction (and the analogous introduction of a protected ketone function by reaction with 2-alkyl-1,3-dioxolanes) was used for the functionalization of such sensitive substrates as α,β-unsaturated aldehydes for the preparation of 1,4-monoprotected dialdehydes and ketoaldehydes.58
POCs such as benzophenone disodium disulfonate (67) or the sodium salt of 4-benzoylbenzoic acid (20 mol%) were used in a mixed aqueous solvent with good results and the additional advantage that the products could be recovered from the mixture containing the catalyst by a mere extraction.58 Terebic acid (69) was obtained from isopropanol and maleic acid (68) by using 67, a synthesis that was carried out on a 10 g scale by 10–15 hours (over three days) exposure to sunlight in a solar concentrator (SOLFIN apparatus, Scheme 31).59
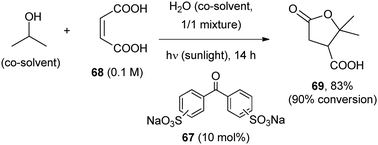 | ||
| Scheme 31 | ||
The labile formyl C–H bond in aldehydes (whether aliphatic or aromatic) was photocatalytically activated by benzophenone (6 mol%), a process that the authors dubbed as “photo Friedel–Crafts acylation” of 1,4-naphthoquinone (70, Scheme 32).60 Naphthohydroquinones (e.g.71) were formed in moderate to good yields and were smoothly oxidized to valuable acylated naphthoquinones. In the absence of the POC some acylation occurred by direct irradiation, but required a long irradiation time and an excess of the aldehyde for completion.60
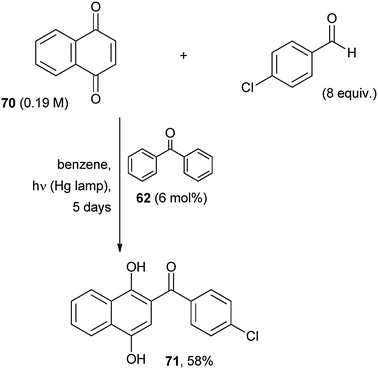 | ||
| Scheme 32 | ||
The activation of strong C–H bonds in alkanes could likewise be obtained by POCs.4a,61–62 The reaction was usually carried out in neat cycloalkane under either homogeneous or heterogeneous conditions. Thus, vinyl cycloalkanes (usually E/Z mixtures) were obtained by benzophenone (14 mol%) photocatalyzed addition of cycloalkanes onto electron-poor alkynes.61 Apparently, the aggressive vinyl radical formed abstracted hydrogen from the alkane, making the reaction efficient, even with a low POC concentration. Adoption of a (potentially recyclable) supported catalyst and of UV-A lamps or sunlight as the light source enhanced the clean/green chemistry character of this alkylation, as in the case of aminopropylsilica bound benzophenone in the synthesis of methyl 3-cyclopentyl-2-propenoate (73, E/Z mixture) starting from cyclopentane and methyl propiolate (72) (see Scheme 33).
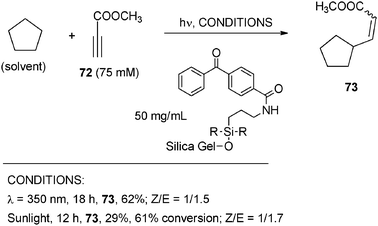 | ||
| Scheme 33 | ||
As for the ET generation of alkyl radicals from aliphatic donors, tetrasubstituted stannanes63 were mainly used due to their low oxidation potential, with UV-B absorbing cyanoarenes or aromatic esters as POCs. As an example, Bu4Sn (74) was used for the alkylation of dimethyl maleate in the presence of tetramethylpyromellitate (76, 20 mol%) to afford succinate 75 in 80% yield (Scheme 34).63a The high reduction potential of cyanoarenes in the excited state allows peculiar reactions, e.g. the use of acetals as electron donors. Thus, the alkylation of dialkyl maleates or acetylenedicarboxylates starting from 2-alkyl-2-phenyl-1,3-dioxolanes took place in the presence of 1,2,4,5-tetracyanobenzene (10 mol%).63a In the last reaction, the presence of a co-catalyst such as phenanthrene or biphenyl was often mandatory for obtaining a satisfactory alkylation yield.
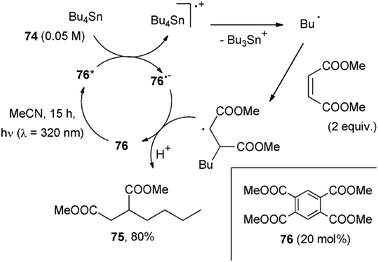 | ||
| Scheme 34 | ||
1,1-Disubstitued olefins such as vinylidene malononitrile or vinylidene cyanoacetate are powerful radical traps and are often used in photocatalyzed alkylation reactions.64,65Scheme 35 reports the functionalization of a 1,1-dicyanoethylene derivative (77) by addition of the ˙CR2CH2CO2Me radical, in turn arising via ring opening of a cyclopropanone silyl acetal, such as 78, induced by oxidative photocatalysis by phenanthrene (81).64a More precisely, excited phenanthrene reduced 77 and Phen˙+ thus formed oxidized 78 and radical 79˙ from it by loss of the Me3Si cation. Radical–radical anion coupling between ˙CR2CH2CO2Me and 77˙− gave dinitrile 80 in 94% yield.
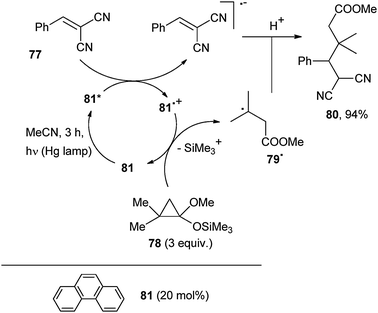 | ||
| Scheme 35 | ||
A related reaction made use of β,β-dialkyl substituted ketene silyl acetals as electron donors, again in the regioselective introduction of R′O2CR2C groups onto electron-poor olefins photocatalyzed by phenanthrene (20 mol%).64b Thus, irradiation of a MeCN solution containing 1-methoxy-2-methyl-1-trimethylsiloxypropene, 1,1-dicyano-2-phenylethene and phenanthrene gave the desired 4,4-dicyano-2,2-dimethyl-3-phenylbutyric acid methyl ester in 95% yield.64b
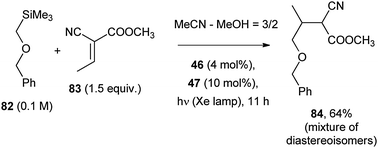
Higher oxidation potential derivatives such as α-silyl ethers (e.g.82), required a more powerful oxidant, such as 9,10-dicyanoanthracene (46; 4 mol%) with biphenyl (47; 10 mol%) as a co-catalyst (Scheme 36). In this case, desilylation of the radical cation (requiring a nucleophilic cosolvent, such as methanol) gave a α-oxy radical that was trapped by an electron-withdrawing substituted olefin, e.g. by 83 to give cyanoester 84 (64% yield).65
 | ||
| Scheme 36 | ||
 | ||
| Scheme 37 | ||
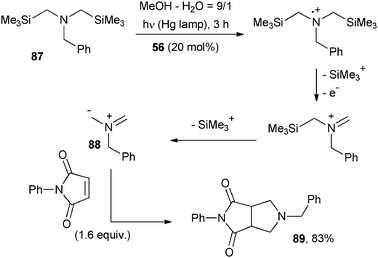
An elegant way for the construction of a five membered ring was developed by Pandey and coworkers by a photocatalyzed 1,3-dipolar cyclization, as shown in Scheme 38.67 Here the 1,3-dipole (a non-stabilized azomethine ylide) was generated starting from bis-silyl amine 87 by two consecutive photooxidations. Again 56 (20 mol%) was used as the POC and initially an iminium cation was generated upon oxidation of 87, TMS cation loss ensued by oxidation. In turn, the cation underwent a second desilylation and the resulting 1,3-dipole 88 was trapped by the dipolarophile N-phenylmaleimide yielding the bicyclic compound 89 in 83% yield.67 Noteworthily, the POC was almost completely recovered at the end of the reaction.
 | ||
| Scheme 38 | ||
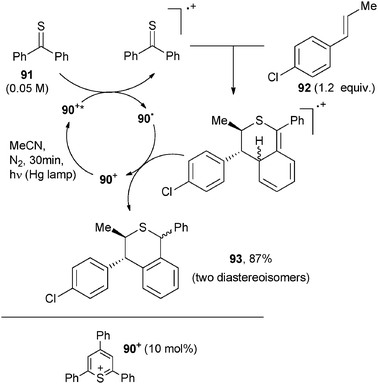
Six-membered rings were obtained through a photocatalyzed Diels–Alder type reaction. Most of the work in this field is due to the group of Prof. Steckhan. A typical example involved exocyclic dienes and indoles for building highly functionalized tetrahydrocarbazole derivatives. The reaction was initiated by oxidation of the indole moiety to the corresponding radical cation followed by reaction with the diene. Triphenylpyrylium tetrafluoroborate and tris(4-methoxyphenyl)pyrylium tetrafluoroborate (5 mol%) were found suitable as POCs.15,68 Analogously, (thia)pyrylium salt 90+ was used as a photoredox photocatalyst in the [4+2] cycloaddition between thiobenzophenone (91) and a 4-substituted-β-methylstyrene (e.g.92, Scheme 39).69 Interestingly, in this case both reagents could be oxidized by the excited photocatalyst but only radical cation 91˙+ was reactive and added to the neutral arylalkene 92 to give the thia-1,2,3,4-tetrahydronaphthalene radical cation. Back electron transfer to this species from the pyranyl radical 90˙ led to the sulfur heterocycle 93, along with the recovered photocatalyst.69
 | ||
| Scheme 39 | ||
A recent application concerns with the intramolecular photocycloaddition between a furan moiety and a non-activated alkene in the diethyl malonate derivative 94.70 The photooxidation was catalyzed by 9,10-dicyanoanthracene (46, 5 mol%) and afforded oxabicyclo[2.2.1]heptane 95 (Scheme 40). With alkyl substituted furans, the cycloadducts were formed with a high stereoselectivity.
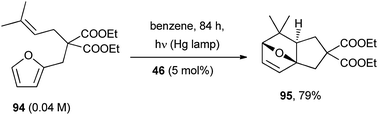 | ||
| Scheme 40 | ||
2.4. Arylations
Photocatalyzed formation of Ar–X bonds (mainly Ar–C) has also been reported (see also Section 2.1.1). An interesting example deals with the synthesis of (+)-2,7-dideoxypancratistatin, a model compound for the preparation of phenanthridone alkaloids. One of the key steps of this synthesis was a 1,4-dicyanonaphthalene (56, 2 mol%) ET initiated carbocyclization (Scheme 41).71 Thus, irradiation of enol silyl ether 96 in aqueous acetonitrile in the presence of 56 gave cyclized product 97 in 68% yield as a single diastereoisomer.71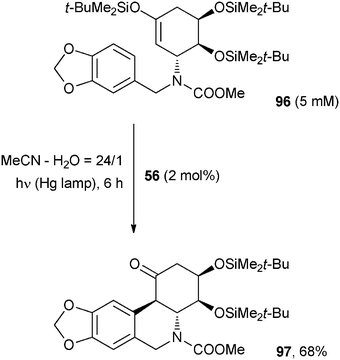 | ||
| Scheme 41 | ||
The same POC was employed in the PET oxidation of aromatics substituted with an enol silyl ether moiety. Depending on the structure of the starting aromatic, the reaction led to different benzannulated as well as benzospiroannulated benzyl ketones with the intermediacy of an arene radical cation.72
The high reducibility of aryl diazonium salts has been recently exploited for the transition-metal-free formation of aryl-heteroaryl or aryl-vinyl bonds.73 The excited photocatalyst (Eosin Y, 23, 1 mol%) was able to reduce the diazonium salt that upon nitrogen loss gave an aryl radical. This in turn added to furan in a regioselective fashion and the resulting radical adduct was oxidized by POC˙+ and the desired 2-arylfuran was finally formed upon proton loss.73a The reaction conditions well tolerated the presence of several functional groups on the aromatic ring. Under the same visible light irradiation conditions, both N-Boc protected pyrroles and thiophenes were likewise smoothly arylated.73a Arylation of unsaturated compounds such as alkenes, alkynes and enones was later developed by the same group by photoredox catalysis (Eosin Y, 7.5 mol%, green LEDs as the light source) in an updated version of the Meerwein arylation.73b In particular, the coupling between an aromatic diazonium salt and styrene in DMSO allowed for the preparation of stilbene derivatives in what can be considered an actual metal-free photo-Heck reaction.
A single case was reported where the formation of Ar–Br bonds occurred in place of Ar–C bonds. Aqueous HBr was used here as the bromine source and the 9-mesityl-10-methylacridinium ion (11, 5 mol%) as the photocatalyst.74Scheme 42 shows the photobromination of 1,2,4-trimethoxybenzene (98) by visible light photolysis of an aerated acetonitrile mixture containing the photocatalyst and HBr to afford the brominated derivative 99 in 61% yield. The photocatalytic bromination started by an intramolecular photoinduced electron transfer from the mesitylene moiety to the singlet excited state of the acridinium ion. An electron transfer from aromatic compounds to the mesitylene radical cation moiety and from the acridinyl radical moiety to oxygen then ensued. The selective monobromination took place by bromide addition onto the resulting radical cation of the starting aromatic derivative (Scheme 42).74 Other methoxysubstituted aromatics were likewise well suited for this reaction.
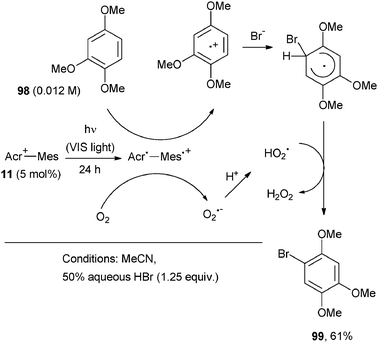 | ||
| Scheme 42 | ||
2.5. Miscellaneous
The ring opening of an epoxide by MeOH addition using 2,3-dichloro-5,6-dicyano-1,4-benzoquinone (100) as the POC has been likewise reported (Scheme 43).75 An initial ET between 100* and α-epoxyketone 101 was envisaged. The thus formed intermediate 101+ added methanol and gave the corresponding α-hydroxy-β-methoxyketone 102 as a mixture of diastereoisomers through Cβ–O bond cleavage in excellent yields.75 Notice however that the same process took place, indeed in a higher yield, under microwave irradiation.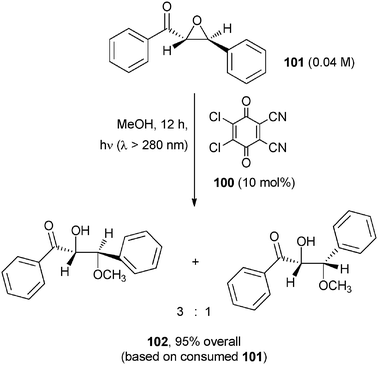 | ||
| Scheme 43 | ||
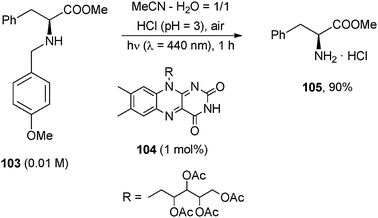
Photoorganocatalysis has also been found useful for the photodeprotection of the amine group, when protected as a methoxybenzyl derivative (Scheme 44).76 The reaction is based on the flavin-mediated aerobic photooxidation of benzyl amines such as 103. Thus, 1 h photolysis of an acidic acetonitrile–water solution containing 103 and riboflavin tetraacetate (104, 1 mol%) allowed for the isolation of the hydrochloride salt of phenylalanine methyl ester (105) in 90% yield.
 | ||
| Scheme 44 | ||
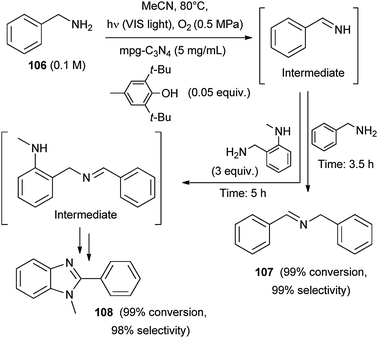
Aerobic oxidative coupling of benzyl amines (e.g.106) was carried out by mpg-C3N4 under visible light irradiation.77 Thus, the corresponding imine was initially formed that upon reaction with the starting benzyl amine afforded the corresponding N-benzylidene benzylamine (107, a second imine), within 3.5 h irradiation (Scheme 45). When a substituted aromatic amine was added to the reaction mixture various heteroaromatics including benzoxazoles, benzimidazoles (108) and benzothiazoles were obtained via intramolecular cyclization and subsequent oxidation of the imine formed. (Scheme 45).77
 | ||
| Scheme 45 | ||
Another emerging application of photoorganocatalysts is their use in radical or cationic polymerizations through a photoredox process.78 In this case, POCs are mainly polycyclic aromatics such as pyrene, anthracene, naphthacene, and pentacene derivatives78a,b albeit mesoporous graphitic carbon nitride was used as well.78c The process starts with an ET reaction between the POC* and an organic molecule under visible light irradiation. The radicals formed in the process (from an amine, an alkyl halide, a trialkylsilane) initiate the polymerization.
3. Conclusion and perspective
Photoorganocatalysis fundamentally differs from organocatalysis. The key idea of organocatalysis is that the substrate becomes covalently bound or at least complexed to a small organic molecule. This enhances the reactivity or changes it (frequently causing the Umpolung of the substrate) and quite often directs the stereochemistry of the ensuing attack, so that in the mind of synthesis practitioners organocatalysis is classified essentially as a method for asymmetric synthesis. This does not apply in any way to photoorganocatalysis (as defined here), because the short lifetime of electronically excited states makes possible only very fast reactions that are unlikely to be affected by a chiral moiety present in the catalyst. It is unlikely that during the ns to μs lifetime of the excited state a strong interaction develops, let alone arriving at a significant stereoselectivity. Rather, the field of action of POC involves, just as in the PC case, the activation of the substrate through a chemical reaction and the formation of a reactive intermediate, such as a radical, an ion or a radical ion. Thus, the two phenomena are essentially unrelated, but this will not hinder, in our opinion, that the two disciplines grow independently. In fact, POC takes advantage of the high energy of excited states of ‘small’ organic molecules for generating intermediates under otherwise unattainable mild conditions either via atom transfer or via a redox process. The high energy incorporated into the excited states makes accessible reaction paths that have no role in thermal reactions, catalytic or not, unless extremely aggressive reagents are used, whereas here these are generated in the neat solvent or in a medium conveniently devised for trapping them. Furthermore, the two actions may be combined. As an example, in the process shown in Scheme 28, the nucleophile is generated in situ through thermal organocatalysis and the electrophilic radical through independent POC. This is a promising way to profit from the characteristics of both phenomena. A case where a single photoorganocatalyst (44) complexes the reagent and is involved in the activation step (through the operation of two different moieties of the molecule) is illustrated in Scheme 23. This may be another development path (scarcely trodden at present). Whether the present POC definition should be widened to include the formation of a ground state complex may be an argument of debate.Advancement in the area requires that robust photocatalysts are developed that rival inorganic complexes and semiconductor oxides in terms of light fastness and can thus exploit their high and varied reactivity for activating chemical processes much beyond the sparse examples present in the literature at this moment. New compounds and materials that could be tested as POCs may become available also from different fields, such as organic compounds applied in solar cells, for water splitting or photocatalytic depollution as well as from other photoresponsive materials and devices. In particular carbon based materials such as nanocarbons79 or graphene-based derivatives80 or other aromatic compounds such as acene-based derivatives, oligothiophenes, porphyrins and phthalocyanines81 need to be considered as new POCs. Furthermore, as mentioned, it is possible that more general interactions between organocatalysis and photo(organo)catalysis develop in the future and exploit the opposite characteristics of the two methods.
References
- M. Beller and C. Bolm, Transition Metals for Organic Synthesis, 2nd Ed, Wiley-VCH, Weinheim, 2004 Search PubMed.
- A. Berkessel and H. Gröger, Asymmetric Organocatalysis, Wiley-VCH, Weinheim, 2005 RSC; R. C. Wende and P. R. Schreiner, Green Chem., 2012, 14, 1821 RSC.
- X. Bugaut and F. Glorius, Chem. Soc. Rev., 2012, 41, 3511 RSC.
- (a) M. Fagnoni, D. Dondi, D. Ravelli and A. Albini, Chem. Rev., 2007, 107, 2725 CrossRef CAS and references quoted therein; (b) D. Ravelli, D. Dondi, M. Fagnoni and A. Albini, Chem. Soc. Rev., 2009, 38, 1999 RSC; (c) G. Palmisano, V. Augugliaro, M. Pagliaro and L. Palmisano, Chem. Commun., 2007, 3425 RSC; (d) N. Hoffmann, ChemSusChem, 2012, 5, 352 CrossRef CAS.
- (a) J. W. Tucker and C. R. J. Stephenson, J. Org. Chem., 2012, 77, 1617 CrossRef CAS; (b) J. M. R. Narayanam and C. R. J. Stephenson, Chem. Soc. Rev., 2011, 40, 102 RSC; (c) T. P. Yoon, M. A. Ischay and J. Du, Nat. Chem., 2010, 2, 527 CrossRef CAS; (d) F. Teply, Collect. Czech. Chem. Commun., 2011, 76, 859 CrossRef CAS.
- N. Hoffmann, J. Photochem. Photobiol., C, 2008, 9, 43 CrossRef CAS.
- D. Ravelli and M. Fagnoni, ChemCatChem, 2012, 4, 169 CrossRef CAS.
- M. L. Marin, L. Santos-Juanes, A. Arques, A. M. Amat and M. A. Miranda, Chem. Rev., 2012, 112, 1710 CrossRef CAS.
- See for example: H. Sundén, M. Engqvist, J. Casas, I. Ibrahem and A. Córdova, Angew. Chem., Int. Ed., 2004, 43, 6532 CrossRef.
- See for example: P. Wessig, Angew. Chem., Int. Ed., 2006, 45, 2168 CrossRef CAS.
- C. Müller, A. Bauer, M. M. Maturi, M. C. Cuquerella, M. A. Miranda and T. Bach, J. Am. Chem. Soc., 2011, 133, 16689 CrossRef.
- L. Cui, N. Tada, H. Okubo, T. Miura and A. Itoh, Green Chem., 2011, 13, 2347 RSC.
- K. Tahara and Y. Hisaeda, Green Chem., 2011, 13, 558 RSC.
- J. Pérez-Prieto, R. E. Galian and M. A. Miranda, Mini-Rev. Org. Chem., 2006, 3, 117 CrossRef.
- M. A. Miranda and H. Garcia, Chem. Rev., 1994, 94, 1063 CrossRef CAS.
- Y. Wang, X. Wang and M. Antonietti, Angew. Chem., Int. Ed., 2012, 51, 68 CrossRef CAS.
- H. Liu, W. Feng, C. W. Kee, Y. Zhao, D. Leow, Y. Pan and C.-H. Tan, Green Chem., 2010, 12, 953 RSC.
- H. Kotani, K. Ohkubo and S. Fukuzumi, J. Am. Chem. Soc., 2004, 126, 15999 CrossRef CAS.
- K. Ohkubo, T. Nanjo and S. Fukuzumi, Org. Lett., 2005, 7, 4265 CrossRef CAS.
- Y.-Q. Zou, J.-R. Chen, X.-P. Liu, L.-Q. Lu, R. L. Davis, K. A. Jørgensen and W.-J. Xiao, Angew. Chem., Int. Ed., 2012, 51, 784 CrossRef CAS.
- H.-J. Xu, F.-F. Zhu, Y.-Y. Shen, X. Wan and Y.-S. Feng, Tetrahedron, 2012, 68, 4145 CrossRef CAS.
- A. G. Griesbeck, M. Reckenthäler and J. Uhlig, Photochem. Photobiol. Sci., 2010, 9, 775 CAS.
- D. Prasad Hari and B. König, Org. Lett., 2011, 13, 3852 CrossRef.
- J. Xuan, Z.-J. Feng, S.-W. Duan and W.-J. Xiao, RSC Adv., 2012, 2, 4065 RSC.
- N. Tada, Y. Ikebata, T. Nobuta, S.-i. Hirashima, T. Miura and A. Itoh, Photochem. Photobiol. Sci., 2012, 11, 616 CAS.
- N. Tada, K. Hattori, T. Nobuta, T. Miura and A. Itoh, Green Chem., 2011, 13, 1669 RSC.
- K. Ohkubo, K. Mizushima, R. Iwata, K. Souma, N. Suzuki and S. Fukuzumi, Chem. Commun., 2010, 46, 601 RSC.
- A. G. Griesbeck and M. Cho, Org. Lett., 2007, 9, 611 CrossRef CAS.
- K. Ohkubo, K. Suga and S. Fukuzumi, Chem. Commun., 2006, 2018 RSC.
- H. J. Xu, X. L. Xu, Y. Fu and Y. S. Feng, Chin. Chem. Lett., 2007, 18, 1471 CrossRef CAS.
- R. Lechner, S. Kümmel and B. König, Photochem. Photobiol. Sci., 2010, 9, 1367 CAS.
- F. Su, S. C. Mathew, G. Lipner, X. Fu, M. Antonietti, S. Blechert and X. Wang, J. Am. Chem. Soc., 2010, 132, 16299 CrossRef CAS.
- P. Zhang, Y. Wang, J. Yao, C. Wang, C. Yan, M. Antonietti and H. Li, Adv. Synth. Catal., 2011, 353, 1447 CrossRef CAS.
- S. M. Bonesi, E. Carbonell, H. Garcia, M. Fagnoni and A. Albini, Appl. Catal., B, 2008, 79, 368 CrossRef CAS.
- P. Zhang, Y. Wang, H. Li and M. Antonietti, Green Chem., 2012, 14, 1904 RSC.
- M. Neumann, S. Füldner, B. König and K. Zeitler, Angew. Chem., Int. Ed., 2011, 50, 951 CrossRef CAS.
- M. Neumann and K. Zeitler, Org. Lett., 2012, 14, 2658 CrossRef CAS.
- S. Gazi and R. Ananthakrishnan, Appl. Catal., B, 2011, 105, 317 CrossRef CAS.
- S. Bertrand, C. Glapski, N. Hoffmann and J.-P. Pete, Tetrahedron Lett., 1999, 40, 3169 CrossRef CAS.
- S. Bertrand, N. Hoffmann, S. Humbel and J.-P. Pete, J. Org. Chem., 2000, 65, 8690 CrossRef CAS.
- S. Bertrand, N. Hoffmann and J.-P. Pete, Tetrahedron Lett., 1999, 40, 3173 CrossRef CAS.
- D. Harakat, J. Pesch, S. Marinković and N. Hoffmann, Org. Biomol. Chem., 2006, 4, 1202 CAS.
- S. Marinković, C. Brulé, N. Hoffmann, E. Prost, J.-M. Nuzillard and V. Bulach, J. Org. Chem., 2004, 69, 1646 CrossRef.
- A. Bauer, F. Westkamper, S. Grimme and T. Bach, Nature, 2005, 436, 1139 CrossRef CAS.
- Y. Miyake, K. Nakajima and Y. Nishibayashi, J. Am. Chem. Soc., 2012, 134, 3338 CrossRef CAS.
- M. Jonas, S. Blechert and E. Steckhan, J. Org. Chem., 2001, 66, 6896 CrossRef CAS.
- J. Santamaria, M. T. Kaddachi and J. Rigaudy, Tetrahedron Lett., 1990, 31, 4735 CrossRef CAS.
- J. Santamaria, D. Herlem and F. Khuong-Huu, Tetrahedron, 1977, 33, 2389 CrossRef CAS.
- Y. Pan, S. Wang, C. W. Kee, E. Dubuisson, Y. Yang, K. P. Loh and C.-H. Tan, Green Chem., 2011, 13, 3341 RSC.
- G. Pandey and S. R. Gadre, Arkivoc, 2003, 45 CAS.
- Y. Pan, C. W. Kee, L. Chen and C.-H. Tan, Green Chem., 2011, 13, 2682 RSC.
- Q. Liu, Y.-N. Li, H.-H. Zhang, B. Chen, C.-H. Tung and L.-Z. Wu, Chem.–Eur. J., 2012, 18, 620 CrossRef CAS.
- K. Fidaly, C. Ceballos, A. Falguiéres, M. Sylla-Iyarreta Veitia, A. Guy and C. Ferroud, Green Chem., 2012, 14, 1293 RSC.
- G. Pandey and R. Sochanchingwung, J. Chem. Soc., Chem. Commun., 1994, 1945 RSC.
- (a) B. Fraser-Reid, N. L. Holder, D. R. Hicks and D. L. Walker, Can. J. Chem., 1977, 55, 3978 CrossRef CAS; (b) B. Fraser-Reid, R. C. Anderson, D. R. Hicks and D. L. Walker, Can. J. Chem., 1977, 55, 3986 CrossRef CAS; (c) Z. Benko, B. Fraser-Reid, P. S. Mariano and A. L. J. Beckwith, J. Org. Chem., 1988, 53, 2066 CrossRef CAS.
- C. Manfrotto, M. Mella, M. Freccero, M. Fagnoni and A. Albini, J. Org. Chem., 1999, 64, 5024 CrossRef CAS.
- H. Graalfs, R. Fröhlich, C. Wolff and J. Mattay, Eur. J. Org. Chem., 1999, 1057 CrossRef CAS.
- D. Dondi, I. Caprioli, M. Fagnoni, M. Mella and A. Albini, Tetrahedron, 2003, 59, 947 CrossRef CAS.
- D. Dondi, S. Protti, A. Albini, S. Mañas Carpio and M. Fagnoni, Green Chem., 2009, 11, 1653 RSC.
- M. Oelgemöller, C. Schiel, R. Fröhlich and J. Mattay, Eur. J. Org. Chem., 2002, 2465 CrossRef.
- R. A. Doohan, J. J. Hannan and N. W. A. Geraghty, Org. Biomol. Chem., 2006, 4, 942 CAS.
- R. A. Doohan and N. W. A. Geraghty, Green Chem., 2005, 7, 91 RSC.
- (a) M. Fagnoni, M. Mella and A. Albini, J. Org. Chem., 1998, 63, 4026 CrossRef CAS; (b) M. Fagnoni, M. Mella and A. Albini, J. Am. Chem. Soc., 1995, 117, 7877 CrossRef CAS.
- (a) K. Mizuno, T. Nishiyama, N. Takahashi and H. Inoue, Tetrahedron Lett., 1996, 37, 2975 CrossRef CAS; (b) K. Mizuno, N. Takahashi, T. Nishiyama and H. Inoue, Tetrahedron Lett., 1995, 36, 7463 CrossRef CAS.
- G. Gutenberger, E. Steckhan and S. Blechert, Angew. Chem., Int. Ed., 1998, 37, 660 CrossRef CAS.
- K. Nakanishi, K. Mizuno and Y. Otsuji, J. Chem. Soc., Chem. Commun., 1991, 90 RSC.
- G. Pandey, G. Lakshmaiah and G. Kumaraswamy, J. Chem. Soc., Chem. Commun., 1992, 1313 RSC.
- T. Peglow, S. Blechert and E. Steckhan, Chem. Commun., 1999, 433 RSC.
- J. E. Arguello, R. Pérez-Ruiz and M. A. Miranda, Org. Lett., 2007, 9, 3587 CrossRef.
- N. Arai, K. Tanaka and T. Ohkuma, Tetrahedron Lett., 2010, 51, 1273 CrossRef CAS.
- G. Pandey, A. Murugan and M. Balakrishnan, Chem. Commun., 2002, 624 RSC.
- G. Pandey, M. Karthikeyan and A. Murugan, J. Org. Chem., 1998, 63, 2867 CrossRef CAS.
- (a) D. Prasad Hari, P. Schroll and B. König, J. Am. Chem. Soc., 2012, 134, 2958 CrossRef; (b) P. Schroll, D. Prasad Hari and B. König, ChemistryOpen, 2012, 1, 130 CrossRef CAS.
- K. Ohkubo, K. Mizushima, R. Iwata and S. Fukuzumi, Chem. Sci., 2011, 2, 715 RSC.
- H. R. Memarian and A. Saffar-Teluri, J. Mol. Catal. A: Chem., 2007, 274, 224 CrossRef CAS.
- R. Lechner and B. König, Synthesis, 2010, 1712 CAS.
- F. Su, S. C. Mathew, L. Möhlmann, M. Antonietti, X. Wang and S. Blechert, Angew. Chem., Int. Ed., 2011, 50, 657 CrossRef CAS.
- (a) J. P. Fouassier and J. Lalevée, RSC Adv., 2012, 2, 2621 RSC; (b) M.-A. Tehfe, J. Lalevée, F. Morlet-Savary, B. Graff, N. Blanchard and J.-P. Fouassier, Macromolecules, 2012, 45, 1746 CrossRef CAS; (c) B. Kiskan, J. Zhang, X. Wang, M. Antonietti and Y. Yagci, ACS Macro Lett., 2012, 1, 546 CrossRef CAS.
- F. D′Souza and O. Ito, Chem. Soc. Rev., 2012, 41, 86 RSC.
- Q. Xiang, J. Yu and M. Jaroniec, Chem. Soc. Rev., 2012, 41, 782 RSC.
- H. Dong, H. Zhu, Q. Meng, X. Gong and W. Hu, Chem. Soc. Rev., 2012, 41, 1754 RSC.
| This journal is © The Royal Society of Chemistry 2013 |