Targeting and delivery of platinum-based anticancer drugs
Xiaoyong
Wang
*a and
Zijian
Guo
*b
aState Key Laboratory of Pharmaceutical Biotechnology, School of Life Sciences, Nanjing University, State Key Laboratory of Analytical Chemistry for Life Science, Nanjing 210093, P. R. China. E-mail: boxwxy@nju.edu.cn; Fax: +86 25 83314502; Tel: +86 25 83594549
bState Key Laboratory of Coordination Chemistry, School of Chemistry and Chemical Engineering, Nanjing University, Nanjing 210093, P. R. China. E-mail: zguo@nju.edu.cn; Fax: +86 25 83314502; Tel: +86 25 83594549
First published on 5th October 2012
Abstract
Platinum-based anticancer drugs occupy a crucial role in the treatment of various malignant tumours. However, the efficacy and applicability of platinum drugs are heavily restricted by severe systemic toxicities and drug resistance. Different drug targeting and delivery (DTD) strategies have been developed to prevent the shortcomings of platinum-based chemotherapy. These approaches can be roughly categorized into two groups; namely, active and passive tactics. Active DTD is realized through specific molecular interactions between the drugs and cell or tissue elements, while passive DTD is achieved by exploiting the enhanced permeability and retention effect in tumour tissues. The principal methods for active DTD include conjugation of platinum drugs with selective targeting moieties or encapsulation of platinum drugs in host molecules. Bioactive substances such as hormones, carbohydrates, bisphosphonates, peptides and proteins are commonly used in active DTD. Passive DTD generally involves the fabrication of functionalized polymers or nanoparticles and the subsequent conjugation of platinum drugs with such entities. Polymeric micelles, liposomes, nanotubes and nanoparticles are frequently used in passive DTD. In some cases, both active and passive mechanisms are involved in one DTD system. This review concentrates on various targeting and delivery techniques for improving the efficacy and reducing the side effects of platinum-based anticancer drugs. The content covers most of the related literatures published since 2006. These innovative tactics represent current state-of-the-art developments in platinum-based anticancer drugs.
 Xiaoyong Wang | Xiaoyong Wang was born in Gansu, China, in 1963. He received his BSc degree in Chemistry in 1986 at Northwest Normal University, and his MMSc degree in Medicinal Chemistry in 1994 at Shandong University, where he worked as a Lecturer from 1994 to 2000. He obtained his PhD degree in 2003 at Nanjing University and stayed there as a postdoctoral research fellow from 2003 to 2005. He is now an Associate Professor in the School of Life Sciences, Nanjing University. His research is mainly in the fields of Inorganic Medicinal Chemistry and Biochemistry, particularly the design and mechanism of metallodrugs. |
 Zijian Guo | Zijian Guo was born in Hebei, China, in 1961. He received his PhD degree from the University of Padua in 1994. After completing postdoctoral research at the University of London, the University of British Columbia and the University of Edinburgh, he joined Nanjing University in 1999. He is currently a Professor of Chemistry in the State Key Laboratory of Coordination Chemistry, and the dean of the School of Chemistry and Chemical Engineering, Nanjing University. His research interests include the design and mechanism of action of metal-based anticancer complexes, biological investigations of fluorescent metal sensors, and metal-based artificial nucleases and proteases. |
1 Introduction
Platinum-based anticancer agents are a mainstay of clinical drugs for the treatment of various solid tumors such as genitourinary, colorectal, and non-small cell lung cancers.1–3 The leading anticancer drug, cisplatin, has been used for more than three decades in standard chemotherapy regimens either as a single therapeutic modality or in combination with other cytotoxic agents or radiotherapy.4,5 However, platinum-based anticancer chemotherapy is associated with severe side effects because of poor specificity.6 In the case of cisplatin, systemic toxicities like nephrotoxicity, neurotoxicity, ototoxicity, and emetogenesis inflict serious disorders or injuries on the patients during the treatment, which badly restrict its efficacy.7–9In addition to systemic toxicities, the efficacy of cisplatin is often limited by the intrinsic and acquired resistance possessed by various cancers.10 Multiple mechanisms have been proposed to elucidate the cellular resistance to cisplatin and its analogues in preclinical models.11,12 The four representatives include: (i) decreased drug accumulation or increased drug efflux;13 (ii) increased detoxification of the drug by sulfur-containing molecules within the cells;14,15 (iii) enhanced repair and increased tolerance to DNA damage;16 and (iv) changes in molecular pathways involved in the regulation of cell survival or cell death.17 Since these mechanisms have been expounded in a series of reviews,18–22 we shall skip the details here for brevity.
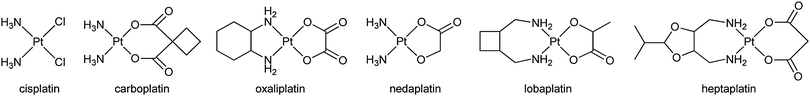
The disadvantages of cisplatin have created a sustained momentum for the improvement of platinum-based anticancer drugs.23 Over the last 40 years, thousands of platinum complexes have been prepared in the hope of finding new drugs with a more tolerable toxicological profile and higher efficacy.24,25 These efforts have brought five more drugs into clinical use, i.e. carboplatin, oxaliplatin, nedaplatin, lobaplatin, and heptaplatin, and about 10 other complexes are currently under clinical trials.26–28 Each of the latecomers shows some qualities that are not revealed by cisplatin. For example, nedaplatin displays less nephrotoxicity and neurotoxicity than cisplatin and carboplatin,29,30 and oxaliplatin demonstrates less toxicity and little or no cross resistance to cisplatin or carboplatin.31 However, since most of these drugs operate via a similar non-specific mechanism of action, some defects of cisplatin are consequently retained, albeit to a lesser extent. Thus, simple modification of the ligands seems unlikely to bring about a quality leap from an indiscriminative drug to a “magic bullet”.
Systemic toxicity and drug resistance are the main concerns in current development of platinum anticancer agents. Ideally, future platinum drugs should attack exclusively cancerous cells without affecting normal ones, and enter the former more readily than the latter. However, this goal is virtually unattainable for such a complicated disease as cancer. Nonetheless, it is possible to approach the ideal situation by developing platinum-based prodrugs that are safe in the administered form but are cytotoxic within the cancer cells after being activated under certain conditions. Obviously, the realization of this ideal is determined by the tumour selectivity of platinum complexes.
Generally speaking, at least three options are viable in the design of new platinum drugs: (i) constructing complexes that display different DNA-binding modes; (ii) exploiting prodrugs that can be activated only in the tumour tissues; and (iii) improving drug accumulation at the tumour site by means of an accurate targeting and delivery strategy.32 The first category comprises polynuclear platinum complexes, trans-platinum complexes, and monofunctional platinum complexes.33 The second category includes complexes that exploit the unique features of solid tumours, such as acidic pH,34 and hypoxic or reducing conditions. For instance, inert platinum(IV) complexes can be reduced to cytotoxic platinum(II) complexes with the loss of the two axial ligands under hypoxic conditions in the tumour tissue, and thus, act as prodrugs.35,36 This review will focus on the third category, that is, ameliorating the selective accumulation of platinum drugs in tumour tissues through targeting and delivery techniques. Since many valuable reviews and books on this topic have appeared over the years,37–46 the materials of this review are exclusively sourced from literature published since 2006.
2 Drug targeting and delivery
Drug targeting and delivery (DTD) represents a highly active field of research for drugs that can go straight to their biological targets as “magic bullets”.47 In comparison with traditional chemotherapy, targeted therapy for cancers has two major advantages: avoidance of damage to normal tissues, and restraint of drug resistance. In recent years, various DTD approaches have been developed in an attempt to minimize the systemic toxicity and drug resistance of platinum-based anticancer drugs.48,49 The ultimate goal of these endeavors is to obtain platinum drugs that are highly selective for tumour tissues and can be administered at lower doses with fewer side effects and an improved therapeutic index.DTD can be active or passive. Active DTD is based on the specific biomolecular interactions between drugs and cell or tissue elements. This approach can be applied to tumours containing biochemical entities whose quantity or functionality differs from those of normal tissues. In a typical active DTD system, the pharmacophore is bound to the targeting moiety via a spacer and a linker; the specific functionality of the transporter-, antigen- or receptor-based conjugate drives the drug towards the tumour tissue by virtue of its specific binding affinity.50 Bioactive substances, such as hormones, sugars, amino acids, proteins, and bis-phosphonates, are commonly employed to fulfil the targeting function. Additionally, biodegradable molecules, such as polysaccharides, polyamino acids, proteins, and water soluble poly(ethylene glycol) (PEG), are adopted to perform the delivery function. The addition of targeting functionality to the drug makes it possible to distinguish cancerous cells or tissues form healthy ones, and thereby ensures the high efficacy and low side effects of the drug.51
Passive DTD is achieved by taking advantage of the enhanced permeability and retention (EPR) effect in tumour tissues.52 Tumour vasculature is conspicuously disorganized and twisted as compared with the vasculature in normal tissues. The vascular endothelium in tumours proliferates rapidly and discontinuously, resulting in a higher number of fenestrations and open junctions than normal vessels, ranging from 200 nm to 1.2 μm.53 Consequently, particles that are small enough, typically measuring on the order of a few hundred nanometers, can passively cross the tumour endothelial barrier through fenestrations. Moreover, the lack of a functional lymphatic network prevents the efficient removal of excess fluid from the solid tumour tissue. The combination of these two effects makes tumours hyperpermeable to the circulating macromolecules, which extravasate and accumulate in the solid tumour tissue because of inefficient drainage by the lymphatic system, and remain there for substantial periods of time (Fig. 1).54 For platinum-based anticancer drugs, carriers used in passive DTD usually contain donor groups capable of coordinating with the platinum moiety.
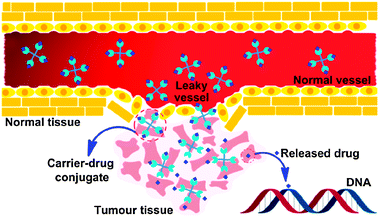 | ||
| Fig. 1 Schematic representation of the EPR effect: normal vessels have a tight endothelium, while tumour vessels are disorganized and leaky, allowing preferential extravasation of circulating macromolecules. In tumour tissues, the carrier-drug conjugate is cleaved to generate the active platinum species, leading to the formation of cell-lethal DNA adducts. | ||
The EPR effect is evident in a large number of tumour types; it is applicable for any biocompatible macromolecular compounds above 40 kDa. The drug concentration in tumour tissue can be 10–30 times higher than that in the blood. It is noteworthy that the EPR effect not only brings on the momentary passive DTD for tumour tissues, but also prolongs the drug retention for more than several weeks or longer.55 In fact, to exert a substantial EPR effect, drugs need to circulate in the bloodstream for at least 6 h. With the help of the EPR effect, novel cancer-specific platinum drugs can be created by exploiting the properties of nanoparticles and macromolecules such as micelles and liposomes.
The microenvironmental difference between normal and tumour cells plays a key role in some DTD systems to generate the active platinum species. Normal cells typically use mitochondrial oxidative phosphorylation to metabolize glucose and switch over to glycolysis only when there is little or no oxygen, producing lactate as a byproduct. By contrast, the metabolism of cancer cells is characterized by the increase in aerobic glycolysis and the dependency on the glycolytic pathway for ATP generation, known as the Warburg effect. Glycolysis is a series of metabolic processes by which one molecule of glucose is catabolized into two molecules of pyruvate, a net gain of two ATP, and two protons:56
| Glucose + 2P |
| i |
| + 2ADP + 2NAD |
| + |
| → 2Pyruvate + 2ATP + 2NADH + 2H |
| + |
| + 2H |
| 2 |
| O |
3 Active targeting and delivery
3.1 Estrogens as carriers
Estrogens can easily cross the cellular membrane by passive diffusion because of high lypophilicity, bind to the estrogen receptor (ER) in the cytoplasm and then be transferred to the nucleus, hence they are attractive carriers for the active DTD. Tissues and the corresponding cell lines that express ERs are defined as estrogen responsive ER(+). In addition to the classical estrogen receptor α (ERα), a new estrogen receptor β (ERβ) is also involved in the growth modulation of normal and malignant breasts and other tissues. ERα mediates the proliferative effect of estrogen in breast cancer cells, whereas ERβ seems to be antiproliferative. Since the distribution of the two ERs is not uniform in the target cells, the hormone derivatives could lead to either agonistic or antagonistic biological effects.57ERβ is an interesting therapeutic target in breast cancer cells and ERβ-selective agonists are potential drugs for the therapy of ERα(+)/ERβ(+) breast cancer due to their antiproliferative and antiangiogenic properties.58 Tissues rich in ERs, such as breast and ovarian cancers, tend to accumulate molecules that have high binding affinity for these receptors. Therefore, molecules that bind to the ER and have favorable cellular transport properties can be incorporated into platinum complexes to target these cancers. For example, the biological affinity between 17β-estradiol and its cognate receptor can theoretically be used to direct a platinum agent to the target cells. Conjugates of platinum complexes with natural or synthetic estrogens and antiestrogens targeting the nuclear ERs have been reviewed recently.59 These conjugates are targeted cytotoxic agents for ER-rich tissues such as hormone-dependent breast cancer.60 At least two benefits are possible from attaching estrogen to platinum complexes: increasing cellular uptake of the drug, and sensitizing cells to the drug. The following examples are some new platinum complexes designed on such principles.
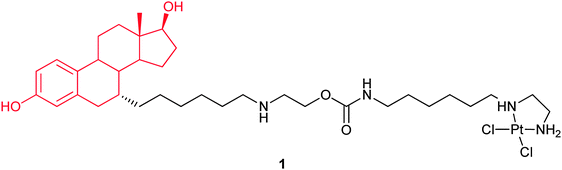
In complex 1, a DNA damaging warhead, [Pt(ethylenediamine)Cl2], is tethered through a carbamate-containing linker to the steroid residue. The ligand has 28% relative binding affinity for the ER as compared to 17β-estradiol. After covalent binding to a synthetic DNA duplex 16-mer, the affinity of 1 for the ER is still retained. Complex 1 shows higher cytotoxicity against the ER(+) ovarian cancer cell line CAOV3 than the control compound. It is also more toxic to the ER(+) MCF-7 than to the ER(−) MDA-MB-231 breast cancer cell lines. These results indicate that both the presence of the estradiol moiety in 1 and the expression of the ER in target cells contribute to the enhanced activity.61
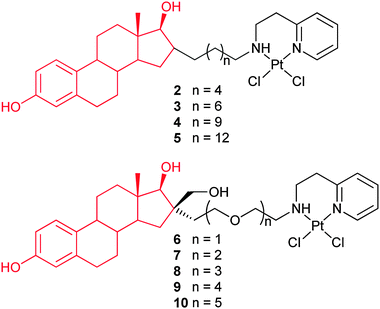
Complexes 2–5 are made of 17β-estradiol and a cisplatin analogue linked together by a 6–14 carbon atom chain. These complexes are 4–9 times more cytotoxic than cisplatin on the ER(+) MCF-7 and ER(−) MDA-MB-231 breast cancer cell lines. Among them, 4 exhibits a promising cytotoxicity towards all the tested cancer cells, while 2 shows the least cytotoxicity. This may be due to the short linkage which causes additional steric hindrance around the 17-hydroxyl group of the steroid, which is necessary for binding to the ER. As expected, these complexes display similar binding affinities for the ERα and ERβ, close to that of 17β-estradiol, and cisplatin shows no affinity for the ERs. Their strong cytotoxic activity may be partly attributed to their interactions with the ERα and ERβ. The length of the alkyl chain separating the estradiol from the platinum moiety does not seem to play a crucial role for the ER binding affinity and cytotoxicity. Unexpectedly, these complexes show no clear specific action on estrogen-dependent cells as compared to estrogen-independent cells in vitro. However, several in vivo assays with 4 on nude mouse xenografted model show strong and selective anticancer activity on hormone-dependent breast and ovarian cancers. Consequently, complexes 2–5 have the potential to target the ER in vivo and reduce the systemic toxicities.62 In analogues 6–10, the alkyl linking chain is replaced with a PEG linking chain of various lengths. The length of the PEG chain does not influence the solubility of the complexes. The most active complex 10 contains 5 ethylene glycol units and is equipotent to cisplatin against breast cancer cell lines MCF-7 and MDA-MB-231. However, they also present no specific toxicity towards ER(+) MCF-7 cells in vitro.63
Estradiol-linked carboplatin and oxaliplatin analogues have also been prepared. Their antiproliferative activities on MCF-7 and MDA-MB-231 cell lines are at the micromolar range and more active than carboplatin and oxaliplatin alone but less active than their cisplatin counterparts. Oxaliplatin derivatives show a high affinity for ERα whereas carboplatin derivatives show a very low affinity for ERα, suggesting that the nature of the platinum(II) unit is important for the antiproliferative activity and estrogen-selectivity of the vector complexes.64
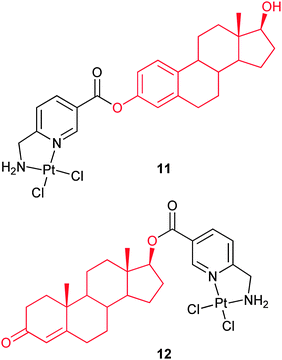
The cellular accumulation and inhibitory effect of several dichloro(6-aminomethylnicotinate)platinum(II) steroid conjugates have been investigated on ER(+)/ER(−) MCF-7 and MDA-MB-231 breast cancer cells. Complex 11, with 3-O-linked estrogen, distinctly inhibits the growth of ER(+) MCF-7 cells, but has little if any effect on ER(−) MCF-7 cells. The same behavior of complex 12 with 17-O-linked androgen against ER(+) and ER(−) breast cancer cell lines is less effective. These complexes bind strongly to sex hormone-binding globulin, and 11 also binds strongly and agonistically to the nuclear ERα. The results suggest that the accumulation of 11 and 12 in ER(+) breast cancer cells is a receptor-mediated process. Interestingly, the most cytotoxic complex 11 exerts no DNA distorting effect, suggesting that its mode of action is different from that of cisplatin.65
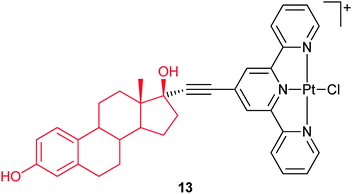
Steroids are effective delivery vehicles, even for charged platinum complexes. In complex 13, the ligand terpyridine is attached to the 17α-position on the estrogen via an ethynyl link. The aim of this design is to add the targeting quality of estrogen to the platinum moiety and thereby to increase the drug accumulation in tumour cells. A whole cell ER assay on ER(+) MCF-7 breast cancer cells confirms the binding of 13 to the ER. Complex 13 also binds to both human and bovine serum albumin (SA) and to DNA, in each case the biomolecule is linked to the platinum(II) center through coordination with the displacement of the labile chloride ligand. Circular dichroism indicates that a termolecular entity involving 13, SA and DNA is formed.66
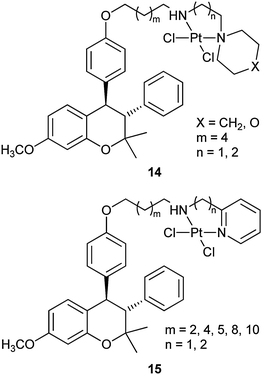
Nonsteroidal estrogens have also been used to selectively target mammary tumour cells.67 For example, benzopyran-based platinum complexes such as 14 and 15 are synthesized as hybrids of selective ER modulators and cytotoxic anticancer agents. The diamminodichloroplatinum(II) group functions as the cytostatic moiety, whereas 3,4-diarylbenzopyran nucleus, well known for its affinity for ERs with selectivity for ERα, acts as the carrier moiety. The platinum(II) moiety is linked with the carrier through a linker of various lengths at a position corresponding to the 7α- or 11β-position of the 17β-estradiol. These complexes are expected to reach the target site efficiently and the possible free amine metabolite could act as a selective ER modulator. Actually, they show significant in vitro cytotoxic activity against different ER-dependent and -independent breast cancer cell lines. Furthermore, complexes from aromatic amines are more potent than their aliphatic amine analogues. However, the length of the linker chain has little effect on the biological activity. These complexes may potentially be used as cytotoxic agents and selective ER modulators in cancer treatment.68 The selective ER modulator tamoxifen, a leading agent in the adjuvant treatment of breast cancer, has also been linked via a spacer to the platinum anticancer group, but no particular benefit is obtained.69
3.2 Carbohydrates as carriers
Carbohydrates such as mono- and polysaccharides are fundamental components of glycolipids and glycoproteins, and are building blocks of nucleotides. Cancer cells commonly display altered sugar metabolism, for instance, increased glucose consumption for energy production via glycolysis to survive in the profoundly hypoxic environment of malignant lesions. Therefore, carbohydrates can be exploited as carriers for platinum-based anticancer drugs. Natural carbohydrates and synthetic derivatives possess manifold donors endowing them with the ability to coordinate with metal centers and provide some advantages over other ligands, for example, biocompatibility, membrane permeability, non-toxicity, enantiomeric purity, and water solubility. More importantly, the existence of specific saccharide receptors exclusively expressed by some cancer cells could enhance the specificity and recognition of platinum drugs. In recent years, a number of platinum complexes with carbohydrates suitably functionalized with mono- and bidentate amine ligands, phosphines or other P-donor groups, S- or Se-donor ligands or alcohols have been synthesized, and some of them are promising anticancer drugs.70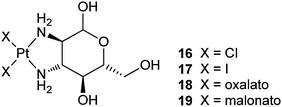
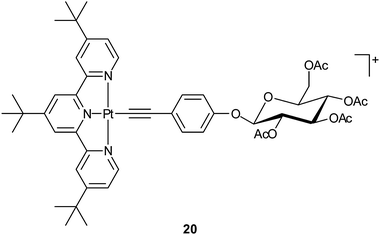
A water-soluble [Pt(terpy)(glycosylated arylacetylide)]+ complex 20 shows up to ∼100 times higher cytotoxicity against human cancer cell lines than cisplatin, and is at least 8-fold more cytotoxic than [Pt(terpy)Cl]+. It is approximately 10-times more active than the non-glycosylated analogous complex. Judged from the morphology and cell membrane integrity, complex 20 induces 52% apoptosis with only 5% necrosis in the large cell lung cancer cells (NCI-H460). Besides, treatment of NCI-H460 cells with 20 results in significant alteration in 111 types of gene expressions. The Pt(II)-glycosylated arylacetylide moiety remains intact in aqueous solution for 72 h at room temperature. This organometallic compound represents a rare example of platinum complexes linked to sugar molecules exhibiting lower IC50 values than cisplatin in cell culture studies.72
 | ||
| Fig. 2 Molecular model of β-cyclodextrin. | ||
In complex 21, an adamantane-tethered carboplatin analogue has been encapsulated in the cavity of βCD. This inclusion complex exhibits higher cytotoxicity towards human neuroblastoma SK-N-SH cells and a higher binding to plasmid pBR322 DNA than carboplatin. However, the cellular uptake rate of carboplatin is about 4 times higher than the inclusion complex. The result suggests that the higher cytotoxicity of 21 is not related to the amount of platinum that enters the cell, but perhaps to the more effective transport of 21 to the nucleus via the appended βCD moiety and thereby more efficient binding to nuclear DNA. Factors such as intracellular transport, receptor binding, and heterogeneous distributions inside cells seem to play more important roles than just the cellular uptake for the cytotoxic effect of the drug.74 Water-insoluble trans-dichloro(dipyridine)platinum(II) is difficult to be used in biological settings. The encapsulation of this complex with βCD increases its solubility to 1.6 mg mL−1. Moreover, the cytotoxicity of the inclusion complex 22in vitro is much higher than that of trans-dichloro(dipyridine)platinum(II) and cisplatin against CT26 colon carcinoma and B16F10 melanoma cell lines.75 These examples demonstrate that βCD is an effective carrier for improving the DNA binding activity and other pharmaco-properties of platinum-based anticancer drugs.
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000000 Da in vivo. Increased presence and uptake of HA have been correlated with the progression and metastasis of prostate and breast cancers.76,77 The carboxyl groups on HA are appropriate for the binding of platinum units. For example, HA could form stable conjugate with cisplatin.78 Since breast cancer cells are known to have greater uptake of HA than normal tissues, and invasive breast cancer cells overexpress CD44, the primary receptor for HA, and are dependent on high concentrations of CD44-internalized HA for proliferation,79 cisplatin–HA conjugates may be efficacious against lymphatic metastases.
000000 Da in vivo. Increased presence and uptake of HA have been correlated with the progression and metastasis of prostate and breast cancers.76,77 The carboxyl groups on HA are appropriate for the binding of platinum units. For example, HA could form stable conjugate with cisplatin.78 Since breast cancer cells are known to have greater uptake of HA than normal tissues, and invasive breast cancer cells overexpress CD44, the primary receptor for HA, and are dependent on high concentrations of CD44-internalized HA for proliferation,79 cisplatin–HA conjugates may be efficacious against lymphatic metastases.
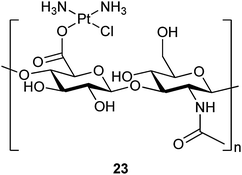
The tissue distribution and anticancer activity of a cisplatin–HA conjugate 23 have been tested to determine whether the targeted cisplatin can increase the localized dose in lymphatic metastases without systemic toxicities. Conjugate 23 releases drug with a half-life of 10 h in saline and shows high anticancer activity in vitro similar to cisplatin in highly metatstatic MCF-7 and MDA-MB-231 human breast cancer cells. In addition, the conjugate is well tolerated in rodents with no signs of injection site morbidity or major organ toxicity after 96 h. Fluorescence imaging confirms the accumulation of 23 in the lymph nodes.80 This is the first in vivo study of cisplatin–HA conjugate and the first examination of HA-drug conjugates designed for lymphatic deposition and retention.
Recently, oxaliplatin has been conjugated to HA-coupled chitosan nanoparticles for targeted delivery to colorectal tumours. In murine models, the drug delivery system results in relatively high local drug concentration in colonic tumours with prolonged exposure time, reflecting its targeting potential with enhanced antitumor efficacy and low systematic toxicity.81
3.3 Bisphosphonates as carriers
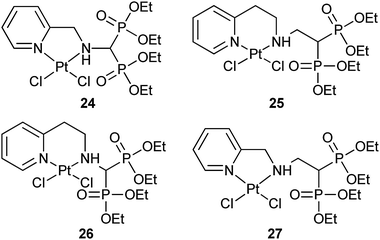
Bisphosphonates (BPs) have shown a high affinity for bone and other calcified tissues, and have widely been used as therapeutic agents for several bone-related diseases.82 The ability to chelate calcium ions is the basis for the bone-targeting property of BPs. Since BPs can be absorbed onto bone surfaces and show significant inhibition to osteoclastic resorption or antitumor effects in preclinical models,83 they are potential bone-targeting carriers for platinum drugs.In our attempt to seek more specific platinum-based anticancer drugs, analogues of picoplatin (ZD0473), a developing drug candidate at Phase III stage, are linked to a bisphosphonate tetraethyl ester targeting carrier. The resultant complexes 24–27 exhibit excellent solubility in both organic and aqueous solutions. In cytotoxicity assay against human osteosarcoma MG-63 and ovarian cancer COC1 cell lines, complexes 25 and 27 demonstrate much higher activity than 24 and 26, which can be correlated to the length of the linkers between the platinum moiety and the targeting bisphosphonate ester. The apoptotic assay with the most cytotoxic complex 27 reveals a different mode of cell death compared to cisplatin. In accordance with this, complexes 24–27 hardly bind to DNA, which is again very different from cisplatin. Thus, these complexes represent a new class of non-classical platinum anticancer agents with promising bone-targeting property.84
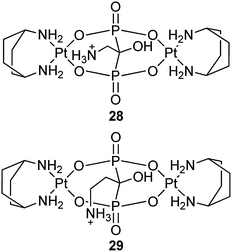
A series of dinuclear platinum(II) complexes with bridging geminal BPs have been reported as prodrugs with potential activity at the bone surface after embedment in inorganic matrices and implantation at the tumour site.85,86 For example, complexes 28 and 29 show a cytotoxic activity with mean IC50 values of 22.46 and 13.57 μM, respectively, towards a panel of 13 human tumor cell lines. More impressively, they are able to overcome the cisplatin-resistance with mean resistance factors of 0.92 for A431 and A431/Pt cervical carcinoma cells and of 0.93 for 2008 and cisplatin-resistant C13* ovarian adenocarcinoma cells, which are roughly 3 and 15 times lower than those calculated for cisplatin in cervical and ovarian carcinoma cells, respectively. Complexes 28 and 29 can also circumvent the multi-drug resistance in the colorectal cell line pair LoVo/LoVo-MDR, with a resistance factor of 1.25.87 Nevertheless, BPs attached to a platinum center as leaving groups are likely to be detached during the delivery in physiological conditions, leading to the loss of bone-targeting property.
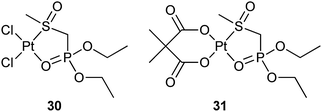
In addition to the bone-targeting ability, some BP-tethered platinum complexes also show an inhibition effect against matrix metalloproteinases (MMPs), which are zinc-dependent endopeptidases mediating the homeostasis of the extracellular matrix and are upregulated in almost every type of human cancers. MMPs are expressed mainly by cancer cells and their overexpression has been correlated with tumour progression; therefore, they are attractive therapeutic targets.88 Platinum complexes 30 and 31 with diethyl[(methylsulfinyl)methyl]phosphonate as the carrier ligand have been proved to selectively inhibit MMP-9, -3, and -12 through a noncompetitive mechanism. In contrast, cisplatin, carboplatin, and the ligand are inactive. The growth inhibitory effects of 30 and 31 are markedly lower than those of cisplatin and carboplatin toward cisplatin-sensitive A2780 ovarian cancer cells, and maintain their activity toward cisplatin-resistant A2780cisR cells. These results demonstrate that BP-modifications to platinum complexes can be exploited to target biological substrates distinct from DNA.89 Such a non-DNA-binding mode is also observed in platinum-pyrophosphato complexes.90
3.4 Peptides and proteins as carriers
Tumour growth and metastasis are driven by angiogenesis; meanwhile, the αvβ3 and αvβ5 integrins and aminopeptidase N (APN) are upregulated in endothelial cells, thus, these cell surface proteins may serve as targets for chemotherapy. Integrins αvβ3 and αvβ5 and APN can recognize the peptide motifs RGD (Arg-Gly-Asp) and NGR (Asn-Gly-Arg), respectively. Therefore, peptide motifs containing RGD, NGR, cyclic pentapeptide (CRGDC)c or (RGDfK)c have been appended to a series of mono- and difunctionalized platinum(IV) complexes 32 and 33 to specifically target the tumour vasculature. Cyclic peptides are chosen because they target angiogenic endothelial cells more efficiently compared to their linear counterparts. Cell inhibition assays show that the Pt(IV)–RGD conjugates are highly and specifically cytotoxic to cell lines containing the integrins, approaching the activity of cisplatin; the Pt(IV)–NGR conjugates are less active than Pt(IV)–RGD complexes but are more active than the nonspecific Pt-peptide controls.91 These results suggest that some surface-protein-recognizable peptide motifs could be exploited as targeting devices for selective delivery of platinum-based anticancer drugs.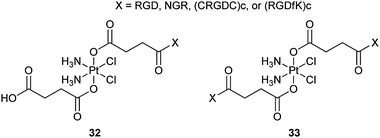
Chlorotoxin (CTX) is a 36-amino-acid peptide with four disulfide bridges. This peptide binds preferentially to glioma cells and many non-glioma tumour cell lines derived from lung, prostate, and melanoma cancers, but not to normal non-transformed cells. Thus, CTX could be integrated into platinum complexes as a targeting agent for the tumour-specific chemotherapy. In order to deliver cisplatin selectively to cancer cells, a conjugate structurally similar to 32 (X = CTX) was prepared recently. Like most platinum(IV) derivatives, the cytotoxicity of the conjugate is lower in cell cultures than that of cisplatin, but greater than those of its Pt(IV) precursor and CTX in several cancer cell lines.92
The native iron-storage protein ferritin (Ft) could be a promising vehicle for the active DTD since the binding sites and endocytosis of Ft have been identified in some tumour cells, and the internalization is associated with membrane-specific receptors. Ft receptors have shown potential value in the delivery of anticancer drugs to the brain. Ft can be easily demineralized into apoferritin (AFt), a hollow protein cage with internal and external diameters of 8 and 12 nm, respectively. This protein cage can be employed to deliver platinum drugs, which may enhance the drug selectivity for cell surfaces that express Ft receptors. On these grounds, we developed an active, as well as a passive, DTD system using AFt to improve the specificity of platinum drugs. Cisplatin, carboplatin, and oxaliplatin have been successfully encapsulated in the cavity of AFt. The encapsulation was achieved through manipulating the pH-dependent unfolding–refolding process of AFt at pH 2.0 and 7.4, respectively, in saturated drug solution (Scheme 1).93 The structural integrity of the protein shell remains intact after encapsulation and hence the potential recognition nature should not be affected. In vitro assays on the rat pheochromocytoma PC12 cell line show that AFt–cisplatin inhibits the cells in a slow but sustaining mode and the cellular uptake of platinum is enhanced by AFt.94 These protein-coated drugs are expected to improve the toxicity profiles of the naked ones and finally to overcome the detrimental effects of platinum-based drugs.
 | ||
| Scheme 1 Schematic illustration of the pH-mediated encapsulation of cisplatin (CDDP), carboplatin (CBDCA), or oxaliplatin (LOHP) by apoferritin (AFt) via an unfolding–refolding process. | ||
3.5 Cucurbiturils as carriers
Cucurbit[n]urils (n = 6, 7, 8) have become the most attractive macrocycles used for encapsulating platinum anticancer drugs in recent years.95 Such systems have shown particular potential as protective carriers in drug delivery. Cucurbit[n]urils contain two symmetrical hydrophilic carbonyl lined portals, capping a central hydrophobic cavity, thus imparting an amphipathic nature to the macrocycles (Fig. 3). The hydrophobic inner cavity provides a favorable binding site for non-polar molecules, while the carbonyl units of the macrocycles are sites for hydrogen bonding and electrostatic interactions with cationic moieties.96 These barrel-shaped molecules could be used as molecular hosts for neutral and charged mono- and multinuclear platinum anticancer agents.97–99![Chemical structure (left), X-ray crystal structure (middle) and electrostatic potential map (right) of cucurbit[7]uril.](/image/article/2013/CS/c2cs35259a/c2cs35259a-f3.gif) | ||
| Fig. 3 Chemical structure (left), X-ray crystal structure (middle) and electrostatic potential map (right) of cucurbit[7]uril. | ||
Partial or full encapsulation within cucurbit[n]urils creates steric hindrance to drug degradation by peptides and proteins, and allows for the tuning of drug release rates, cytotoxicity and toxicity.100 For example, dinuclear platinum complex 34 has been included inside a cucurbit[7]uril macrocycle. Its cytotoxicity against the L1210 cell line and the corresponding cisplatin-resistant L1210/DDP sub-line is not affected heavily by the encapsulation, but the reactivity of the platinum center is reduced at least 3-fold.101 Complex 35 is a DNA intercalator that displays cytotoxicity against a panel of human cancer cell lines, with some activity significantly higher, e.g. up to 100-fold greater in the L1210 and A-427 cell lines, than that of cisplatin. Partial encapsulation of 35 by cucurbit[6]uril barely changes its cytotoxicity;102–105 however, such encapsulation by cucurbit[n]urils (n = 6, 7, 8) can drastically reduce the deactivation by glutathione.106 The size of the cavity and the binding affinity are closely relevant to the cytotoxicity, in that small changes of the size could either decrease or increase the activity (up to 2.5 fold) of the platinum complexes. The decrease in activity may result from the protective effects of the macrocycles on the encapsulated complexes. Nevertheless, in vitro results may not be sufficient to determine the fate of the encapsulated complexes. Recently, it is demonstrated that although the encapsulation of cisplatin in cucurbit[7]uril exhibits no effect on the in vitro cytotoxicity of cisplatin in the human ovarian carcinoma cell line A2780 and its cisplatin-resistant sub-lines A2780/cp70 and MCP1, a significant effect on the in vivo cytotoxicity is observed using human tumour xenografts, in that both A2780 and A2780/cp70 tumours are sensitive to the host–guest complex. The total concentration of the circulating complex over a period of 24 h is significantly higher than that of free cisplatin when administered at the equivalent dose, implying that the improved pharmacokinetics plays a key role in overcoming the drug resistance.107
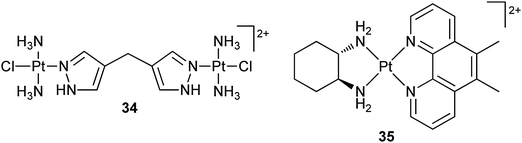
In some cases, encapsulation by cucurbit[n]urils can significantly affect the cytotoxicity and limit the water solubility of platinum complexes.108 For these reasons, a number of other macrocycles such as β-cyclodextrin (vide ante) and calix[4]arene are investigated as potential alternatives. For instance, encapsulation of 35 in these macrocycles increases its stability to glutathione threefold, but shows no significant enhancement of the cytotoxicity against the LoVo human colorectal cancer cell line.109 Similar result is also observed for 34 after such encapsulation.110
3.6 Other alternative carriers
Dichloroacetate (DCA) can reverse the Warburg effect (vide ante) by inhibiting a key enzyme, pyruvate dehydrogenase kinase (PDK), for the process, thereby causing cancer cells to commit suicide by apoptosis.111 In complex 36 (Mitaplatin), two DCA units are appended to the axial positions of a platinum(IV) center. After crossing the plasma membrane, 36 is reduced to release the active drugs cisplatin and DCA. DCA inhibits mitochondrial PDK while cisplatin enters the nucleus to form 1,2-intrastrand d(GpG) DNA cross-links. By this unique mechanism, the complex attacks both nuclear DNA and mitochondria. Complex 36 is equally or more cytotoxic than all known platinum(IV) complexes and is comparable to cisplatin in a variety of cancer cell lines, but is nontoxic in normal cells. These properties demonstrate that a DCA-modified platinum(IV) complex is only effective in cancer cells and therefore could be an alternative avenue for active DTD of platinum anticancer agents.112 A recent study indicates that 36 induces more apoptosis in cisplatin-resistant human epidermoid adenocarcinoma KB-CP 20 and hepatoma BEL 7404-CP 20 cancer cells compared with cisplatin on an equal molar basis, accumulates more than cisplatin in these cells due to enhanced transmembrane permeability, and shows special targeting to mitochondria. As a result, 36 is able to circumvent cisplatin resistance via the dual mechanism.113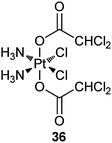
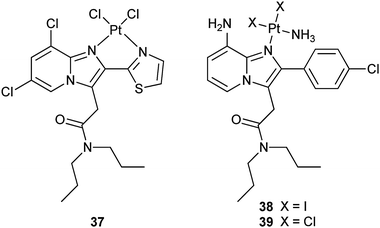
Peripheral benzodiazepine receptor (PBR) is overexpressed in many tumour types, such as brain, liver, breast, and ovary cancers, with its overexpression grade correlating with the malignancy of the tumour. Thus, PBR-binding ligands have been widely explored as carriers for receptor-mediated drug delivery. For example, complex 37 contains a ligand with specific affinity for PBR; as a result, it possesses high affinity and selectivity for the PBR, which makes this compound a potential selective drug for tumours.114 Complexes 38 and 39 have a similar carrier ligand with a nanomolar affinity for PBR. In vitro studies on human and rat glioma cells show that 38 and 39 keep high affinity and selectivity for PBR (nanomolar concentration) and are as cytotoxic as cisplatin. Moreover, they appear to be equally active against cisplatin-sensitive and -resistant A2780 cells. Similar to cisplatin, these complexes induce apoptosis but show a favorable 10- to 100-fold enhanced accumulation in the glioma cells.115
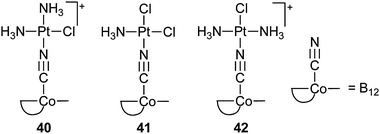
Fast proliferating cells require a higher amount of vitamin B12 than normal cells; therefore, vitamin B12 is an attractive DTD carrier for platinum complexes to enhance the tumour accumulation via the receptor-mediated uptake system. For example, the cyanide group in vitamin B12 can coordinate to various square-planar PtII complexes, forming the central {B12–CN–PtII} motif. These {B12–CN–PtII} conjugates are still recognizable by the intracellular adenosylation enzyme. Release of the platinum complexes from the conjugates is driven by the intracellular reduction of CoIII to CoII to CoI and subsequent adenosylation catalyzed by the adenosyltransferase. Thus, {B12–CN–PtII} conjugates can be considered as prodrugs suitable for targeted delivery of platinum complexes.116 However, preliminary in vitro cytotoxicity studies using the {B12–CN–PtII} conjugates 40–42 indicate that they are less active than cisplatin against the human breast carcinoma cell line MCF-7 and human ovarian cancer cell line A2780, probably owing to a low uptake of the conjugates.117
Cisplatin is usually administered intravenously as a short-term infusion. This yields a high drug concentration in the injection area and quick distribution in the body, leading to high local and systemic toxicity. Carbonated hydroxyapatite crystals (HACs) are similar to the porous structure in bones and hence, can be used to deliver cisplatin. This method has resulted in tumour inhibition and lower systemic toxicity. Cisplatin is adsorbed in the crystals instead of being included as solids. The adsorption depends on the physical and chemical properties of HACs such as the composition, the morphology, the surface area or the size; while the release of the drug depends on temperature, chloride concentration in the medium and crystallinity of HACs. Lower crystallinity leads to higher adsorption and slower release, and temperature slightly increases the drug release rate. The shape of HACs is important for the adsorption and desorption of cisplatin. Although both plate- and needle-shaped HACs have similar Ca/P bulk ratios, the surface areas and Ca/P surface ratios are different. The lower amount of calcium in the surface of needle-shaped HACs allows easier loading of the positively charged aquated platinum species. However, cisplatin release is the same for both shapes.118
4 Passive targeting and delivery
4.1 General concerns
Polymer-drug conjugates are emerging as an important class of anticancer nanomedicines,119 particularly as potential passive DTD systems for platinum-based anticancer drugs.120 Advantages of polymer-drug conjugates include: (i) enhanced cellular uptake because of the EPR effect and dynamic endocytosis characteristic of tumour cells;121 (ii) prolonged circulation time in blood vessels and drug retention time in tumours; and (iii) high drug loading capacity and water solubility. Besides, polymer-drug conjugates can be modified with targeting moieties to actively target the tumour cells or vasculature.122 These properties are helpful in overcoming the multidrug resistance and systemic toxicities that are inherent in current platinum anticancer drugs.Both natural and synthetic polymers could be used as polymeric carriers for the delivery of platinum drugs.123 Polymeric carriers used for DTD should be biodegradable and nontoxic,124 commonly contain binding groups, and may also contain solubilising and targeting units. The binding groups can form covalent links with platinum moieties, and the solubilising and targeting units make the polymers more bioavailable and specific for cancers.125 Targeting units could be antibodies, proteins, peptides or other small molecules.126 In a typical polymer-platinum conjugate, the platinum moiety is linked to the polymeric backbone by a cleavable spacer, and a solubilising or hydrophilic group and a cellular targeting moiety may be attached at different points on the polymer backbone. The polymer-platinum linker must be stable during transport, but capable of releasing the platinum moiety at the therapeutic target. In general, the cleavage of the spacer is accomplished by some enzymes up-regulated in the tumour environment or by pH-sensitive hydrolytic reactions.
In addition to polymer-drug conjugates, passive DTD systems for platinum anticancer drugs also involve nanoparticulate drug delivery systems such as polymeric micelles, liposomes, lipids, dendrimers, nanospheres, and nanoparticles.127–129 Compared to polymer-drug conjugates with size generally around 10 nm or less, the size of these systems is typically in a range of 20–300 nm.130 Ideally, the size of an engineered particle should be in the range of 100–200 nm in diameter. Particles over 300 nm are rapidly recognized and sequestered by the reticuloendothelial system, resulting in either poor drug accumulation at the target site or a short circulation half-life.131 Examples of approved polymers include poly(ethylene glycol) (PEG), poly(vinylpyrolidone) (PVP), poly(N-(2-hydroxypropyl)methacrylamide) (PHPMA), and poly(ethylene oxide) (PEO).132
4.2 Polymer-drug conjugates
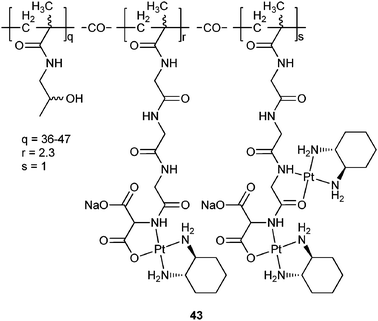
Polymer-platinum conjugates are formed between a polymer with suitable donor groups and a platinum moiety through coordination bonds. Polymers such as poly(amino acids), poly(amidoamine) dendrimers, and PHPMA are generally used as carriers because they contain inherent, pendant or terminal ligating groups.So far, PHPMA is one of the most successful polymers that have been used to construct polymer-platinum conjugates. One of such conjugates, 43 (AP5346), have entered clinical trials. In 43, 1,2-diaminocyclohexaneplatinum(II) (DACHPt) moiety, a fragment of oxaliplatin, is bound to a hydrophilic biocompatible polymer with pH-sensitive triglycine side chains and an aminomalonic acid terminal group.133 The conjugate is much more effective than oxaliplatin with exceptional tolerability in a large number of murine tumour models. The greater tolerability is attributed to the improved drug delivery toward the tumour, in that the platinum release rate of 43 is only 3.5% in 24 h in buffer at pH 7.4, but it is 7-fold higher in a slightly acidic environment (pH 5.4).134 This means that 43 is not active until it reaches the tumour tissues where the environment is more acidic than that of normal tissues. In fact, 43 is capable of delivering 16-fold more platinum to the tumour than oxaliplatin, and 14-fold more platinum-DNA adducts are formed in the nucleus of tumour cells when it is administered at a dose of toxicity equal to that of oxaliplatin. Moreover, at least five times more diaminocyclohexane-platinum units could be administered to patients with 43 compared to oxaliplatin.135 Phase II results indicate that a clinically effective stabilization of disease has been achieved with 43, and there is no indication of the acute neurotoxicity associated with oxaliplatin. Sustained levels of activity are also observed in patients with known resistance to platinum drugs.136 Compound 43 has progressed through phase I trials and a phase II study in patients with recurrent ovarian cancer has been completed under the commercial name of Prolindac™.137
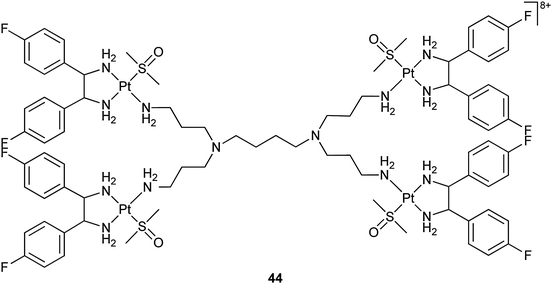
Dendrimers are highly branched tree-like polymers with multiple end groups. The functional groups on the surface of a dendrimer can be used to link platinum drugs via a cleavable chemical bond. Besides, attachment of various moieties to the backbone can provide targeting and other properties to the dendrimer. Poly(amidoamine) dendrimers are commercially available and have been investigated as potential drug delivery systems for several drugs including cisplatin.138 A interesting example is the relatively simple polyimine dendrimer complex 44 in which 4 platinum(II) moieties are bonded to the linker N,N,N′,N′-tetrakis(3-aminopropyl)butane-1,4-diamine. This complex strongly binds to human serum albumin by hydrophobic and electrostatic interactions. It shows a 20-fold higher cellular uptake and a ca. 700-fold higher DNA binding than cisplatin in MCF-7 breast cancer cells without serum medium. The complex crosses the cell membrane through a passive transport and the polyimine dendrimer seems to serve as a carrier for the shuttling of platinum into the cell nuclei. As a result, 44 exhibits a relatively high cytotoxicity in MCF-7 cells.139 Mechanistic studies on the cellular uptake with a dinuclear analogue of 44 suggest that the drug accumulation in the MCF-7 breast cancer cells is caused by macropinocytosis, which is only expressed shortly in normal cells, but is responsible for increased motility and metastasis in cancer cells. Thus, such polynuclear platinum complexes may target cancer cells more selectively than conventional platinum drugs.140
Hyperbranched polyglycerols (PG2) and aliphatic polyesters (H40) based on 2,2-bishydroxymethylpropionic acid are commercially available and have many hydroxyl terminal groups. Both PG2 and H40 have been modified with carboxylic acid groups capable of acting as ligands for platinum complexes derived from cisplatin. An advantage of these polyethers and polyesters over dendrimers is their easy availability. Modified PG2 form strongly bound platinum complexes and give controlled release of the drug over 7 days; while modified H40 forms a higher proportion of weakly bound platinum complexes and give similar release profiles in both water and saline. Modified PG2 appears to be more suitable as a drug delivery carrier for cisplatin.141
4.3 Polymeric micelles
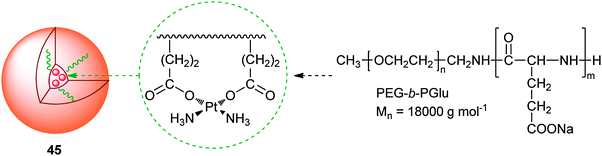
Polymeric micelles are expected to enhance the accumulation of platinum drugs in tumour tissues by the EPR effect. Typically, platinum drugs are incorporated into the inner core of polymeric micelles by chemical conjugation or physical entrapment with relatively high stability. To prevent the micelles passing through normal vessels, their size can be controlled within the range of 20–100 nm, which is helpful in reducing the side effects of platinum anticancer drugs. Polymeric micelles could protect platinum complexes from degradation and achieve controlled release in the delivery, substantially improve the efficacy of platinum-based chemotherapy.A representative example of polymeric micelle carrier systems for platinum drugs is NC-6004 (45), which is a block copolymer of PEG and poly(glutamic acid) (PGlu) coordinated with cis-diammineplatinum moieties. The hydrophilic PEG chain constitutes the outer shell of the micelles, and the PGlu-Pt complex chain comprises the inner core of the micelles. The molecular weight of PEG-b-PGlu as a sodium salt is approximately 18000. The platinum release rates of 45 are 19.6 and 47.8% at 24 and 96 h, respectively.142 A phase I clinical trial of 45 is under way in the UK on patients with solid tumors. The starting dose is 10 mg m−2, and administered once every 3 weeks with only 1000 mL water loading. In general, 45 is well tolerated with minimal nephrotoxicity and no significant myelosuppression, emesis or neurotoxicity but a high rate of hypersensitivity reactions. Disease stabilization has been seen in heavily pre-treated patients.143 Similarly, DACHPt has also been incorporated into the PEG-b-PGlu block copolymer with different lengths of the PGlu block (20, 40, and 70 U). The resulting polymeric micelles are ca. 40 nm in diameter and have a narrow size distribution. In vivo distribution and antitumor activity experiments on CDF1 mice bearing the murine colon adenocarcinoma C-26 show that DACHPt-micelles accumulate at the tumour site 20-fold greater than oxaliplatin and achieve substantially higher antitumor efficacy. DACHPt-micelles also show a very strong antitumor activity against the multiple metastases generated from injected bioluminescent HeLa (HeLa-Luc) cells. These results suggest that DACHPt-micelles could be an outstanding DTD system for oxaliplatin in the treatment of solid tumours, especially the PEG-b-PGlu micelles prepared with 20 U of PGlu.144 Such DACHPt-micelles, with 30 nm diameter, efficiently penetrate and accumulate in an orthotopic scirrhous gastric cancer model, leading to the inhibition of the tumour growth. Moreover, the elevated localization of systemically injected DACHPt-micelles in metastatic lymph nodes can inhibit the growth of metastatic tumours.145
DACHPt and magnetic resonance imaging contrast agent gadolinium–diethylenetriaminepentaacetic acid (Gd-DTPA) have been incorporated into PEG-b-PGlu through reversible complexation of platinum, forming core-shell polymeric micelles. Both DACHPt and Gd-DTPA are released from the micelles in a sustained manner under physiologic conditions and colocalize in the tumour interior. Simultaneous therapy and imaging are achieved in an orthotopic animal model of intractable human pancreatic tumour by this nontoxic formulation. This study provides an effective design of theranostic micelles with high contrast enhancement and site-specific clinical potential.146
Another type of micelles are synthesized using block copolymer ionomers of PEO, poly(methacrylic acid) (PMA) and divalent metal cations as templates. The core of the micelles comprises a network of the cross-linked polyanions, which is surrounded by the shell of hydrophilic PEO chains. Cisplatin has been incorporated into the ionic core of the micelles with remarkably high efficiency (22%, w/w). The drug-loaded micelles are stable in aqueous dispersions without aggregation or precipitation for a prolonged period of time. Platinum complexes release in a slow and sustained manner from the micelles in physiological saline. In vitro studies using human A2780 ovarian carcinoma cells demonstrate that the cross-linked micelles can be rapidly internalized into cells.147 These results indicate that polymeric micelles with cross-linked ionic cores are promising DTD carriers for platinum anticancer drugs.
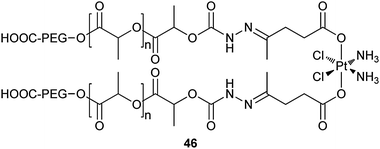
The acid-responsive polymer–platinum conjugate 46 is a newly reported drug delivery vehicle for cisplatin. This nanoparticulate system is constructed by a covalent conjugation of the platinum(IV) prodrug to the hydrophobic segment of two biocompatible diblock copolymer chains through a pH-sensitive hydrazone bond. The conjugate can readily precipitate to form sub-100 nm nanoparticles in aqueous solution due to their low critical micelle concentration. The uniqueness of 46 lies in its highly differential drug release profile at different environmental acidity. During circulation in the blood, the nearly neutral pH (7.4) prevents any release of the drug from 46; upon entering the cancer cells by endocytosis, the acidic intracellular pH (∼5.6) stimulates a rapid release of the drug from 46. The rapid release of drugs in high doses inside cancer cells could suppress the chemoresistance of cancer cells and thereby improve the therapeutic efficacy of the drug. The conjugate shows well-controlled platinum loading yield, excellent drug release kinetics, and an enhanced in vitro cytotoxicity against A2780 ovarian cancer cells as compared to free cisplatin.148
4.4 Liposomal formulations
Formulation is an attractive option for improving the efficacy and reducing side effects of platinum-based anticancer drugs. This approach releases the therapeutic moiety at a tumour target in a controllable manner whilst masking the drugs from deactivation by plasma proteins or macrophages. Lipoplatin™ and Aroplatin™ (L-NDDP) are two successful liposomal formulations of platinum anticancer drugs. Lipoplatin™ is a formulation of cisplatin composed primarily of dipalmitoyl phosphatidyl glycerol (DPPG), soy phosphatidylcholine, and methoxypolyethylene glycol-distearoylphosphatidylethanolamine (cisplatin 8.9%, lipids 91.1%, w/w), with an average diameter of 110 nm (Fig. 4). The preparation of Lipoplatin™ begins with the formation of reverse micelles of DPPG with cisplatin and subsequent conversion to liposomes upon interactions with neutral lipids. PEG units are introduced onto the phospholipid bilayers to sterically stabilise the liposomes, thereby extending the circulation time and improving the uptake, accumulation and retention in tumour tissues due to the EPR effect.149,150 The solubility of cisplatin is enhanced from 1 to 3 mg mL−1 in saline, and the circulation time is prolonged from 6 to 117 h with the aid of liposomes, which is necessary for targeted extravasation into the permeable blood vessels of tumour tissues. In human studies, Lipoplatin™ preferentially concentrates in the primary tumours and their metastases because of the EPR effect and the subsequent avid uptake by the tumour cells either via phagocytosis or by direct fusion with the cell membrane, which lead to a 200-fold higher damage to cancer tissues compared to normal tissues and contribute to the low side effects of the drug. In animal studies, Lipoplatin™ kills not only tumour cells but also endothelial cells of the tumour vasculature after systemic delivery; therefore, it may act both as a chemotherapy drug and an antiangiogenesis agent.151 In preclinical studies, Lipoplatin™ shows a superior cytotoxicity against non-small cell lung cancer (NSCLC) and renal cell carcinoma cell lines and a much lower toxicity in normal cells compared with cisplatin.152 In phase I–III clinical studies, Lipoplatin™ alone or in combination with other anticancer drugs such as gemcitabine, 5-fluorouracil, and paclitaxel has demonstrated substantially reduced systemic toxicities like nephrotoxicity, neurotoxicity and ototoxicity, with an efficacy higher than or similar to that of cisplatin. Lipoplatin™ has finished successfully phase III clinical trials as a first line treatment against NSCLC, and has been granted phase II/III studies on pancreatic cancer as an orphan drug by the European Medicines Agency.153,154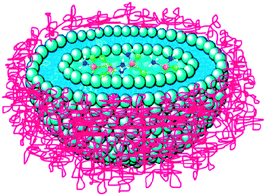 | ||
| Fig. 4 An illustration of Lipoplatin™ shows the encapsulation of cisplatin in the lipid bilayer with the surface of the liposome being coated by hydrophilic PEG molecules. | ||
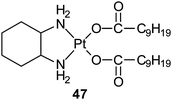
Aroplatin™ is a liposomal formulation of cis-bis(neodecanoato)(trans-R,R-1,2-diaminocyclohexane)platinum(II) (47), a structural analogue of oxaliplatin. Preclinical data show that Aroplatin™ has different biodistribution and toxicity profiles than cisplatin or carboplatin, and does not share cross-resistance with cisplatin. In a phase I trial, its maximum tolerated dose is 312.5 mg m−2 with myelosuppression as the dose limiting toxicity. Aroplatin™ has reached phase II trials in refractory metastatic colorectal cancer.155
Transferrin (TF) receptors are overexpressed on tumour cells and TF receptor-mediated endocytosis is a normal physiological process through which TF delivers iron ions into cells. Therefore, TF receptors could be a viable target for cancer therapy. Liposomes modified with TF-conjugated PEG (TF–PEG-liposomes) have been used as both an active and a passive DTD carrier to realize the tumour-selective delivery of oxaliplatin. This approach has significantly reduced oxaliplatin partitioning to erythrocytes and improved the circulation time of oxaliplatin in Colon 26 tumour-bearing mice, resulting in enhanced extravasation of liposomes into tumours. Intravenously administered oxaliplatin encapsulated within TF–PEG-liposomes has been shown to maintain a high concentration in tumours for over 72 h and suppress tumour growth more effectively than free oxaliplatin.156 Since selectivity and membrane permeability are inherent features of TF–PEG-liposomes and additional affinity for tumour cells is unnecessary for the delivered species, this DTD system may be applicable to other platinum anticancer drugs targeting at various types of tumours that overexpress TF receptors.
Hybrid molecules derived from nucleotides and lipids are ideal candidates for the encapsulation of platinum drugs. Their distinctive supramolecular capabilities and nontoxic properties have been exploited to develop a novel approach to the delivery of cisplatin. The method involves two steps: (i) encapsulation of the cisplatin nanoprecipitate via an anionic nucleotide-lipid; and (ii) stabilization of the resulting anionic nanoparticles using a cationic nucleoside-lipid. The nucleoside polar heads guide the self-assembly of the aggregates into highly loaded and stable nanoparticles (Fig. 5). The cytotoxic activity of the nanoparticles is significantly higher than that of free cisplatin. More impressively, the nanoparticles are much more effective than the free drug against cisplatin-resistant cell lines.157
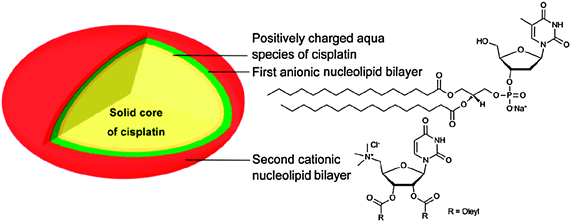 | ||
| Fig. 5 Schematic diagram of a nucleotide-lipid-based nanoparticulate DTD system for cisplatin. | ||
4.5 Carbon nanotubes
Carbon nanotubes are attractive carriers for DTD because of their unique physical, chemical and physiological properties. The structural stability of carbon nanotubes could prolong the circulation time and bioavailability of the loaded drugs.158 The functionalized soluble single-walled carbon nanotubes (SWNTs) and single-walled carbon nanohorns (SWNHs) are two kinds of the most used nanotubes for the delivery of platinum-based anticancer drugs. SWNTs and SWNHs have plenty of inner spaces that render the incorporation of the drugs possible; moreover, the tube walls can physically adsorb the drugs and various functional molecules. The exterior surface or the edges of the tube holes have oxidized functional groups where further chemical modifications are feasible. More importantly, carbon nanotubes show low toxicity in vitro and have negligible impact on a living body.159 In view of the shuttling capacity of SWNTs, a platinum(IV) complex 48 has been tethered to SWNTs through covalent bonding. Upon intracellular reduction of the conjugate, a lethal dose of cisplatin is released. On average, 65 platinum(IV) centers are attached to each SWNT and they enter the cell through endocytosis, leading to higher levels of platinum in the cell than the untethered complex or cisplatin. The SWNT-PtIV conjugate shows a substantial increase in cytotoxicity against the testicular carcinoma cell line NTera-2 as compared with that of the free complex and cisplatin.160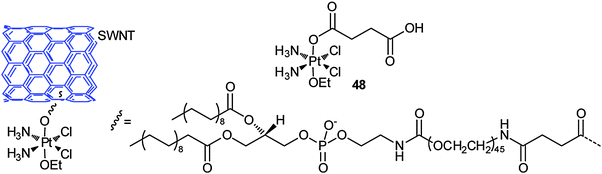
Increased cellular uptake of folic acid and overexpression of folate receptor (FR) are well-known biochemical characters of many tumour types. Therefore, FR, a glycoprotein on the cellular membrane with high affinity for folic acid, is a proper target for anticancer chemotherapeutic agents. Folate-like molecules represent an intriguing class of carriers for DTD specific to FR(+) tumour cells. Folic acid, linked via one of its carboxyl groups to a molecule, can enter cancer cells through FR-mediated endocytosis. An example of such a conjugate is shown in Fig. 6, where the platinum(IV) complex bearing succinates as its axial ligands is tethered to the amine functionalised SWNT and a folic acid derivative. The folate moiety serves as the targeting agent and SWNT as the delivery molecule. The PEG spacer between the platinum centre and folate group makes the conjugate more water soluble and biocompatible. The conjugate indeed delivers the platinum(IV) pharmacophore selectively into the FR(+) cancer cells that overexpress the FR on their surface and releases cisplatin upon intracellular reduction of PtIV to PtII, forming a high level of 1,2-d(GpG) intrastrand cross-link with nuclear DNA. The IC50 values of the conjugate towards FR(+) human choriocarcinoma JAR and human nasopharyngeal carcinoma KB cell lines are significantly lower than those of cisplatin or the free complex.161 This is a telling example for an ideal DTD structure mode, that is, to incorporate the targeting and delivery moieties into a single molecule.
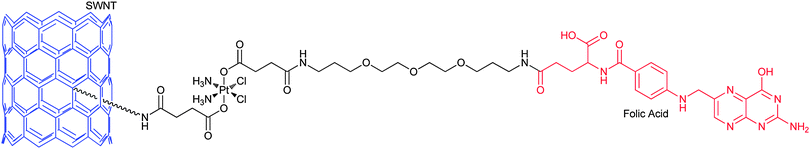 | ||
| Fig. 6 SWNT-tethered platinum(IV) prodrug with targeting property for folate receptor, which releases cisplatin upon intracellular reduction in tumour cells. | ||
Epidermal growth factor receptor (EGFR) is overexpressed in a number of cancers such as ovary and lung cancers.162,163 Accordingly, SWNT-tethered cisplatin is attached to epidermal growth factor (EGF) to specifically target head and neck squamous carcinoma cells (HNSCC) that overexpress EGFR (Fig. 7). The uptake of platinum in both in vitro and in vivo systems is higher for the targeted conjugate than for the untargeted controls. The conjugate enters into the cell through EGFR-directed endocytosis, as demonstrated by the lack of uptake in the absence of EGFR or EGF. In short-term studies on mice, the conjugate mainly distributes in tumours, with only small amounts distributing in various vital organs. The conjugate kills cancer cells more efficiently and inhibit tumour growth in mice more rapidly than cisplatin and the untargeted SWNT-cisplatin. This is the first SWNT-tethered platinum DTD system showing selective anticancer activity in vivo.164
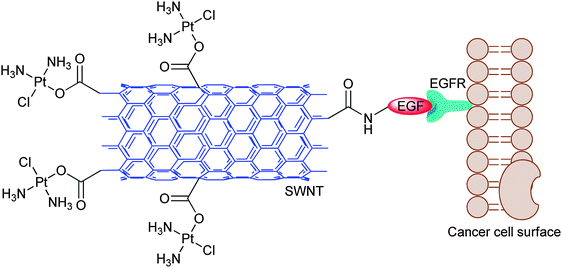 | ||
| Fig. 7 Schematic diagram of the cisplatin-SWNT-EGF conjugate targeting the cell surface EGFR on a single HNSCC cell. | ||
SWNHs have a spherical structure between 80 and 100 nm assembled by several hundreds of SWNTs, and the size is adequate for drug delivery through vascular EPR effect. Cisplatin has been loaded into the SWNHs through a selective precipitation process in water. The amount of incorporated cisplatin is 46%, and the total released quantity of cisplatin is 100% over 48 h. As a result, in vitro anticancer efficiency of the drug-loaded SWNHs is 4–6 times greater than that of free cisplatin, and in vivo anticancer activity against the growth of transplanted tumours in mice is also better than cisplatin and remains for a long time (25 days). Since cisplatin-SWNHs adhere to the cell surfaces in vitro and stay within the tumour tissues in vivo, the released cisplatin can reach high concentrations locally in cells and in tissues, leading to an efficient attack against the tumour cells.165
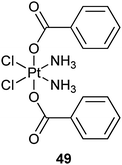
Platinum(II) complexes tethered to the surface of SWNTs may risk being prematurely released from the carrier and binding non-specifically to endogenous nucleophiles. To address this problem, multiwalled carbon nanotubes are adopted as a protective shell to entrap stable hydrophobic platinum(IV) prodrug 49 within its inner cavity via hydrophobic–hydrophobic interactions. Upon chemical reduction by cellular reductants, 49 is converted to its hydrophilic and cytotoxic PtII form and released from the carrier due to the drastic reversal in hydrophobicity. In this way, controlled release of PtII species can be achieved intracellularly to exert its cytotoxic activity.166
4.6 Nanoparticles
Nanoparticles could reach certain solid tumours via the EPR effect and thereby change the tissue distribution and pharmacokinetics of the loaded drugs, which would significantly improve their specificity for tumour tissues.167,168 In comparison with other delivery strategies, nanoparticle-based DTD possesses many advantages, such as enhanced drug accumulation in tumour tissues, reduced systemic toxicity, and sustained drug release in an environmentally responsive manner.169 Besides, nanoparticles could protect the loaded drug from degradation before reaching cancerous cells, and thereby prolong the blood circulation time of the drug and shield the body from undesired side effects. Surface-functionalized nanoparticles by peptides, antibodies or aptamers can further increase the specificity for particular cancerous cells.170 Nanoparticles used for platinum DTD in vivo should be biocompatible, biodegradable, in appropriate size, and have high affinity for the platinum drug to avoid premature release before entering tumour cells. Several nanoparticle-based anticancer drugs have been approved by the FDA, and an interest in the development of nanoparticle formulations for effective delivery of platinum anticancer drugs has increased persistently.171 For example, phospholipid nanocapsules have been exploited to deliver both cisplatin and carboplatin.172–174 Polymeric nanoparticles as sequential release carriers are becoming more and more valuable in the drug delivery for cancer therapy.175 Nanoparticles made of chitosan or N-trimethyl chitosan,176 glycol chitosan,177 poly(lactide-co-glycolide)-methoxy-poly(ethylene glycol),178 poly[2-(N,N-diethylamino)ethyl methacrylate]-block-poly(ethylene glycol),179 and glucosamine-functionalized polyisobutylene-maleic acid180 have been examined as carriers for the delivery of cisplatin in various cancer cells or tumour-bearing mice.Poly(D,L-lactic-co-glycolic acid) (PLGA)-PEG polymers are particularly useful in encapsulating platinum drugs for targeted delivery because of their safety in clinical use and systemic clearance times.184 Prostate-specific membrane antigen (PSMA) is abundantly expressed in prostate cancer, its metastatic form, the hormone-refractory form and the neovascularture of many non-prostate solid tumours, offering a suitable target for cancer chemotherapy. To construct the PSMA-targeted nanoparticles, a hydrophobic platinum(IV) prodrug 50 is encapsulated in PLGA-PEG polymers by nanoprecipitation and subsequent conjugation of PSMA aptamers (Apt) (Fig. 8). The PSMA targeting aptamers on the surface of the nanoparticles direct 50 specifically to the human PSMA-overexpressing LNCaP prostate cancer cells. Upon internalization through endocytosis and intracellular reduction, a lethal dose of cisplatin is released from the polymeric nanoparticles. Controlled release of 50 from the nanoparticles extends over 60 h. The nanoparticles are highly cytotoxic to the LNCaP cells (IC50 = 0.03 μM); under the same conditions, they are 80 times more effective than cisplatin.185 Recently, in vivo studies in different normal and PSMA-expressing animal models reveal that the pharmacokinetics, biodistribution, tolerability, and efficacy of Pt-Apt-NPs are enhanced when compared to cisplatin. Prolonged drug persistence in blood circulation and decreased accumulation of Pt in the kidneys are observed. Pt-Apt-NPs display a significant dose-sparing character of the drug, with equivalent antitumor efficacy in LNCaP xenografts at 1/3 the dose of cisplatin. This system provides a remarkable improvement in the therapeutic index of cisplatin for prostate cancer chemotherapy.186
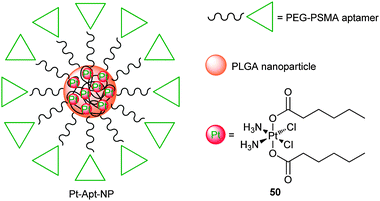 | ||
| Fig. 8 The chemical structure of the encapsulated platinum(IV) prodrug within the PLGA-PEG nanoparticle. | ||
Integrins are heterodimeric transmembrane proteins involved in cell adhesion and cell signaling, and their expression is commonly upregulated in cancers. The αvβ3 integrin is differentially upregulated on angiogenic endothelial cells as well as on many cancer cells. RGD motif (Arg-Gly-Asp) is a tumour vasculature-homing peptide existing in many extracellular matrix components and capable of binding integrins on the cell surface (vide ante). Recently, a PLGA-PEG nanoparticulate system comprising encapsulated 50 and cyclic pentapeptide c(RGDfK) as αvβ3-targeting moieties has been developed for anticancer therapy. The RGD-modified Pt(IV)-PLGA-PEG nanoparticles display enhanced cytotoxicity as compared to cisplatin in prostate and breast cancer epithelial cells in vitro; moreover, they are more efficacious and better tolerated in comparison to cisplatin in an orthotopic human breast cancer xenograft model in vivo.187 This system combines both active and passive targeting approaches, resulting in an enhanced antitumor efficacy and reduced toxicity for platinum drugs.
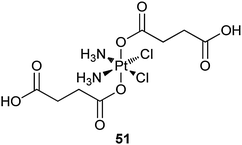
On the same concept, 51 is encapsulated into nanoscale coordination polymers (NCPs) for targeted delivery to cancer cells. NCPs are constructed from TbIII cations and 51 simply by precipitating from their aqueous solution via the addition of a poor solvent, where TbIII ions act as connectors to form the metal–ligand polymers. The Pt-loaded NCPs are stabilized with shells of amorphous silica to prevent rapid dissolution and to effectively control the release of the platinum species. The silica shells extend the half-release time to 5.5 or 9 h, depending on the size of the coating (2 or 7 nm). These release rates would allow sufficient time for the Pt-loaded NCPs to circulate throughout the body and accumulate in tumour tissue. In order to enhance the cellular uptake of Pt-NCPs in vitro, silyl-derived c(RGDfK) is grafted onto the surface, which could enhance the binding affinity for the αvβ3 integrin. The targeted Pt-NCPs show IC50 values lower than that of cisplatin for angiogenic human colon carcinoma cell line HT29. The cytotoxicity against the MCF-7 cell line that does not overexpress the αvβ3 integrin is similar to that of cisplatin.188
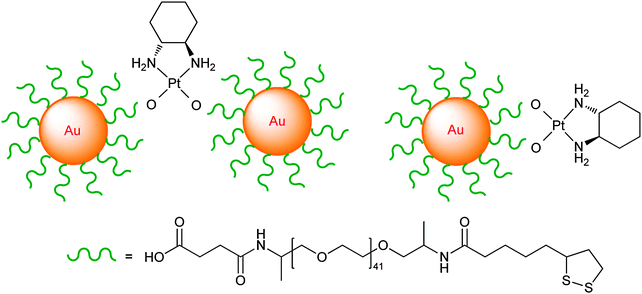 | ||
| Fig. 9 The linking mode of thiolate-PEG-Au nanoparticles with the oxaliplatin fragment. | ||
A novel DTD vehicle involving platinum(IV) polyvalent oligonucleotide gold nanoparticle conjugates has been developed. The gold nanoparticles are functionalized with thiolated 28-mer oligonucleotides containing a terminal dodecyl amine for platinum conjugation. A platinum(IV) complex, cis,cis,trans-[Pt(NH3)2Cl2(OH)(O2CCH2CH2CO2H)], is tethered to the amine-functionalized DNA–Au nanoparticle surface through amide linkages, resulting in Pt(IV)–DNA–Au nanoparticles (Scheme 2). The conjugates allow the platinum(IV) prodrug to travel safely in the bloodstream before reaching the tumour site. They are internalized by tumour cells and reduced to release cisplatin, which enters the nucleus and forms 1,2-d(GpG) intrastrand cross-links with DNA. This nanoparticulate conjugate shows a killing ability superior to cisplatin against human lung carcinoma A549, human prostate cancer PC3, cervical cancer HeLa, and human osteosarcoma U2OS cell lines. In contrast, the parent prodrug displays no significant killing under the same conditions.197 The appealing characteristics of such nanoparticles include high levels of cellular uptake in a number of cell types, nontoxicity and resistance to enzymatic degradation.198,199
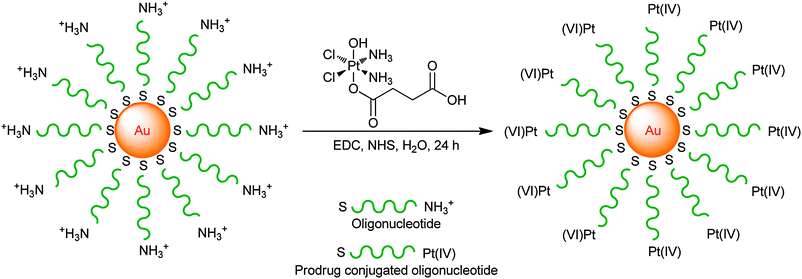 | ||
| Scheme 2 The synthesis of Pt(IV)–DNA–Au nanoparticles through peptide bond formation. | ||
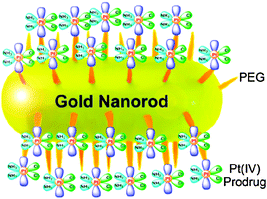
The cellular uptake of gold nanoparticles is not only dependent on their size, but also on their shape.200,201 A peculiar class of gold nanoparticles is the nonspherical gold nanorods (GNRs). In comparison with spherical gold nanoparticles, GNRs have longer circulation time in vivo, which enhances the efficiency of nanoparticle accumulation in tumours.202 Recently, platinum(IV) prodrug cis,cis,trans-[Pt(NH3)2Cl2(O2CCH2CH2CO2H)2] has been tethered to the amine-functionalized PEG-GNRs (Fig. 10). This conjugate is stable under physiological conditions, but is liable to be reduced by cellular reductants, releasing the active PtII species. Compared with cisplatin, the conjugate shows an enhanced cellular platinum accumulation and superior cytotoxicity against cervical cancer HeLa, human lung carcinoma A549 and human breast adenocarcinoma MCF-7 cells lines.203 More importantly, the conjugate can overcome the drug resistance in cisplatin-resistant A549R cells because it gets into cells through endocytosis and low expression of copper transport protein (Ctr1) in A549R cells does not affect its uptake. In addition, the platinum(IV) prodrug attached to PEG-GNRs is more inert than cisplatin, which would reduce the deactivation induced by glutathione and metallothionein.204 These results suggest that PEG-GNRs are effective carriers for the delivery of platinum drugs.
Recently, we fabricated the carboxymethylcellulose-modified superparamagnetic magnetite nanocrystal clusters (CMC–SPMNCs) as nanocarriers for the delivery of platinum drugs. Dechlorinated cisplatin, namely cis-monochlorodiammineplatinum(II) (CMDP), was loaded onto the clusters through the abundant carboxylate groups on the surface of the nanoparticles, forming CMDP–CMC–SPMNC conjugates with a mean diameter of 290.6 nm (Scheme 3). The conjugates display excellent dimensional uniformity, good aqueous dispersibility and strong magnetisability. In comparison with cisplatin, the conjugates can more readily enter cancer cells and exert higher cytotoxicity towards the human cervical cancer HeLa cells and the human hepatocarcinoma HepG2 cells. These nanoparticles are likely to be used as targeted carriers to deliver platinum anticancer drugs.215
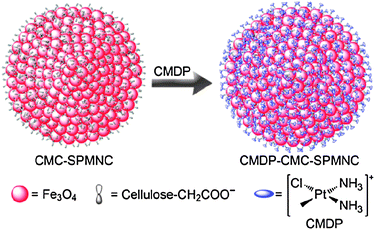 | ||
| Scheme 3 Fabrication route of the CMDP–CMC–SPMNC conjugate. | ||
5 Conclusions
The lack of tumour specificity is one of the major problems for platinum-based anticancer drugs. The nonselective distribution of platinum drugs in normal and cancer cells not only induces excessive systemic toxicity, but also reduces drug accumulation in tumour cells, resulting in tumour resistance to platinum drugs. In addition, unrestrained interactions of the drugs with plasma and tissue proteins may lead to rapid inactivation of platinum drugs and thereby suboptimal treatment for the tumour. Therefore, targeting platinum anticancer drugs to specific tumour tissues is an important issue in platinum-based chemotherapy.216,217 The use of special delivery carriers to selectively transport platinum agents to tumours is very attractive to address these problems. A variety of DTD approaches have been developed to improve the efficacy of platinum anticancer drugs.218 These approaches are classified as active or passive strategies. Active DTD contributes to the selectivity of a platinum drug towards a specific kind of tumour according to its biochemical properties, while passive DTD leads to the beneficial accumulation of a platinum drug in the tumour mass because of the EPR effect. The rational design of DTD systems for platinum complexes has resulted in a number of “magic bullets” with therapeutic indexes better than those of cisplatin and its derivatives. This review presents the major achievements in this area in the past few years. These creative designs may inspire even more ingenious inventions come into being in the future development of platinum-based anticancer drugs.Acknowledgements
We are thankful to the financial support from the National Natural Science Foundation of China (Grants 21271101, 21131003, 21021062, 90713001) and the National Basic Research Program of China (Grant 2011CB935800). We are also grateful to Dr Xiaohui Wang for collecting many of the references.References
- L. Kelland, Nat. Rev. Cancer, 2007, 7, 573–584 CrossRef CAS.
- M. J. Hannon, Pure Appl. Chem., 2007, 79, 2243–2261 CrossRef CAS.
- S. P. Fricker, Dalton Trans., 2007, 4903–4917 RSC.
- A. S. Abu-Surrah and M. Kettunen, Curr. Med. Chem., 2006, 13, 1337–1357 CrossRef CAS.
- X. Y. Wang and Z. J. Guo, Dalton Trans., 2008, 1521–1532 RSC.
- C. A. Rabik and M. E. Dolan, Cancer Treat. Rev., 2007, 33, 9–23 CrossRef CAS.
- A. A. Argyriou, P. Polychronopoulos, G. Iconomou, E. Chroni and H. P. Kalofonos, Cancer Treat. Rev., 2008, 34, 368–377 CrossRef CAS.
- S. R. McWhinney, R. M. Goldberg and H. L. McLeod, Mol. Cancer Ther., 2009, 8, 10–16 CrossRef CAS.
- X. Yao, K. Panichpisal, N. Kurtzman and K. Nugent, Am. J. Med. Sci., 2007, 334, 115–124 CrossRef.
- P. Heffeter, U. Jungwirth, M. Jakupec, C. Hartinger, M. Galanski, L. Elbling, M. Micksche, B. Keppler and W. Berger, Drug Resist. Updates, 2008, 11, 1–16 CrossRef CAS.
- D. J. Stewart, Crit. Rev. Oncol. Hematol., 2007, 63, 12–31 CrossRef.
- J. J. Yu, Current Drug Therapy, 2009, 4, 19–28 CrossRef CAS.
- M. D. Hall, M. Okabe, D.-W. Shen, X.-J. Liang and M. M. Gottesman, Annu. Rev. Pharmacol. Toxicol., 2008, 48, 495–535 CrossRef CAS.
- M. Knipp, Curr. Med. Chem., 2009, 16, 522–537 CrossRef CAS.
- X. Y. Wang and Z. J. Guo, Anti-Cancer Agents Med. Chem., 2007, 7, 19–34 CrossRef.
- L. P. Martin, T. C. Hamilton and R. J. Schilder, Clin. Cancer Res., 2008, 14, 1291–1295 CrossRef CAS.
- V. Benedetti, P. Perego, G. L. Beretta, E. Corna, S. Tinelli, S. C. Righetti, R. Leone, P. Apostoli, C. Lanzi and F. Zunino, Mol. Cancer Ther., 2008, 7, 679–687 CrossRef CAS.
- V. O’Brien and R. Brown, Carcinogenesis, 2006, 27, 682–692 CrossRef.
- P. Yang, J. O. Ebbert, Z. Sun and R. M. Weinshilboum, J. Clin. Oncol., 2006, 24, 1761–1769 CrossRef CAS.
- L. Gossage and S. Madhusudan, Cancer Treat. Rev., 2007, 33, 565–577 CrossRef CAS.
- W. Sakai, E. M. Swisher, B. Y. Karlan, M. K. Agarwal, J. Higgins, C. Friedman, E. Villegas, C. Jacquemont, D. J. Farrugia, F. J. Couch, N. Urban and T. Taniguchi, Nature, 2008, 451, 1116–1120 CrossRef CAS.
- L. Galluzzi, L. Senovilla, I. Vitale, J. Michels, I. Martins, O. Kepp, M. Castedo and G. Kroemer, Oncogene, 2012, 31, 1869–1883 CrossRef CAS.
- K. S. Lovejoy and S. J. Lippard, Dalton Trans., 2009, 10651–10659 RSC.
- J. Reedijk, Eur. J. Inorg. Chem., 2009, 1303–1312 CrossRef CAS.
- G. N. Kaluđerović and R. Paschke, Curr. Med. Chem., 2011, 18, 4738–4752 CrossRef.
- I. Kostova, Recent Pat. Anti-Cancer Drug Discovery, 2006, 1, 1–22 CAS.
- Á. M. Montaña and C. Batalla, Curr. Med. Chem., 2009, 16, 2235–2260 CrossRef.
- N. J. Wheate, S. Walker, G. E. Craig and R. Oun, Dalton Trans., 2010, 39, 8113–8127 RSC.
- K. Hanada, K. Asano, T. Nishimura, T. Chimata, Y. Matsuo, M. Tsuchiya and H. Ogata, J. Pharm. Pharmacol., 2008, 60, 317–322 CrossRef CAS.
- G. Momekov and D. Momekova, Expert Opin. Ther. Pat., 2006, 16, 1383–1403 CrossRef CAS.
- B. Stordal, N. Pavlakis and R. Davey, Cancer Treat. Rev., 2007, 33, 347–357 CrossRef CAS.
- M. A. Jakupec, M. Galanski, V. B. Arion, C. G. Hartinger and B. K. Keppler, Dalton Trans., 2008, 183–194 RSC.
- X. Y. Wang, Anti-Cancer Agents Med. Chem., 2010, 10, 396–411 CAS.
- S. Zorbas-Seifried, C. G. Hartinger, K. Meelich, M. Galanski, B. K. Keppler and H. Zorbas, Biochemistry, 2006, 45, 14817–14825 CrossRef CAS.
- M. D. Hall, H. R. Mellor, R. Callaghan and T. W. Hambley, J. Med. Chem., 2007, 50, 3403–3411 CrossRef CAS.
- C. F. Chin, D. Y. Q. Wong, R. Jothibasu and W. H. Ang, Curr. Top. Med. Chem., 2011, 11, 2602–2612 CrossRef CAS.
- S. Dhar and S. J. Lippard, Current Status and Mechanism of Action of Platinum-Based Anticancer Drugs, in Bioinorganic Medicinal Chemistry, ed. E. Alessio, WILEY-VCH Verlag GmbH & Co. KGaA, Weinheim, 2011, pp. 79–95 Search PubMed.
- X. Y. Wang and Z. J. Guo, New Trends and Future Developments of Platinum-Based Antitumor Drugs, in Bioinorganic Medicinal Chemistry, ed. E. Alessio, WILEY-VCH Verlag GmbH & Co. KGaA, Weinheim, 2011, pp. 97–149 Search PubMed.
- M. Galanski, Recent Pat. Anti-Cancer Drug Discovery, 2006, 1, 285–295 CrossRef CAS.
- Y. W. Jung and S. J. Lippard, Chem. Rev., 2007, 107, 1387–1407 CrossRef CAS.
- A. Rebillard, D. Lagadic-Gossmann and M.-T. Dimanche-Boitrel, Curr. Med. Chem., 2008, 15, 2656–2663 CrossRef CAS.
- H. Choy, C. Park and M. Yao, Clin. Cancer Res., 2008, 14, 1633–1638 CrossRef CAS.
- E. Gabano, M. Ravera and D. Osella, Curr. Med. Chem., 2009, 16, 4544–4580 CrossRef CAS.
- A. V. Klein and T. W. Hambley, Chem. Rev., 2009, 109, 4911–4920 CrossRef CAS.
- N. P. Farrell, Curr. Top. Med. Chem., 2011, 11, 2623–2631 CrossRef CAS.
- H. Burger, W. J. Loos, K. Eechoute, J. Verweij, R. H. J. Mathijssen and E. A. C. Wiemer, Drug Resist. Updates, 2011, 14, 22–34 CrossRef CAS.
- K. Strebhardt and A. Ullrich, Nat. Rev. Cancer, 2008, 8, 473–480 CrossRef CAS.
- P. C. A. Bruijnincx and P. J. Sadler, Curr. Opin. Chem. Biol., 2008, 12, 197–206 CrossRef CAS.
- C. Sanchez-Cano and M. J. Hannon, Dalton Trans., 2009, 10702–10711 RSC.
- J. E. Dancey and H. X. Chen, Nat. Rev. Drug Discovery, 2006, 5, 649–659 CrossRef CAS.
- G. Momekov and D. Momekova, Expert Opin. Ther. Pat., 2006, 16, 1383–1403 CrossRef CAS.
- M. Galanski and B. K. Keppler, Anti-Cancer Agents Med. Chem., 2007, 7, 55–73 CrossRef CAS.
- A. K. Lyer, G. Khaled, J. Fang and H. Maeda, Drug Discovery Today, 2006, 11, 812–818 CrossRef.
- J. Fang, H. Nakamura and H. Maeda, Adv. Drug Delivery Rev., 2011, 63, 136–151 CrossRef CAS.
- H. Maeda, G. Y. Bharate and J. Daruwalla, Eur. J. Pharm. Biopharm., 2009, 71, 409–419 CrossRef CAS.
- H. Pelicano, D. S. Martin, R.-H. Xu and P. Huang, Oncogene, 2006, 25, 4633–4646 CrossRef CAS.
- K. Dahlman-Wright, V. Cavailles, S. A. Fuqua, V. C. Jordan, J. A. Katzenellenbogen, K. S. Korach, A. Maggi, M. Muramatsu, M. G. Parker and J.-Å. Gustafsson, Pharmacol. Rev., 2006, 58, 773–781 CrossRef CAS.
- J. Hartman, K. Lindberg, A. Morani, J. Inzunza, A. Ström and J.-Å. Gustafsson, Cancer Res., 2006, 66, 11207–11213 CrossRef CAS.
- R. Gust, W. Beck, G. Jaouen and H. Schönenberger, Coord. Chem. Rev., 2009, 253, 2742–2759 CrossRef CAS.
- I. Ott and R. Gust, Anti-Cancer Agents Med. Chem., 2007, 7, 95–110 CrossRef CAS.
- E. Kim, P. T. Rye, J. M. Essigmann and R. G. Croy, J. Inorg. Biochem., 2009, 103, 256–261 CrossRef CAS.
- C. Descôteaux, V. Leblanc, G. Bélanger, S. Parent, É. Asselin and G. Bérubé, Steroids, 2008, 73, 1077–1089 CrossRef.
- J. Provencher-Mandeville, C. Descôteaux, S. K. Mandal, V. Leblanc, É. Asselin and G. Bérubé, Bioorg. Med. Chem. Lett., 2008, 18, 2282–2287 CrossRef CAS.
- P. Saha, C. Descôteaux, K. Brasseur, S. Fortin, V. Leblanc, S. Parent, É. Asselin and G. Bérubé, Eur. J. Med. Chem., 2012, 48, 385–390 CrossRef CAS.
- R. Schobert, G. Bernhardt, B. Biersack, S. Bollwein, M. Fallahi, A. Grotemeier and G. L. Hammond, ChemMedChem, 2007, 2, 333–342 CrossRef CAS.
- M. J. Hannon, P. S. Green, D. M. Fisher, P. J. Derrick, J. L. Beck, S. J. Watt, S. F. Ralph, M. M. Sheil, P. R. Barker, N. W. Alcock, R. J. Price, K. J. Sanders, R. Pither, J. Davis and A. Rodger, Chem.–Eur. J., 2006, 12, 8000–8013 CrossRef CAS.
- R. Gust, W. Beck, G. Jaouen and H. Schönenberger, Coord. Chem. Rev., 2009, 253, 2760–2779 CrossRef CAS.
- A. Gupta, S. K. Mandal, V. Leblanc, C. Descôeaux, É. Asselin and G. Bérubé, Bioorg. Med. Chem. Lett., 2008, 18, 3982–3987 CrossRef CAS.
- A. Vessières, S. Top, W. Beck, E. Hilllard and G. Jaouen, Dalton Trans., 2006, 529–541 RSC.
- C. G. Hartinger, A. A. Nazarov, S. M. Ashraf, P. J. Dyson and B. K. Keppler, Curr. Med. Chem., 2008, 15, 2574–2591 CrossRef CAS.
- I. Berger, A. A. Nazarov, C. G. Hartinger, M. Groessl, S.-M. Valiahdi, M. A. Jakupec and B. K. Keppler, ChemMedChem, 2007, 2, 505–514 CrossRef CAS.
- R. W.-Y. Sun, D.-L. Ma, E. L.-M. Wong and C.-M. Che, Dalton Trans., 2007, 4884–4892 Search PubMed.
- A. I. Rosenbaum, G. Zhang, J. D. Warren and F. R. Maxfield, Proc. Natl. Acad. Sci. U. S. A., 2010, 107, 5477–5482 CrossRef CAS.
- D. Prashar, Y. Shi, D. Bandyopadhyay, J. C. Dabrowiak and Y.-Y. Luk, Bioorg. Med. Chem. Lett., 2011, 21, 7421–7425 CrossRef CAS.
- G. Horvath, T. Premkumar, A. Boztas, E. Lee, S. Jon and K. E. Geckeler, Mol. Pharmaceutics, 2008, 5, 358–363 CrossRef CAS.
- A. G. Bharadwaj, J. L. Kovar, E. Loughman, C. Elowsky, G. G. Oakley and M. A. Simpson, Am. J. Pathol., 2009, 174, 1027–1036 CrossRef.
- A. G. Bharadwaj, K. Rector and M. A. Simpson, J. Biol. Chem., 2007, 282, 20561–20572 CrossRef CAS.
- Y.-I. Jeong, S.-T. Kim, S.-G. Jin, H.-H. Ryu, Y.-H. Jin, T.-Y. Jung, I.-Y. Kim and S. Jung, J. Pharm. Sci., 2008, 97, 1268–1276 CrossRef CAS.
- M. Götte and G. W. Yip, Cancer Res., 2006, 66, 10233–10237 CrossRef.
- S. Cai, Y. Xie, T. R. Bagby, M. S. Cohen and M. L. Forrest, J. Surg. Res., 2008, 147, 247–252 CrossRef CAS.
- A. Jain, S. K. Jain, N. Ganesh, J. Barve and A. M. Beg, Nanomed.: Nanotechnol., Biol. Med., 2010, 6, 179–190 CrossRef CAS.
- S. E. Papapoulos, Bone, 2006, 38, 613–616 CrossRef.
- V. Stresing, F. Daubiné, I. Benzaid, H. Mönkkönen and P. Clézardin, Cancer Lett., 2007, 257, 16–35 CrossRef CAS.
- Z. Q. Xue, M. X. Lin, J. H. Zhu, J. F. Zhang, Y. Z. Li and Z. J. Guo, Chem. Commun., 2010, 46, 1212–1214 RSC.
- N. Margiotta, R. Ostuni, D. Teoli, M. Morpurgo, N. Realdon, B. Palazzo and G. Natile, Dalton Trans., 2007, 3131–3139 RSC.
- N. Margiotta, F. Capitelli, R. Ostuni and G. Natile, J. Inorg. Biochem., 2008, 102, 2078–2086 CrossRef CAS.
- N. Margiotta, R. Ostuni, V. Gandin, C. Marzano, S. Piccinonna and G. Natile, Dalton Trans., 2009, 10904–10913 RSC.
- C. M. Overall and O. Kleifeld, Nat. Rev. Cancer, 2006, 6, 227–239 CrossRef CAS.
- R. Sasanelli, A. Boccarelli, D. Giordano, M. Laforgia, F. Arnesano, G. Natile, C. Cardellicchio, M. A. M. Capozzi and M. Coluccia, J. Med. Chem., 2007, 50, 3434–3441 CrossRef CAS.
- R. N. Bose, L. Maurmann, R. J. Mishur, L. Yasui, S. Gupta, W. S. Grayburn, H. Hofstetter and T. Salley, Proc. Natl. Acad. Sci. U. S. A., 2008, 105, 18314–18319 CrossRef CAS.
- S. Mukhopadhyay, C. M. Barnés, A. Haskel, S. M. Short, K. R. Barnes and S. J. Lippard, Bioconjugate Chem., 2008, 19, 39–49 CrossRef CAS.
- N. Graf, T. E. Mokhtari, I. A. Papayannopoulos and S. J. Lippard, J. Inorg. Biochem., 2012, 110, 58–63 CrossRef CAS.
- Z. Yang, X. Y. Wang, H. J. Diao, J. F. Zhang, H. Y. Li, H. Z. Sun and Z. J. Guo, Chem. Commun., 2007, 3453–3455 RSC.
- R. M. Xing, X. Y. Wang, C. L. Zhang, Y. M. Zhang, Q. Wang, Z. Yang and Z. J. Guo, J. Inorg. Biochem., 2009, 103, 1039–1044 CrossRef CAS.
- A. M. Krause-Heuer, M. P. Grant, N. Orkey and J. R. Aldrich-Wright, Aust. J. Chem., 2008, 61, 675–681 CrossRef CAS.
- I. Ghosh and W. M. Nau, Adv. Drug Delivery Rev., 2012, 64, 764–783 CrossRef CAS.
- N. J. Wheate, D. P. Buck, A. I. Day and J. G. Collins, Dalton Trans., 2006, 451–458 RSC.
- M. S. Bali, D. P. Buck, A. J. Coe, A. I. Day and J. G. Collins, Dalton Trans., 2006, 5337–5344 RSC.
- Y. Zhao, M. S. Bali, C. Cullinane, A. I. Day and J. G. Collins, Dalton Trans., 2009, 5190–5198 RSC.
- N. J. Wheate, J. Inorg. Biochem., 2008, 102, 2060–2066 CrossRef CAS.
- A. R. Kennedy, A. J. Florence, F. J. McInnes and N. J. Wheate, Dalton Trans., 2009, 7695–7700 RSC.
- A. M. Krause-Heuer, R. Grünert, S. Kühne, M. Buczkowska, N. J. Wheate, D. D. Le Pevelen, L. R. Boag, D. M. Fisher, J. Kasparkova, J. Malina, P. J. Bednarski, V. Brabec and J. R. Aldrich-Wright, J. Med. Chem., 2009, 52, 5474–5484 CrossRef CAS.
- N. J. Wheate, R. I. Taleb, A. M. Krause-Heuer, R. L. Cook, S. Wang, V. J. Higgins and J. R. Aldrich-Wright, Dalton Trans., 2007, 5055–5064 RSC.
- N. J. Wheate, C. R. Brodie, J. G. Collins, S. Kemp and J. R. Aldrich-Wright, Mini-Rev. Med. Chem., 2007, 7, 627–648 CrossRef CAS.
- D. M. Fisher, P. J. Bednarski, R. Grünert, P. Turner, R. R. Fenton and J. R. Aldrich-Wright, ChemMedChem, 2007, 2, 488–495 CrossRef CAS.
- S. Kemp, N. J. Wheate, M. J. Pisani and J. R. Aldrich-Wright, J. Med. Chem., 2008, 51, 2787–2794 CrossRef CAS.
- J. A. Plumb, B. Venugopal, R. Oun, N. Gomez-Roman, Y. Kawazoe, N. S. Venkataramanan and N. J. Wheate, Metallomics, 2012, 4, 561–567 RSC.
- S. Kemp, N. J. Wheate, S. Wang, J. G. Collins, S. F. Ralph, A. I. Day, V. J. Higgins and J. R. Aldrich-Wright, JBIC, J. Biol. Inorg. Chem., 2007, 12, 969–979 CrossRef CAS.
- A. M. Krause-Heuer, N. J. Wheate, M. J. Tilby, D. G. Pearson, C. J. Ottley and J. R. Aldrich-Wright, Inorg. Chem., 2008, 47, 6880–6888 CrossRef CAS.
- N. J. Wheate, G. M. Abbott, R. J. Tate, C. J. Clements, R. Edrada-Ebel and B. F. Johnston, J. Inorg. Biochem., 2009, 103, 448–454 CrossRef CAS.
- E. D. Michelakis, L. Webster and J. R. Mackey, Br. J. Cancer, 2008, 99, 989–994 CrossRef CAS.
- S. Dhar and S. J. Lippard, Proc. Natl. Acad. Sci. U. S. A., 2009, 106, 22199–22204 CrossRef CAS.
- X. Xue, S. You, Q. Zhang, Y. Wu, G.-Z. Zou, P. C. Wang, Y.-L. Zhao, Y. Xu, L. Jia, X. N. Zhang and X.-J. Liang, Mol. Pharmaceutics, 2012, 9, 634–644 CrossRef CAS.
- N. Margiotta, R. Ostuni, R. Ranaldo, N. Denora, V. Laquintana, G. Trapani, G. Liso and G. Natile, J. Med. Chem., 2007, 50, 1019–1027 CrossRef CAS.
- N. Margiotta, N. Denora, R. Ostuni, V. Laquintana, A. Anderson, S. W. Johnson, G. Trapani and G. Natile, J. Med. Chem., 2010, 53, 5144–5154 CrossRef CAS.
- P. Ruiz-Sánchez, S. Mundwiler, B. Spingler, N. R. Buan, J. C. Escalante-Semerena and R. Alberto, JBIC, J. Biol. Inorg. Chem., 2008, 13, 335–347 CrossRef.
- P. Ruiz-Sánchez, C. König, S. Ferrari and R. Alberto, JBIC, J. Biol. Inorg. Chem., 2011, 16, 33–44 CrossRef.
- B. Palazzo, M. Iafisco, M. Laforgia, N. Margiotta, G. Natile, C. L. Bianchi, D. Walsh, S. Mann and N. Roveri, Adv. Funct. Mater., 2007, 17, 2180–2188 CrossRef CAS.
- R. Duncan, Nat. Rev. Cancer, 2006, 6, 688–701 CrossRef CAS.
- M. Galanski and B. K. Keppler, Anti-Cancer Agents Med. Chem., 2007, 7, 55–73 CrossRef CAS.
- R. Haag and F. Kratz, Angew. Chem., Int. Ed., 2006, 45, 1198–1215 CrossRef CAS.
- R. Satchi-Fainaro, R. Duncan and C. M. Barnes, Adv. Polym. Sci., 2006, 193, 1–65 CrossRef CAS.
- K. J. Haxton and H. M. Burt, J. Pharm. Sci., 2009, 98, 2299–2316 CrossRef CAS.
- L. Y. Qiu and Y. H. Bae, Pharm. Res., 2006, 23, 1–30 CrossRef CAS.
- F. Kratz, I. A. Müller, C. Ryppa and A. Warnecke, ChemMedChem, 2008, 3, 20–53 CrossRef CAS.
- M. E. Davis, Z. Chen and D. M. Shin, Nat. Rev. Drug Discovery, 2008, 7, 771–782 CrossRef CAS.
- K. Letchford and H. Burt, Eur. J. Pharm. Biopharm., 2007, 65, 259–269 CrossRef CAS.
- M. E. Gindy and R. K. Prud’homme, Expert Opin. Drug Delivery, 2009, 6, 865–878 CrossRef CAS.
- S. H. Bai, C. Thomas, A. Rawat and F. Ahsan, Crit. Rev. Ther. Drug Carrier Syst., 2006, 23, 437–495 CrossRef CAS.
- R. Tong and J. J. Cheng, Polym. Rev., 2007, 47, 345–381 CrossRef CAS.
- L. Zhang, F. Yu, A. J. Cole, B. Chertok, A. E. David, J. Wang and V. C. Yang, AAPS J., 2009, 11, 693–699 CrossRef CAS.
- Nanoparticulate Drug Delivery Systems, ed. D. Thassu, M. Deleers and Y. Pathak, Informa Healthcare USA, Inc., New York, 2007, pp. 1–31 Search PubMed.
- P. Sood, K. B. Thurmond, II, J. E. Jacob, L. K. Waller, G. O. Silva, D. R. Stewart and D. P. Nowotnik, Bioconjugate Chem., 2006, 17, 1270–1279 CrossRef CAS.
- J. R. Rice, J. L. Gerberich, D. P. Nowotnik and S. B. Howell, Clin. Cancer Res., 2006, 12, 2248–2254 CrossRef CAS.
- S. C. van der Schoot, B. Nuijen, P. Sood, K. B. Thurmond, II, D. R. Stewart, J. R. Rice and J. H. Beijnen, Pharmazie, 2006, 61, 835–844 CAS.
- D. P. Nowotnik and E. Cvitkovic, Adv. Drug Delivery Rev., 2009, 61, 1214–1219 CrossRef CAS.
- M. Campone, J. M. Rademaker-Lakhai, J. Bennouna, S. B. Howell, D. P. Nowotnik, J. H. Beijnen and J. H. M. Schellens, Cancer Chemother. Pharmacol., 2007, 60, 523–533 CrossRef CAS.
- D. A. Tomalia, L. A. Reyna and S. Svenson, Biochem. Soc. Trans., 2007, 35, 61–67 CrossRef CAS.
- T. Kapp, A. Dullin and R. Gust, J. Med. Chem., 2006, 49, 1182–1190 CrossRef CAS.
- T. Kapp, S. Müller and R. Gust, ChemMedChem, 2006, 1, 560–564 CrossRef CAS.
- K. J. Haxton and H. M. Burt, Dalton Trans., 2008, 5872–5875 RSC.
- Y. Matsumura, Jpn. J. Clin. Oncol., 2008, 38, 793–802 CrossRef.
- Y. Matsumura, Adv. Drug Delivery Rev., 2008, 60, 899–914 CrossRef CAS.
- H. Cabral, N. Nishiyama and K. Kataoka, J. Controlled Release, 2007, 121, 146–155 CrossRef CAS.
- M. Rafi, H. Cabral, M. R. Kano, P. Mi, C. Iwata, M. Yashiro, K. Hirakawa, K. Miyazono, N. Nishiyama and K. Kataoka, J. Controlled Release, 2012, 159, 189–196 CrossRef CAS.
- S. Kaida, H. Cabral, M. Kumagai, A. Kishimura, Y. Terada, M. Sekino, I. Aoki, N. Nishiyama, T. Tani and K. Kataoka, Cancer Res., 2010, 70, 7031–7041 CrossRef CAS.
- S. Bontha, A. V. Kabanov and T. K. Bronich, J. Controlled Release, 2006, 114, 163–174 CrossRef CAS.
- S. Aryal, C.-M. J. Hu and L. F. Zhang, ACS Nano, 2010, 4, 251–258 CrossRef CAS.
- S. C. White, P. Lorigan, G. P. Margison, J. M. Margison, F. Martin, N. Thatcher, H. Anderson and M. Ranson, Br. J. Cancer, 2006, 95, 822–828 CrossRef CAS.
- D. J. Bharali, M. Khalil, M. Gurbuz, T. M. Simone and S. A. Mousa, Int. J. Nanomed., 2009, 4, 1–7 CrossRef CAS.
- T. Boulikas, Cancer Ther., 2007, 5, 349–376 Search PubMed.
- C. Arienti, A. Tesei, A. Ravaioli, M. Ratta, S. Carloni, S. Mangianti, P. Ulivi, S. Nicoletti, D. Amadori and W. Zoli, Anti-Cancer Drugs, 2008, 19, 983–990 CrossRef CAS.
- M. E. Froudarakis, A. Pataka, P. Pappas, S. Anevlavis, E. Argiana, M. Nikolaidou, G. Kouliatis, S. Pozova, M. Marselos and D. Bouros, Cancer, 2008, 113, 2752–2760 CrossRef CAS.
- T. Boulikas, Expert Opin. Invest. Drugs, 2009, 18, 1197–1218 CrossRef CAS.
- T. Dragovich, D. Mendelson, S. Kurtin, K. Richardson, D. Von Hoff and A. Hoos, Cancer Chemother. Pharmacol., 2006, 58, 759–764 CrossRef CAS.
- R. Suzuki, T. Takizawa, Y. Kuwata, M. Mutoh, N. Ishiguro, N. Utoguchi, A. Shinohara, M. Eriguchi, H. Yanagie and K. Maruyama, Int. J. Pharm., 2008, 346, 143–150 CrossRef CAS.
- S. Khiati, D. Luvino, K. Oumzil, B. Chauffert, M. Camplo and P. Barthélémy, ACS Nano, 2011, 5, 8649–8655 CrossRef CAS.
- C. Fabbro, H. Ali-Boucetta, T. D. Ros, K. Kostarelos, A. Bianco and M. Prato, Chem. Commun., 2012, 48, 3911–3926 RSC.
- J. Miyawaki, M. Yudasaka, T. Azami, Y. Kubo and S. Iijima, ACS Nano, 2008, 2, 213–226 CrossRef CAS.
- R. P. Feazell, N. Nakayama-Ratchford, H. Dai and S. J. Lippard, J. Am. Chem. Soc., 2007, 129, 8438–8439 CrossRef CAS.
- S. Dhar, Z. Liu, J. Thomale, H. Dai and S. J. Lippard, J. Am. Chem. Soc., 2008, 130, 11467–11476 CrossRef CAS.
- E. M. Bublil and Y. Yarden, Curr. Opin. Cell Biol., 2007, 19, 124–134 CrossRef CAS.
- D. T. Merrick, J. Kittelson, R. Winterhalder, G. Kotantoulas, S. Ingeberg, R. L. Keith, T. C. Kennedy, Y. E. Miller, W. A. Franklin and F. R. Hirsch, Clin. Cancer Res., 2006, 12, 2281–2288 CrossRef CAS.
- A. A. Bhirde, V. Patel, J. Gavard, G. Zhang, A. A. Sousa, A. Masedunskas, R. D. Leapman, R. Weigert, J. S. Gutkind and J. F. Rusling, ACS Nano, 2009, 3, 307–316 CrossRef CAS.
- K. Ajima, T. Murakami, Y. Mizoguchi, K. Tsuchida, T. Ichihashi, S. Iijima and M. Yudasaka, ACS Nano, 2008, 2, 2057–2064 CrossRef CAS.
- J. Li, S. Q. Yap, C. F. Chin, Q. Tian, S. L. Yoong, G. Pastorin and W. H. Ang, Chem. Sci., 2012, 3, 2083–2087 RSC.
- S. M. Nie, Y. Xing, G. J. Kim and J. W. Simons, Annu. Rev. Biomed. Eng., 2007, 9, 257–288 CrossRef CAS.
- V. Wagner, A. Dullaart, A.-K. Bock and A. Zweck, Nat. Biotechnol., 2006, 24, 1211–1217 CrossRef CAS.
- L. Zhang, F. X. Gu, J. M. Chan, A. Z. Wang, R. S. Langer and O. C. Farokhzad, Clin. Pharmacol. Ther., 2008, 83, 761–769 CrossRef CAS.
- W. Jiang, B. Y. S. Kim, J. T. Rutka and W. C. W. Chan, Expert Opin. Drug Delivery, 2007, 4, 621–633 CrossRef CAS.
- D. Peer, J. M. Karp, S. Hong, O. C. Farokhzad, R. Margalit and R. Langer, Nat. Nanotechnol., 2007, 2, 751–760 CrossRef CAS.
- I. H. L. Hamelers, R. W. H. M. Staffhorst, J. Voortman, B. de Kruijff, J. Reedijk, P. M. P. van Bergen en Henegouwen and A. I. P. M. de Kroon, Clin. Cancer Res., 2009, 15, 1259–1268 CrossRef CAS.
- I. H. L. Hamelers and A. I. P. M. de Kroon, J. Liposome Res., 2007, 17, 183–189 CrossRef CAS.
- I. H. L. Hamelers, E. van Loenen, R. W. H. M. Staffhorst, B. de Kruijffl and A. I. P. M. de Kroon, Mol. Cancer Ther., 2006, 5, 2007–2012 CrossRef CAS.
- K. Cho, X. Wang, S. M. Nie, Z. Chen and D. M. Shin, Clin. Cancer Res., 2008, 14, 1310–1316 CrossRef CAS.
- S. Cafaggi, E. Russo, R. Stefani, R. Leardi, G. Caviglioli, B. Parodi, G. Bignardi, D. De Totero, C. Aiello and M. Viale, J. Controlled Release, 2007, 121, 110–123 CrossRef CAS.
- J.-H. Kim, Y.-S. Kim, K. Park, S. Lee, H. Y. Nam, K. H. Min, H. G. Jo, J. H. Park, K. Choi, S. Y. Jeong, R.-W. Park, I.-S. Kim, K. Kim and I. C. Kwon, J. Controlled Release, 2008, 127, 41–49 CrossRef CAS.
- E. C. Gryparis, M. Hatziapostolou, E. Papadimitriou and K. Avgoustakis, Eur. J. Pharm. Biopharm., 2007, 67, 1–8 CrossRef CAS.
- P. Xu, E. A. Van Kirk, W. J. Murdoch, Y. Zhan, D. D. Isaak, M. Radosz and Y. Shen, Biomacromolecules, 2006, 7, 829–835 CrossRef CAS.
- A. S. Paraskar, S. Soni, K. T. Chin, P. Chaudhuri, K. W. Muto, J. Berkowitz, M. W. Handlogten, N. J. Alves, B. Bilgicer, D. M. Dinulescu, R. A. Mashelkar and S. Sengupta, Proc. Natl. Acad. Sci. U. S. A., 2010, 107, 12435–12440 CrossRef CAS.
- Y. Zhou, D. C. Drummond, H. Zou, M. E. Hayes, G. P. Adams, D. B. Kirpotin and J. D. Marks, J. Mol. Biol., 2007, 371, 934–947 CrossRef CAS.
- L. Yang, H. Mao, Y. A. Wang, Z. H. Cao, X. H. Peng, X. X. Wang, H. W. Duan, C. C. Ni, Q. Yuan, G. Adams, M. Q. Smith, W. C. Wood, X. H. Gao and S. M. Nie, Small, 2009, 5, 235–243 CrossRef CAS.
- X.-H. Peng, Y. Q. Wang, D. H. Huang, Y. X. Wang, H. J. Shin, Z. J. Chen, M. B. Spewak, H. Mao, X. Wang, Y. Wang, Z. Chen, S. M. Nie and D. M. Shin, ACS Nano, 2011, 5, 9480–9493 CrossRef CAS.
- F. Alexis, E. Pridgen, L. K. Molnar and O. C. Farokhzad, Mol. Pharmaceutics, 2008, 5, 505–515 CrossRef CAS.
- S. Dhar, F. X. Gu, R. Langer, O. C. Farokhzad and S. J. Lippard, Proc. Natl. Acad. Sci. U. S. A., 2008, 105, 17356–17361 CrossRef CAS.
- S. Dhar, N. Kolishetti, S. J. Lippard and O. C. Farokhzad, Proc. Natl. Acad. Sci. U. S. A., 2011, 108, 1850–1855 CrossRef CAS.
- N. Graf, D. R. Bielenberg, N. Kolishetti, C. Muus, J. Banyard, O. C. Farokhzad and S. J. Lippard, ACS Nano, 2012, 6, 4530–4539 CrossRef CAS.
- W. J. Rieter, K. M. Pott, K. M. L. Taylor and W. Lin, J. Am. Chem. Soc., 2008, 130, 11584–11585 CrossRef CAS.
- P. Ghosh, G. Han, M. De, C. K. Kim and V. M. Rotello, Adv. Drug Delivery Rev., 2008, 60, 1307–1315 CrossRef CAS.
- D. A. Giljohann, D. S. Seferos, W. L. Daniel, M. D. Massich, P. C. Patel and C. A. Mirkin, Angew. Chem., Int. Ed., 2010, 49, 3280–3294 CrossRef CAS.
- W. Lu, A. K. Singh, S. A. Khan, D. Senapati, H. Yu and P. C. Ray, J. Am. Chem. Soc., 2010, 132, 18103–18114 CrossRef CAS.
- G. E. Craig, S. D. Brown, D. A. Lamprou, D. Graham and N. J. Wheate, Inorg. Chem., 2012, 51, 3490–3497 CrossRef CAS.
- A. Verma and F. Stellacci, Small, 2010, 6, 12–21 CrossRef CAS.
- L. Ren, X.-L. Huang, B. Zhang, L.-P. Sun, Q.-Q. Zhang, M.-C. Tan and G.-M. Chow, J. Biomed. Mater. Res., Part A, 2008, 85A, 787–796 CrossRef CAS.
- S. Bhattacharyya, M. Gonzalez, J. D. Robertson, R. Bhattacharya and P. Mukherjee, Chem. Commun., 2011, 47, 8530–8532 RSC.
- S. D. Brown, P. Nativo, J. A. Smith, D. Stirling, P. R. Edwards, B. Venugopal, D. J. Flint, J. A. Plumb, D. Graham and N. J. Wheate, J. Am. Chem. Soc., 2010, 132, 4678–4684 CrossRef CAS.
- S. Dhar, W. L. Daniel, D. A. Giljohann, C. A. Mirkin and S. J. Lippard, J. Am. Chem. Soc., 2009, 131, 14652–14653 CrossRef CAS.
- D. A. Giljohann, D. S. Seferos, P. C. Patel, J. E. Millstone, N. L. Rosi and C. A. Mirkin, Nano Lett., 2007, 7, 3818–3821 CrossRef CAS.
- N. L. Rosi, D. A. Giljohann, C. S. Thaxton, A. K. R. Lytton-Jean, M. S. Han and C. A. Mirkin, Science, 2006, 312, 1027–1030 CrossRef CAS.
- B. D. Chithrani, A. A. Ghazani and W. C. W. Chan, Nano Lett., 2006, 6, 662–668 CrossRef CAS.
- W. Jiang, B. Y. S. Kim, J. T. Rutka and W. C. W. Chan, Nat. Nanotechnol., 2008, 3, 145–150 CrossRef CAS.
- G. von Maltzahn, J.-H. Park, A. Agrawal, N. K. Bandaru, S. K. Das, M. J. Sailor and S. N. Bhatia, Cancer Res., 2009, 69, 3892–3900 CrossRef CAS.
- Y. Z. Min, C. Q. Mao, D. C. Xu, J. Wang and Y. Z. Liu, Chem. Commun., 2010, 46, 8424–8426 RSC.
- Y. Z. Min, C.-Q. Mao, S. M. Chen, G. L. Ma, J. Wang and Y. Z. Liu, Angew. Chem., Int. Ed., 2012, 51, 6742–6747 CrossRef CAS.
- C. Sun, J. S. H. Lee and M. Q. Zhang, Adv. Drug Delivery Rev., 2008, 60, 1252–1265 CrossRef CAS.
- M. Arruebo, R. Fernández-Pacheco, M. R. Ibarra and J. Santamaría, Nano Today, 2007, 2, 22–32 CrossRef.
- X.-H. Peng, X. M. Qian, H. Mao, A. Y. Wang, Z. Chen, S. M. Nie and D. M. Shin, Int. J. Nanomed., 2008, 3, 311–321 CAS.
- C. J. Sunderland, M. Steiert, J. E. Talmadge, A. M. Derfus and S. E. Barry, Drug Dev. Res., 2006, 67, 70–93 CrossRef CAS.
- M. Guo, Y. Yan, H. K. Zhang, H. S. Yan, Y. J. Cao, K. L. Liu, S. R. Wan, J. S. Huang and W. Yue, J. Mater. Chem., 2008, 18, 5104–5112 RSC.
- V. I. Shubayev, T. R. Pisanic II and S. Jin, Adv. Drug Delivery Rev., 2009, 61, 467–477 CrossRef CAS.
- J. Yang, C.-H. Lee, H.-J. Ko, J.-S. Suh, H.-G. Yoon, K. Lee, Y.-M. Huh and S. Haam, Angew. Chem., Int. Ed., 2007, 46, 8836–8839 CrossRef CAS.
- D. Y. Chen, M. J. Jiang, N. J. Li, H. W. Gu, Q. F. Xu, J. F. Ge, X. W. Xia and J. M. Lu, J. Mater. Chem., 2010, 20, 6422–6429 RSC.
- R. Hao, R. J. Xing, Z. C. Xu, Y. L. Hou, S. Gao and S. H. Sun, Adv. Mater., 2010, 22, 2729–2742 CrossRef CAS.
- C. J. Xu, B. D. Wang and S. H. Sun, J. Am. Chem. Soc., 2009, 131, 4216–4217 CrossRef CAS.
- R. M. Xing, X. Y. Wang, C. L. Zhang, J. Z. Wang, Y. M. Zhang, Y. Song and Z. J. Guo, J. Mater. Chem., 2011, 21, 11142–11149 RSC.
- E. Gabano, M. Ravera and D. Osella, Curr. Med. Chem., 2009, 16, 4544–4580 CrossRef CAS.
- G. Cossa, L. Gatti, F. Zunino and P. Perego, Curr. Med. Chem., 2009, 16, 2355–2365 CrossRef CAS.
- B. W. Harper, A. M. Krause-Heuer, M. P. Grant, M. Manohar, K. B. Garbutcheon-Singh and J. R. Aldrich-Wright, Chem.–Eur. J., 2010, 16, 7064–7077 CAS.
| This journal is © The Royal Society of Chemistry 2013 |