DOI:
10.1039/C2CS35321K
(Tutorial Review)
Chem. Soc. Rev., 2013,
42, 128-142
The organometallic chemistry of cycloheptatrienyl zirconium complexes
Received
8th August 2012
First published on 1st October 2012
Abstract
This tutorial review summarizes the organometallic chemistry derived from the half-sandwich complex [(η7-C7H7)ZrCl(tmeda)], which was used as an efficient and versatile starting material for the incorporation of monoanionic ligands into the cycloheptatrienyl zirconium coordination sphere by conventional salt metathesis reactions. A broad variety of ligands was employed, affording novel and previously inaccessible cycloheptatrienyl (sandwich) complexes of the type [(η7-C7H7)Zr(Y)]; Y comprises pentadienyl, cyclopentadienyl, allyl, phospholyl, boratabenzene, imidazolin-2-iminato and amido systems. The cycloheptatrienyl ring in these systems usually acts as an “innocent spectator ligand”, but reactivity can arise from the second ligand Y or the Lewis acidity of the, formally, Zr(+IV) center, which was probed in selected examples and put in perspective to related studies. The corresponding results emphasize why the use of [(η7-C7H7)ZrCl(tmeda)] is clearly an advancement in the chemistry of the still fairly unexplored area of cycloheptatrienyl transition metal complexes.

Andreas Glöckner
| Andreas Glöckner was born in Germany in 1983 and studied chemistry at the Technische Universität Braunschweig and the University of Utah. In 2008 he worked on his diploma thesis in Salt Lake City under the supervision of Matthias Tamm (Braunschweig) and Richard D. Ernst (Utah) with a project on cycloheptatrienyl–pentadienyl zirconium complexes. In 2009, he continued to study cycloheptatrienyl complexes as part of his PhD work under the guidance of Matthias Tamm in Braunschweig, where he obtained his PhD in July 2012. He had short research visits with Zuowei Xie (Chinese University of Hong Kong) in 2010, and with Moris S. Eisen (Israel Institute of Technology) in 2011. He was awarded several fellowships and prizes, including from the German Academic Exchange Service, the Bayer Science & Education Foundation and the Fonds der Chemischen Industrie. |

Matthias Tamm
| Matthias Tamm studied chemistry at the Technische Universität Berlin, where he received his PhD in 1992 under the supervision of F. Ekkehardt Hahn. After spending one year as a Visiting Research Scientist at DuPont Central Research and Development, Experimental Station, Wilmington, USA with Anthony J. Arduengo, III, he returned to Germany in 1994 to complete his Habilitation. Tamm was appointed to Privatdozent at the Westfälische Wilhelms-Universität Münster in 1999 and held a temporary professorship at the Technische Universität München from 2002 to 2005. He moved to the Technische Universität Carolo-Wilhelmina zu Braunschweig in 2005 to become full professor of inorganic chemistry. Research interests lie in the areas of preparative organometallic chemistry and coordination chemistry with an emphasis on the development of novel, unusual ligand systems and on the application of their transition metal complexes. |
Introduction
Although chemists usually like to foresee the outcome of their reactions based on the current knowledge, great advances and ground-breaking developments were often created by serendipity. One such story started in 1951 with the discovery of the iconic sandwich molecule ferrocene, [Fe(η5-C5H5)2], which has had a tremendous impact on the development of the chemistry of metal–carbon bonds.1
Shortly thereafter, in 1956, Wilkinson turned the attention to group 4 transition metals and reported the synthesis of bis(cyclopentadienyl)titanium, known as titanocene, which was thought to have a structure similar to that of ferrocene.2 About 15 years later, Brintzinger and Bercaw postulated the presence of bridging hydrides based on IR data and they suggested two possible structures for [Ti(C5H5)2].3 NMR spectroscopic investigations and eventually a structural determination in 1992 confirmed the earlier proposal and showed that [Ti(C5H5)2] actually consists of two (η5-C5H5)Ti units bridged by two hydrides and an η5:η5-fulvalene ligand (Fig. 1) resulting from hydrogen abstraction and subsequent dimerization of the titanafulvene hydride intermediate.4 Finally, the first “true” titanocene was isolated and structurally characterized by Lawless and his group in 1998 (Fig. 2): they achieved the stabilization of the sandwich structure with parallel rings by using bulky cyclopentadienyl ligands, which prevented decomposition or rearrangement.5 This, however, was not the first group 4 transition metal sandwich complex known at that time.
![Molecular structure of “[Ti(C5H5)2]” (see ref. 4).](/image/article/2013/CS/c2cs35321k/c2cs35321k-f1.gif) |
| | Fig. 1 Molecular structure of “[Ti(C5H5)2]” (see ref. 4). | |
![Molecular structure of [Ti{η5-C5Me4(SiMe2tBu)}2] (see ref. 5).](/image/article/2013/CS/c2cs35321k/c2cs35321k-f2.gif) |
| | Fig. 2 Molecular structure of [Ti{η5-C5Me4(SiMe2tBu)}2] (see ref. 5). | |
Already in 1982, Liu and Ernst published a seminal paper on the first open titanocene, [Ti(η5-2,4-C7H11)2] (C7H11 = dimethyl-pentadienyl), which is diamagnetic in contrast to Lawless' complex.6 They even managed to synthesize and crystallize a 14-electron open zirconocene, [Zr{η5-1,5-(TMS)2C5H5}2] (TMS = trimethylsilyl), and the corresponding titanium complex shortly afterwards (Fig. 3).7 Both results can be considered as a breakthrough in the chemistry of the so-called open Cp ligands, because they already point at dramatic differences between the ubiquitous cyclopentadienyl ligand and its open congener.
![Molecular structure of [Ti{η5-1,5-(TMS)2C5H5}2] (see ref. 7).](/image/article/2013/CS/c2cs35321k/c2cs35321k-f3.gif) |
| | Fig. 3 Molecular structure of [Ti{η5-1,5-(TMS)2C5H5}2] (see ref. 7). | |
Notably, even more than a decade earlier, van Oven and de Liefde Meijer came up with the syntheses of the mixed cycloheptatrienyl and cyclooctatetraenyl cyclopentadienyl titanium complexes, [(η7-C7H7)Ti(η5-C5H5)] and [(η8-C8H8)Ti(η5-C5H5),8 which both have the typical sandwich structure as revealed by X-ray diffraction analysis (Fig. 4).9,10
![Structure of [(η7-C7H7)Ti(η5-C5H5)] (see ref. 10).](/image/article/2013/CS/c2cs35321k/c2cs35321k-f4.gif) |
| | Fig. 4 Structure of [(η7-C7H7)Ti(η5-C5H5)] (see ref. 10). | |
Consequently, the cycloheptatrienyl ligand participated in the chemistry of group 4 metallocene-type molecules from early on. Despite this early birth, this tutorial review is intended to validate Chirik's recent statement: “Group 4 Transition Metal Sandwich Complexes: Still Fresh after Almost 60 Years” with a strong focus on cycloheptatrienyl zirconium complexes.11
The extensive studies by Green and co-workers in the 1980s and 1990s established the organotransition metal chemistry of the cycloheptatrienyl (Cht) ligand,12 although the first complex, [(η7-C7H7)Mo(CO)3][BF4] (1), had been synthesized by Dauben and Honnen in 1958.13 The recent resurgence of early transition metal complexes containing the C7H7 ligand is manifested by independent reports from the groups of Braunschweig,14 Elschenbroich,15 Girolami,16 and Tamm.17 One reason for the revitalized interest is the landmark discovery by Manners et al. of using strained ansa-ferrocenes for ring-opening polymerization (ROP) leading to metallopolymers,18 which ultimately encouraged the development of non-iron metallocenes for similar applications.19 Among them is a series of ansa-Cht–Cp complexes of titanium, which were prepared from the parent [(η7-C7H7)Ti(η5-C5H5)] complex (2, “troticene”) by lithiation and subsequent reaction with electrophiles (Scheme 1);20 in the presence of N,N′,N′,N′′,N′′-pentamethyldiethylenetriamine (pmdta) the reactive dilithio-intermediate 3 could also be isolated and structurally characterized.21 Interestingly, some of these [n]troticenophanes are not only precursors for ROPs, they also regioselectively insert Pt(0) complexes into the E–C bond to the Cht ligand.
![Synthesis of [n]troticenophanes.](/image/article/2013/CS/c2cs35321k/c2cs35321k-s1.gif) |
| | Scheme 1 Synthesis of [n]troticenophanes. | |
The lithiation/quenching procedure is also the method of choice for the synthesis of mono-functionalized troticenes, which can be performed selectively at either the five- or alternatively at the seven-membered ring.21,22 Unfortunately, this approach is limited to troticene, while analogous protocols have not been realized for [(η7-C7H7)Zr(η5-C5H5)] (4, “trozircene”) and [(η7-C7H7)Hf(η5-C5H5)] (5, “trohafcene”) yet.23 Therefore, the variation of the substituents at the cyclopentadienyl ring is rather tedious, because it builds on the established protocol for the preparation of mixed cycloheptatrienyl–cyclopentadienyl group 4 transition metal complexes.
Typically, [(η5-Cp)MCl3] (Cp = cyclopentadienyl or substituted derivatives thereof; M = Ti, Zr, Hf) or [(η5-Cp)2MCl2] starting materials are reacted with a reducing agent such as Mg or iPrMgCl in the presence of an excess of cycloheptatriene and catalytic amounts of FeCl3 (Scheme 2).8,17,23,24 Several disadvantages are encountered with this approach. (1) The yields are quite low with roughly 25% for both M = Zr and Hf. (2) In some cases, for instance in the synthesis of phosphane substituted complexes of the type [(η7-C7H7)M(η5-C5H4PR2)] (M = Zr, Hf; R = Ph, iPr),23 metallocene dihalides have to be used. Consequently, one of the valuable C5H4PR2 ligands has to be sacrificed during the course of the reaction. (3) Whenever a different substituent (named R in Scheme 2) at the cyclopentadienyl ligand is desired, a new starting material is required. (4) The limitation to (substituted) cyclopentadienyl ligands does not allow for the incorporation of more elaborated 6π-electron donor ligands.
![Common synthesis of [(η7-C7H7)M(η5-C5H5)] complexes (M = Ti, Zr, Hf).](/image/article/2013/CS/c2cs35321k/c2cs35321k-s2.gif) |
| | Scheme 2 Common synthesis of [(η7-C7H7)M(η5-C5H5)] complexes (M = Ti, Zr, Hf). | |
The last argument became particularly evident when we decided to start a collaborative project on complexes of the type [(η7-C7H7)Zr(η5-Pdl)] (Pdl = pentadienyl or substituted pentadienyl) together with Richard D. Ernst (University of Utah). These complexes rely on a pentadienyl (“open Cp”) system as the co-ligand and are referred to as half-open trozircenes,25 because of their relationship to “closed” trozircene. Indeed, [(η7-C7H7)M(η5-c-C7H9)] complexes (M = Ti, Zr, Hf, V, Cr, Mo, W; c-C7H9 = cycloheptadienyl) have long been known and had been synthesized for instance by metal vapor synthesis,12 suggesting that related species represent a feasible target. In analogy to Scheme 2, however, the attempted formation of [(η7-C7H7)Zr(η5-6,6-dmch)] (dmch = dimethylcyclohexadienyl) from [(η5-C5H5)(η5-6,6-dmch)ZrCl2] under reductive conditions failed. Together with the arguments discussed above, it was clear that a versatile starting material was called for!
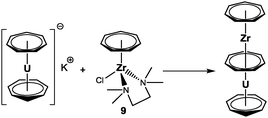
Such a system would already bear the seven-membered ring and provide the possibility to incorporate a broad variety of monoanionic ligands into the cycloheptatrienyl zirconium coordination sphere by salt metathesis reactions. The literature offers some half-sandwich complexes as promising candidates for this task (Fig. 5):12 the η6-cycloheptatriene complex 6 is the only one that has already been employed for the preparation of sandwich complexes, because its reaction with lithium indenide afforded [(η7-C7H7)Zr(η5-C9H7)] (C9H7 = indenyl). In contrast, the analogous reaction with sodium cyclopentadienide did not result in the formation of the expected [(η7-C7H7)Zr(η5-C5H5)] complex. Compounds 7 and 8 were both obtained from [Zr(η6-C7H8)2] and [Et2AlCl]2 or I2, respectively, but in particular the synthesis of the zerovalent bis(η6-cycloheptatriene)zirconium complex appears challenging.12 The most promising candidate in this series is [(η7-C7H7)ZrCl(tmeda)] (9, tmeda = N,N,N′,N′-tetramethylethylene-1,2-diamine), which was first prepared in a three step reaction sequence by Green et al.; later on, a convenient one-pot synthesis was also reported for this compound.26
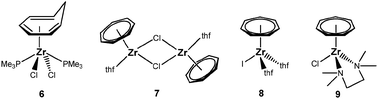 |
| | Fig. 5 Potential versatile starting materials. | |
Upon slight modification of the original procedure, 9 is now synthesized in yields ranging from 31 to 37% (1.34–1.58 g based on 3.00 g ZrCl4) from a sodium amalgam reduction of ZrCl4 in the presence of cycloheptatriene and tmeda followed by extraction with THF (Scheme 3).27 An ORTEP diagram of this key complex is shown in Fig. 6, displaying a three-legged piano stool geometry. Although Green et al. reported that the titanium analogue of 9 can be obtained in a similar fashion,26 reproduction was not possible in our hands despite several attempts.
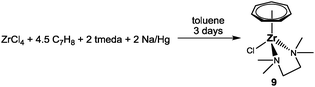 |
| | Scheme 3 Synthesis of 9. | |
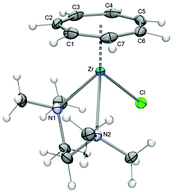 |
| | Fig. 6 ORTEP diagram of 9. | |
Half-open trozircenes – the start of an entirely new approach to cycloheptatrienyl zirconium complexes
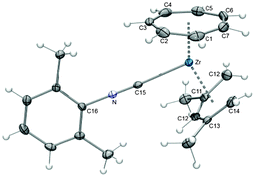
As mentioned above, the groups of Tamm and Ernst first targeted half-open trozircenes, and the reaction of [(η7-C7H7)ZrCl(tmeda)] (9) with a variety of pentadienyl anions indeed resulted in the formation of [(η7-C7H7)Zr(η5-C5H7)] (10), [(η7-C7H7)Zr(η5-2,4-C7H11)] (11, C7H11 = dimethylpentadienyl), [(η7-C7H7)Zr(η5-6,6-dmch)] (12, dmch = dimethylcyclohexadienyl) and [(η7-C7H7)Zr(η5-c-C7H9)] (13, c-C7H9 = cycloheptadienyl) accompanied by the loss of the stabilizing tmeda ligand (Scheme 4). Isolation of these thermally stable, but air-sensitive, diamagnetic 16-electron compounds was achieved by sublimation.27 As a representative example, the molecular structure of 11 with the most widely used pentadienyl ligand, the 2,4-dimethylpentadienyl, is depicted in Fig. 7.
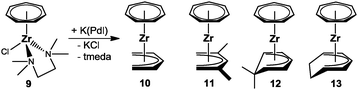 |
| | Scheme 4 Synthesis of half-open trozircenes. | |
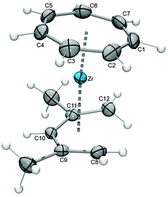 |
| | Fig. 7 ORTEP diagram of 11; the molecule has a mirror plane bisecting C1, Zr and C10. | |
While bis(cyclopentadienyl) and bis(benzene) complexes have been known for a long time, an extrapolation to bis(cycloheptatrienyl) transition metal complexes has not been accomplished to date; the only example, [U(η7-C7H7)2]−, comes from the actinides.28 Recently, Wang and King et al. addressed [M(C7H7)2] complexes (M = Ti, V, Cr, Mn, Fe, Co, Ni) in a theoretical study.29 For titanium, an [(η7-C7H7)Ti(η5-C7H7)] structure was predicted as the global minimum, in which the η5-cycloheptatrienyl ligand has an uncoordinated C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C double bond and is therefore an unsaturated derivative of the cycloheptadienyl ligand found in 13. The corresponding anion, C7H7−, can be formally derived from the deprotonation of cycloheptatriene, but it would most likely circumvent anti-aromaticity by forming a pentadienyl-type anion. Experimentally, however, the attempted “deprotonation” of cycloheptatriene with either nBuLi/tmeda or tBuLi/tmeda resulted in nucleophilic addition in the α-position to the methylene group of C7H8, leading to the formation of Li(C7H8R) (R = nBu, tBu), which were identified based on their hydrolysis products.30 As an alternative, these substituted cycloheptadienide anions were now used as ligands, and their reaction with 9 afforded the complexes 14 and 15, respectively (Scheme 5). It is worth mentioning that two isomers are found in both cases by NMR spectroscopy. The molecular structure of 15 was also confirmed by X-ray diffraction analysis, albeit with poor data.31
C double bond and is therefore an unsaturated derivative of the cycloheptadienyl ligand found in 13. The corresponding anion, C7H7−, can be formally derived from the deprotonation of cycloheptatriene, but it would most likely circumvent anti-aromaticity by forming a pentadienyl-type anion. Experimentally, however, the attempted “deprotonation” of cycloheptatriene with either nBuLi/tmeda or tBuLi/tmeda resulted in nucleophilic addition in the α-position to the methylene group of C7H8, leading to the formation of Li(C7H8R) (R = nBu, tBu), which were identified based on their hydrolysis products.30 As an alternative, these substituted cycloheptadienide anions were now used as ligands, and their reaction with 9 afforded the complexes 14 and 15, respectively (Scheme 5). It is worth mentioning that two isomers are found in both cases by NMR spectroscopy. The molecular structure of 15 was also confirmed by X-ray diffraction analysis, albeit with poor data.31
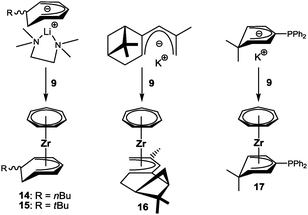 |
| | Scheme 5 Half-open trozircenes with unconventional pentadienyl ligands. | |
Three other unconventional pentadienyl ligands were explored. Salzer et al. recently introduced an optically active pentadiene based on the natural product (−)-myrtenal, which showed a distinct facial selectivity upon coordination to ruthenium, because one of the two sides is blocked by the dimethylmethylene bridge.32 This selectivity was also found in the reaction of the corresponding potassium pentadienide, obtained through deprotonation of the diene with the Schlosser-base, and 9. Thus, the molecular structure of complex 16 displays two methyl groups facing away from the metal, thereby avoiding repulsive interactions with the cycloheptatrienyl ligand.31 Furthermore, complex 17 (Fig. 8) represents the first example of a phosphane substituted pentadienyl system. The corresponding ligand was synthesized from K(6,6-dmch) and ClPPh2, which resulted in selective nucleophilic attack of the central carbon atom, followed by deprotonation.31 Finally, it was not possible to attach the recently introduced novel open indenyl ligand, oIndMe,33 to the (C7H7)Zr fragment; attempts to synthesize [(η7-C7H7)Zr(η5-oIndMe)] failed several times.
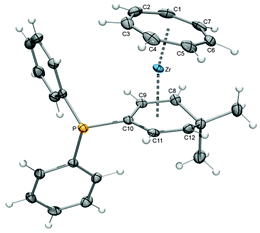 |
| | Fig. 8 ORTEP diagram of 17. | |
Bonding in cycloheptatrienyl complexes of group 4 transition metals
Pentadienyl ligands show a preference for metals in low oxidation states, and detailed studies eventually revealed that they often generate even stronger metal–carbon bonds than cyclopentadienyl ligands, but nevertheless exhibit a significantly higher reactivity, in particular in coupling reactions with unsaturated molecules.34,35 With that in mind, the formal oxidation state in complexes 10–13 definitely had to be addressed.
According to the Hückel-rule, a planar carbocycle is aromatic, if it possesses (4n + 2)π-electrons. For the cycloheptatrienyl ring, two extremes can be envisaged: n = 1 gives a monocationic 6π-electron ligand that is isoelectronic to Cp or benzene, whereas n = 2 results in a 10π-electron ligand as in the cyclooctatetraenyl dianion with a formal −3 charge. Consequently, there has been much controversy about the assignment of the formal oxidation state in group 4 transition metal cycloheptatrienyl complexes. Early theoretical and photoelectron studies in the 1970s already suggested that the metal oxidation state in [(η7-C7H7)M(η5-C5H5)] complexes decreases on going from titanium to chromium with a simultaneous decrease of the negative charge on the C7H7 ligand.36 The advances in technology have more recently initiated similar investigations by Green et al.,37 Menconi and Kaltsoyannis38 and Tamm17 with basically the same conclusion that “the cycloheptatrienyl ring functions more as a −3 ligand than a +1 ligand” in group 4 transition metal cycloheptatrienyl–cyclopentadienyl complexes.38 It must be noted, however, that an experimental electron density study based on high accuracy X-ray data demonstrated that the real electron distribution does not correspond to the formal −3 charge.39 In conclusion, the truth is certainly found somewhere between these two extremes, but if a decision has to be made, the formal +IV oxidation state is in better agreement with computational studies and the observed reactivity.17
This has also an impact on the half-open trozircenes. First of all, the structurally characterized complexes 10–12 display an onset of slippage of the pentadienyl ligands towards the central carbon atom and a distinct short–long–long–short pattern of the C–C bonds along the pentadienyl chain. Thus, the Zr–C10 bond is the shortest, while the Zr–C9/C11 and Zr–C8/C12 become progressively longer (Table 1, the numbering refers to Fig. 7).27 A closer inspection also reveals longer average Zr–C bond lengths (2.53, 2.52 and 2.52 Å) for the pentadienyl ligands compared to the corresponding value found for C5H5 in [(η7-C7H7)Zr(η5-C5H5)] (4, 2.50 Å).17 These observations are typical for pentadienyl ligands in the coordination sphere of high-oxidation state metals, as for instance [(η5-C5H5)(η5-6,6-dmch)ZrCl2], and result from the difficulty of the wide π-system to effectively overlap with contracted metal orbitals. Therefore, high-valent pentadienyl complexes remain quite often elusive, because they prefer lower oxidation states, and comparatively few examples are known.35
Table 1 Pertinent bond lengths (Å) for 10–12
| Complex |
Ligand |
Zr–C8/C12 |
Zr–C9/C11 |
Zr–C10 |
|
10
|
C5H7 |
2.604(3)/2.571(3) |
2.508(3)/2.480(3) |
2.437(3) |
|
11
|
2,4-C7H11 |
2.606(2) |
2.512(2) |
2.431(3) |
|
12
|
6,6-dmch |
2.577(2)/2.595(2) |
2.486(2)/2.493(2) |
2.459(2) |
Not only the bonding parameters clearly indicate the presence of formally +IV metals in the half-open trozircenes, further support was also provided by density functional theory (DFT) calculations.27 As had been expected, the frontier orbitals of [(η7-C7H7)Zr(η5-2,4-C7H11)] (11) resemble those of 4 (Fig. 9).17 The first two pairs, HOMO−5/HOMO−4 and HOMO−3/HOMO−2, represent the largely ionic π interaction of the metal with the Cht and Pdl orbitals, respectively. Of essential relevance for the assignment of the formal oxidation state are HOMO−1 and HOMO, which correspond to the δ interaction with the seven-membered ring. Significant mixing of metal and ligand orbital character is identified, implying strongly covalent interactions, but in agreement with the −3 formalism the contribution of the C7H7 ligand dominates (Zr: 30%; Cht: 64%, Pdl: 6% for the HOMO based on the M05-2X functional). Finally, the LUMO is metal-localized and mainly dz2 in character, because relevant ligand orbitals are too low in energy for efficient interaction.
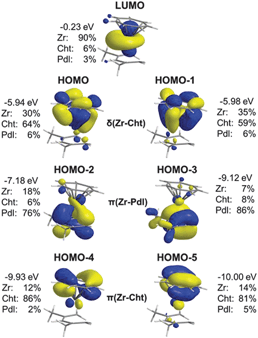 |
| | Fig. 9 Contour plots and composition of the frontier orbitals in 11 based on the M05-2X functional. | |
Reactivity of half-open trozircenes
The Lewis acidic 16-electron complexes [(η7-C7H7)M(η5-C5H5)] (M = Zr, Hf) form (mono)adducts with two-electron donor ligands such as isocyanides, phosphines or N-heterocyclic carbenes (NHCs), which, according to experimental results and DFT calculations, are generally comparatively weak.17,24,40 For related studies on the half-open analogues, [(η7-C7H7)Zr(η5-2,4-C7H11)] (11) was our system of choice, which was reacted in 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 ratios with 2,6-xylyl isocyanide (CN-o-Xy), tert-butyl isocyanide (CN-tBu), trimethylphosphine (PMe3) and 1,3,4,5-tetramethylimidazolin-2-ylidene (IMe) to afford the adducts 18–21, respectively (Scheme 6).27,41 Additionally, the dinuclear complex 22 was obtained by using 1,2-bis(dimethylphosphino)ethane as the bridging ligand. In all cases, coordination occurred exclusively via the closed edge of the pentadienyl ligand as exemplified by the molecular structure of 18 (Fig. 10) and is accompanied by geometric adjustments in order to accommodate the additional ligand. For instance, the Zr–C bond distances are slightly elongated and the two ligand planes now differ significantly from a planar orientation (α = 42.03° for 18vs. 15.74° for 11). In the context of coupling reactions of (low-valent) pentadienyl complexes with unsaturated molecules such as isocyanides,35 it is important to note that 11 lacks a similar reactivity, probably because the extra driving force of achieving a higher oxidation state cannot be provided.
:1 ratios with 2,6-xylyl isocyanide (CN-o-Xy), tert-butyl isocyanide (CN-tBu), trimethylphosphine (PMe3) and 1,3,4,5-tetramethylimidazolin-2-ylidene (IMe) to afford the adducts 18–21, respectively (Scheme 6).27,41 Additionally, the dinuclear complex 22 was obtained by using 1,2-bis(dimethylphosphino)ethane as the bridging ligand. In all cases, coordination occurred exclusively via the closed edge of the pentadienyl ligand as exemplified by the molecular structure of 18 (Fig. 10) and is accompanied by geometric adjustments in order to accommodate the additional ligand. For instance, the Zr–C bond distances are slightly elongated and the two ligand planes now differ significantly from a planar orientation (α = 42.03° for 18vs. 15.74° for 11). In the context of coupling reactions of (low-valent) pentadienyl complexes with unsaturated molecules such as isocyanides,35 it is important to note that 11 lacks a similar reactivity, probably because the extra driving force of achieving a higher oxidation state cannot be provided.
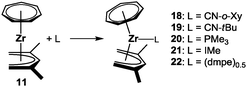 |
| | Scheme 6 Adduct formation of 11. | |
 |
| | Fig. 10 ORTEP diagram of 18. | |
Remarkably, the isocyanide adducts 18 and 19 can be used as an indirect probe for the metal oxidation state. Their IR spectra display well-defined C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) N stretches at 2100 cm−1 and 2139 cm−1, which is fairly close to the values found for free CN-o-Xy (2122 cm−1) and CN–tBu (2132 cm−1), respectively, consistent with the absence of significant π-backbonding effects as expected for formally d0 Zr(+IV). Similarly, the C–N stretch for [(η7-C7H7)Zr(η5-6,6-dmch)(CN-o-Xy)] appears at 2121 cm−1.
N stretches at 2100 cm−1 and 2139 cm−1, which is fairly close to the values found for free CN-o-Xy (2122 cm−1) and CN–tBu (2132 cm−1), respectively, consistent with the absence of significant π-backbonding effects as expected for formally d0 Zr(+IV). Similarly, the C–N stretch for [(η7-C7H7)Zr(η5-6,6-dmch)(CN-o-Xy)] appears at 2121 cm−1.
Furthermore, DFT calculations employing the M05-2X functional suggest exothermic, but rather weak adduct formation of the isocyanide and phosphine complexes compared to the higher stabilization by about 10 kcal mol−1 calculated for the NHC adduct 22 (Table 2).41 This observation can mainly be attributed to the higher propensity of 11 to interact more efficiently with hard, good σ-donor ligands such as NHCs than with σ-donor/π-acceptor ligands such as isocyanides.
Table 2 Calculated standard enthalpies of adduct formation (based on the M05-2X functional); calculated and experimental metal–ligand distances
| Complex |
L |
ΔH (kcal mol−1) |
d(M–L)calc (Å) |
d(M–L)exp (Å) |
|
18
|
CN-o-Xy |
−13.9 |
2.37 |
2.364(2) |
|
20
|
PMe3 |
−11.1 |
2.89 |
2.844(1) |
|
21
|
IMe |
−22.7 |
2.48 |
2.453(1) |
|
22
|
dmpe |
−12.1 |
2.89 |
2.840(6) |
Cycloheptatrienyl–cyclopentadienyl zirconium complexes and the question “How big is a Cp?”
The successful incorporation of pentadienyl ligands into the (C7H7)Zr coordination sphere raises hope that [(η7-C7H7)ZrCl(tmeda)] (9) can also serve as a starting material for a variety of other ligands, which not necessarily have to be monoanionic, 6π-electron ligands as long as they readily undergo salt metathesis reactions. This would allow us to synthesize and characterize novel cycloheptatrienyl zirconium complexes, which previously have not been accessible by established methods (Scheme 2), either because the required metallocene dihalides are not known or do not react under those conditions. The basic idea will remain the same (Scheme 7): a ligand salt M(Y) (M = Li, Na, K) is reacted with 9, which leads to the formation of a new Zr–Y bond and (insoluble) MCl. Whenever the resulting [(η7-C7H7)Zr(Y)] complex is electronically and/or sterically saturated, tmeda might be “lost” in the course of the reaction or replaced by another stabilizing ligand.
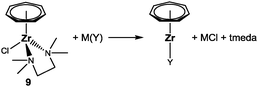 |
| | Scheme 7 General procedure for the preparation of novel (C7H7)Zr complexes. | |
Obviously, if pentadienyl ligands can be incorporated into the cycloheptatrienyl zirconium coordination sphere by the procedure outlined in Scheme 7, there is no reason for conventional cyclopentadienyl ligands not to work. At first, two known trozircenes [(η7-C7H7)Zr(η5-C5H5)] (4) and [(η7-C7H7)Zr(η5-C5H4PPh2)] (23) were successfully tested (Scheme 8). The isolated yields are at least twice as high as those obtained by the established method (Scheme 2),17,23 demonstrating the power of the new approach.
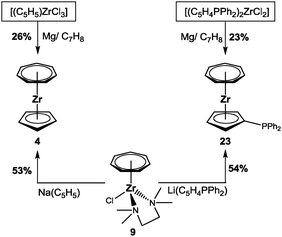 |
| | Scheme 8 Possible synthetic routes to 4 and 23. | |
This is emphasized even more by taking into account the array of trozircenes, which were synthesized from only one starting material, that is [(η7-C7H7)ZrCl(tmeda)] (9). While we first concentrated on monosubstituted cyclopentadienyl ligands,42 we later turned our attention to bulky derivatives as well as some selected indenyl ligands.43 So far, only fluorenyl and the extremely sterically encumbered penta(isopropyl)cyclo-pentadienyl ligand, C5iPr5, did not yield the desired products. Overall, the library comprises twelve structurally characterized trozircenes (Fig. 11); they range from no substitution at the η5-bound ligand (in C5H5) to pentasubstitution (in C5Me5). Only for 30, the molecular structure was calculated, for all other examples, X-ray data are available,43 providing the possibility of systematic comparison.
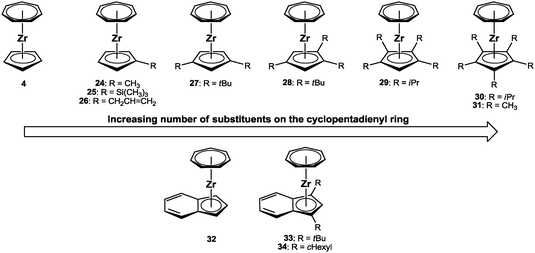 |
| | Fig. 11 Drawings of the cycloheptatrienyl zirconium sandwich complexes 4 and 24–34. | |
A characteristic feature of the cycloheptatrienyl ring in all complexes is a noticeable tilt of the hydrogen substituents out of the C1–C7 plane towards the metal. As representative examples (Fig. 12), the corresponding angles in [(η7-C7H7)Zr(η5-C5H4Me)] (24) and [(η7-C7H7)Zr(η5-C5H3tBu2)] (27, tBu = tert-butyl) are on average 7.2° and 5.6°, respectively. In comparison, significantly smaller values are found for the hydrogen atoms at the cyclopentadienyl ligand (2.1° and 0.8°), which result from the smaller diameter of the π-system that does not require a pronounced improvement of ligand–metal orbital overlap as shown in Scheme 9.43 The obvious effort of the seven-membered ring to develop strong Zr–C bonds is also recognized in the bond lengths, which are ca. 0.15 Å shorter for the C7H7vs. the η5-bound ligand, and agrees with the presence of a formally trianionic cycloheptatrienyl ligand. Consequently, dramatic differences in metal–centroid distances of 1.668 vs. 2.201 Å (averaged values) are found, because a much closer approach of the Cht ligand to the metal is required. This leads to a decent protection of the zirconium atom from one face, while the shielding of the second half depends on the size of the cyclopentadienyl or indenyl ligand. But how big is a Cp?
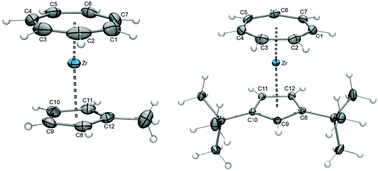 |
| | Fig. 12 ORTEP diagrams of 24 (left) and 27 (right). | |
 |
| | Scheme 9 Improvement of ligand–metal orbital overlap. | |
Early work by Tolman introduced the concept of cone angle measurements to assess the steric demand of group 15 donor ligands, particularly that of phosphines.44 Some related studies, in particular by Coville, have adopted this concept to “smaller” cyclopentadienyl ligands,45 but the data usually rely on molecular modeling and not on crystallographic data. Furthermore, comparison of values obtained from [Fe(η5-C5H5−nRn)2] and [(η5-C5H5−nRn)2ZrCl2] complexes is not valid, because cone angles depend on metal–centroid distances. In contrast, the (C7H7)Zr fragment appears to be a useful reference system for several reasons. (1) An extension of the present library (Fig. 11) should be facile, because the synthesis of these complexes is straightforward and crystallization can usually be carried out easily. (2) The Zr–Cpcent distances are in a narrow range (2.172–2.234 Å), which allows the direct comparison of cone angles. (3) Resulting from the sandwich structure, the substituents on the η5-ligand are not hindered in their expansion in all three dimensions and can adopt the most stable conformation. (4) Free rotation is possible, which is not always given in complexes involving bulky Cp ligands.43
Similar to previous studies,45 cone angle measurements on complexes 4 and 24–34 according to Fig. 13 gave the following order of size for the ligands
| C5H5 < C5H4Me < C9H7 ≈ C5H4TMS ≈ C5H4(allyl) < C5H3tBu2 < C5Me5 < C9H5tBu2 ≈ C9H5(chexyl)2 ≈ C5H2tBu3 < C5HiPr4 < C5iPr5 |
based on the angle
Θ and for the substituents
| hexyl ≈ iso-propyl < TMS < |
based on the angle
Ω.
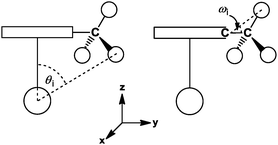 |
| | Fig. 13 Cone angle measurement leading to Θ (left) and Ω (right). | |
The cone angle Ω was introduced to take into account that some of the substituents, for instance in C5HiPr4 (C5HiPr4 = tetra(iso-propyl)cyclopentadienyl), are not rotationally symmetric and their “real” steric demand is consequently overestimated by only considering Θ. Therefore, Ω describes the size in the z-dimension, whereas Θ is a measure for the steric demand of the ligand in the xy-dimension (Table 3). Both angles have to be used together to interpret experimental observations such as rotational barriers.43
Table 3 Measured cone angles (°) for 4 and 24–34
| Complex |
Cp |
Θ
|
Ω
|
|
Θ = 2/5(θ1 + θ2 + θ3 + θ4 + θ5), θi = maximum half cone angle of each substituent with Zr as apex.
Average of the maximum cone angles of each group (=ωi) on the α-C with ipso-C as apex.
Two independent molecules in the asymmetric unit.
|
|
4
|
C5H5 |
88.2 |
0 |
|
24
|
C5H4Me |
95.1 |
49.0 |
|
25
|
C5H4TMS |
104.3 |
95.6 |
|
26
|
C5H4(allyl) |
106.0 |
68.6 |
|
27
|
C5H3tBu2 |
116.2 |
100.7 |
|
28
|
C5H2tBu3 |
132.0 |
99.8 |
|
29
|
C5HiPr4 |
146.4 |
85.9 |
|
30
|
C5Me5 |
122.4 |
51.2 |
|
31
|
C5iPr5 |
167.4 |
88.6 |
| Complex |
Indenyl |
Θ
|
Ω
|
|
32
|
C9H7 |
102.6 |
0 |
|
33
|
C9H5tBu2 |
130.7/131.8 |
101.3/100.2 |
|
34
|
C9H5(chexyl)2 |
131.0/131.7 |
84.9/84.2 |
Cycloheptatrienyl zirconium allyl complexes
In particular Rosenthal and co-workers have developed a beautiful chemistry based on unsaturated 14-electron metallocene fragments generated in situ from [(η5-Cp)2M(bmtsa)] (M = Ti, Zr, Hf; Cp = C5H5 or substituted derivatives thereof; bmtsa = bis(trimethylsilyl)acetylene).46 As an isoelectronic relative to the [(η5-Cp2)M] fragment, [(η7-C7H7)M(η3-C3H5)] can be envisaged by the formal exchange of two Cp ligands by one cycloheptatrienyl and one allyl ligand (Fig. 14). However, it must be noted that the former has a +II oxidation state, while the formal C7H73− ligand results in a +IV oxidation state for the latter, which should lead to some marked differences in their reactivity.
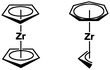 |
| | Fig. 14 Isoelectronic complex fragments. | |
Encouraged by the “Rosenthal chemistry”, we set out to introduce the C3H5 ligand into the cycloheptatrienyl coordination sphere by a reaction of [(η7-C7H7)ZrCl(tmeda)] (9) with (C3H5)MgBr as an allyl transfer reagent. We attributed the failure of that attempt to the small size of the unsubstituted allyl ligand and turned our attention to the sterically encumbered 1,3-bis(trimethylsilyl)allyl ligand. Studies by Hanusa and others have shown that this ligand is capable of stabilizing a number of unusual molecules, including a bis(allyl)nickel complex that does not decompose at room temperature.47,48 As had been desired, the bulky allyl ligand also reacted cleanly with 9 and afforded [(η7-C7H7)Zr{η3-C3H3(TMS)2}(thf)] (35) as a brown solid. Obviously, the reduced electron count of the allyl ligand compared to a pentadienyl or cyclopentadienyl ligand requires the coordination of an additional THF molecule.49 DFT calculations suggest an enthalpy of formation from the hypothetical [(η7-C7H7)Zr{η3-C3H3(TMS)2}] complex and THF of ΔH298 = −18.2 kcal mol−1 (Fig. 15).
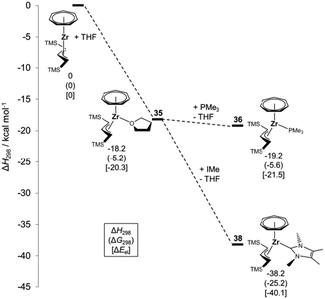 |
| | Fig. 15 Energy diagram based on the B97-D values for the formation of the complexes 35, 36 and 38. | |
The THF ligand in 35 can be replaced by other two-electron donor ligands, and reactions were performed with PMe3, 4-(dimethylamino)pyridine (DMAP), IMe and 1,3,4,5-tetramethyl-2-methyleneimidazoline (H2C–IMe) resulting in 36–39, respectively (Scheme 10).50 A pronounced stabilization of ca. 20 kcal mol−1 compared to the THF or PMe3 complexes was computed for the carbene adduct 38, which once again (vide supra) emphasizes the demand for strong σ-donor ligands. Additionally, a comparison with studies on [(η7-C7H7)Zr(η5-C5H5)(L)] complexes (L = PMe3, IMe) reveals that their calculated enthalpies of formation are at least 10 kcal mol−1 lower than in the present case,24 illustrating the difference in Zr–L bond strengths in an 18- vs. a 16-electron complex. Similarly, a variable temperature NMR study on 36 with a second equivalent PMe3 afforded a free energy of activation for the exchange of coordinated and free phosphane of 13.4(5) kcal mol−1, which is 3.7 kcal mol−1 more than determined under identical conditions for [(η5-C7H7)Zr(η5-C5H5)(PMe3)].40
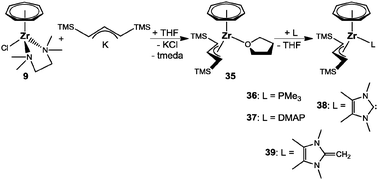 |
| | Scheme 10 Preparation of 35 and subsequent ligand exchange. | |
The molecular structures of all five examples were established by X-ray diffraction analysis and their bonding parameters resemble each other. As shown in Fig. 16 for 35, the ligands are coordinated to zirconium at the open edge of the allyl moiety, corresponding to an exo-conformation. With respect to the central hydrogen atom of the allyl moiety, the trimethylsilyl substituents adopt a syn–syn orientation to avoid steric interactions with the incoming ligand. Worth mentioning, the Zr–C bond distance to the central carbon atom is slightly shorter than those to the terminal carbon atoms; for example, for 35 the corresponding values are 2.466(8) Å and 2.514(6) Å.
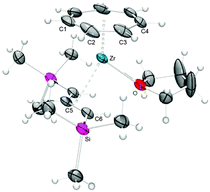 |
| | Fig. 16 ORTEP diagram of 35. The molecule possesses crystallographic mirror symmetry. | |
Hanusa and Layfield recently stated in independent review articles that the area of second and third row transition metal silyl–allyl complexes still remains fairly unexplored.47,48 In particular the capability of the C3H3(TMS)2 ligand to act as a base in certain (intramolecular) deprotonation reactions was completely unknown (vide infra). Furthermore, no structurally characterized bis(trimethylsilyl)allyl complexes of group 4 transition metals had been published until our initial report on the synthesis and characterization of 35,49 followed by extended studies later on.50 The only closely related systems are the two trivalent complexes [{1,3-(tBuMe2Si)2C3H3}2M(μ-Cl)2Li(tmeda)] (M = Ti, Zr) and homoleptic ansa-bis(silylallyl)zirconium- and hafnium complexes.47,48
Phosphatrozircenes – bifunctional trozircenes
Thus far, the focus was on monoanionic ligands, which exclusively form metal–carbon bonds and only have aliphatic substituents. Some new interesting aspects can arise, if functionalized ligands are used instead. Recently, a number of phosphane-substituted complexes of the type [(η7-C7H7)Zr(η5-C5H4PR2)] (M = Ti, Zr, Hf) were introduced and used as metallophosphanes for the preparation of bimetallic complexes. In some cases, the concomitant presence of a Lewis basic substituent and a Lewis acidic metal led to bifunctional reactivity and the development of interesting secondary interactions, e.g. weak Zr–metal or Zr–isocarbonyl contacts.23,51 As a continuation of this concept, we now envisaged to move the phosphorus atom directly from the substituent into the five-membered ring by employing phospholyl ligands. This interesting class of five-membered π-ligands was extensively studied by Mathey and co-workers, among others, and results from the formal exchange of a CH unit by a P atom in cyclopentadienyl rings.52,53 Phospholyl ligands are in an isolobal relationship to cyclopentadienyl, and consequently the η5-coordination mode through the π-system dominates, although (intermediate) coordination via phosphorus (η1-mode) is not impossible.54 The electron lone pair on phosphorus is crucial for the formation of (hetero)bimetallic complexes, and many examples including systems with relevance to catalysis are known.52,53
Salt metathesis reactions are most commonly used for the introduction of phospholyl ligands into metal complexes. This offered the possibility to present the first series of heterotrozircenes, the phosphatrozircenes,25 by reacting different in situ prepared phospholide anions with [(η7-C7H7)ZrCl(tmeda)] (9). Three examples were synthesized and fully characterized: [(η7-C7H7)Zr(η5-C4PMe4)] (40), [(η7-C7H7)Zr(η5-C4PH2Me2)] (41) and [(η7-C7H7)Zr(η5-C4PPhHMe2)] (42) (Scheme 11).55 Remarkably, X-ray diffraction analysis revealed an important difference depending on the substitution pattern. While 42 remains monomeric in the solid state, dimer-formation by μ-η1:η5-bridging phospholyl ligands takes place for 40 and 41 (Fig. 17). These weak intermolecular metal⋯P interactions with Zr–P distances of 2.7567(4) and 2.8664(7) Å, respectively, originate from the adjacency of Lewis acidic metal and Lewis basic phosphorus atoms. In analogy to the aforementioned geometric adjustments upon coordination of additional ligands to [(η7-C7H7)Zr(η5-2,4-C7H11)] (11), a marked deviation from coplanarity is observed for the seven- and the five-membered rings of 40 (α = 34.9°) and 41 (α = 31.7°). For 42, the corresponding tilt (α = 13.7°) results from the asymmetric coordination of the phospholyl ligand, which is found in all three cases as evidenced by significantly shorter Zr–C bond distances compared to the Zr–P bond distances; the former range between 2.506(2) and 2.623(2) Å, while intramolecular Zr–P bond lengths of 2.722(1) to 2.783(1) Å are found. Similar observations have also been made for related [(η5-C4PR4)2ZrCl2] complexes.56
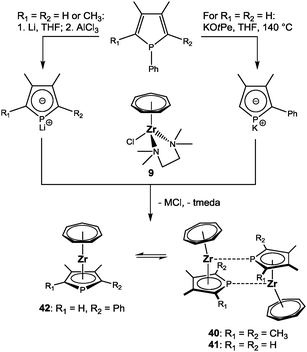 |
| | Scheme 11 Synthesis of phosphatrozircenes; the assignment of mono- or dimeric structures refers to the solid-state only. | |
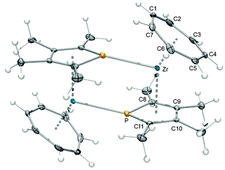 |
| | Fig. 17 ORTEP diagram of dimeric 40. | |
The identification of selected frontier orbitals for (monomeric) 40 by DFT calculations gave a picture similar to that derived for the half-open trozircenes (vide supra) with one important difference. In between the two sets of orbitals representing the largely ionic π interaction of the cycloheptatrienyl and phospholyl rings, a fairly stabilized molecular orbital resides as HOMO−4 (eigenvalue = −9.06 eV), which is essentially the lone pair at the phosphorus atom (Fig. 18). Similar conclusions were recently made for phosphaferrocene,57 but experimental results also showed that the energetically low-lying lone pair can still be used for coordination chemistry.52,53
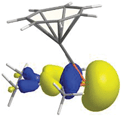 |
| | Fig. 18 Representation of HOMO−4 of 40. | |
In the present case, this was demonstrated by the coordination of 40–42 to the W(CO)5 complex fragment leading to a series of [{L}W(CO)5] complexes (43: L = 40; 44: L = 41; 45: L = 42).55 Additionally, the complex [{(η7-C7H7)Zr(η5-C5H4PPh2)}W(CO5] (46) was prepared for comparison. Fig. 19 shows the molecular structure of 43 as a representative example for these bimetallic complexes with a tungsten atom surrounded by five carbonyl ligands and one metallophosphane in an almost perfectly octahedral fashion. The shortening of the W–CO bond opposite to the W–P bond can be attributed to the trans-effect (Table 4). Moreover, the donor capability of [(η7-C7H7)Zr(η5-C5H4PPh2)] (23) and 40–42 is comparable to that of PMe3 as judged by IR spectroscopy, which suggests that these metallophosphanes could find applications as surrogates for traditional electron-rich phosphanes, for instance in catalytic reactions. A remarkable example of phosphatrozircene coordination is found in the Ni(0) complex 47 (Fig. 19), which was synthesized from [Ni(cod)2] (cod = 1,5-cyclooctadiene). The observation that 40 was able to replace even both chelating dienes further substantiates that their potential as ligands is worth investigating.55
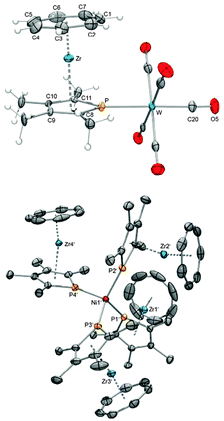 |
| | Fig. 19 ORTEP diagrams of 43 (top) and 47 (bottom). | |
Table 4 Selected bond lengths (Å) and stretching frequencies (cm−1) of the A1(2) IR band for the [{L}W(CO)5] complexes
| Complex |
L |
P–W |
W–COtrans |
W–COcis |
![[small nu, Greek, tilde]](https://www.rsc.org/images/entities/i_char_e0e1.gif)
|
|
43
|
40
|
2.470(2) |
2.000(6) |
2.020(6)–2.053(6) |
2074 |
|
44
|
41
|
2.473(1) |
2.008(6) |
2.037(6)–2.053(6) |
2076 |
|
45
|
42
|
— |
— |
— |
2077 |
|
46
|
23
|
2.539(1) |
1.987(3) |
2.032(3)–2.046(3) |
2069 |
It is of course also possible to have more than only one phosphorus atom in the ring. Such an example is displayed in Fig. 20; [(η7-C7H7)Zr(η5-C2P3tBu2)] (48) was synthesized according to Scheme 7 from the corresponding potassium salt and 9. It has to be noted that 48 is accompanied by about 10% of [(η7-C7H7)Zr(η5-C3P2tBu3)] (49) as judged by NMR spectroscopy, because K(C2P3tBu2) is contaminated with K(C3P2tBu3). Repeated crystallization from pentane, however, gave pure 48 with a significant decrease of the yield.31 Since the three Zr–P bond distances are in a narrow range, 2.7484(5)–2.7581(4) Å, the deviation from coplanarity (α = 7.29°) resembles rather that of [(η7-C7H7)Zr(η5-C5H3tBu2)] (27, α = 5.21°) than that of 42 (α = 13.7°).
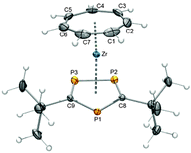 |
| | Fig. 20 ORTEP diagram of 48. | |
Boratatrozircenes: can we switch the hapticity?
Boratabenzene ligands caught our attention for several reasons. (1) They are monoanionic, 6π-electron ligands, which can be used as a replacement for the isoelectronic cyclopentadienyl ligand.58 (2) In contrast to the cyclic ligands used so far, these systems are six-membered rings. (3) Their bonding pattern resembles that of pentadienyl ligands in high oxidation state metal complexes.35
Building on the general procedure (Scheme 7), three boratatrozircenes of the type [(η7-C7H7)Zr(η6-C5H5B–R)] (50: R = H; 51: R = CH3; 52: R = CC-TMS) were prepared and characterized (Scheme 12).59 Structural data for 51 and 52 were obtained and helped to verify the coordination of the boratabenzene ligands in a distorted η6-fashion. Although bonding interaction of the boron atom with zirconium is indicated by no visual displacement out of the C8–C12 plane in each case, a closer inspection reveals a slippage of the metal away from boron, which is accompanied by progressively elongated Zr–C bond distances on going from the central (C10 in Fig. 21) to the terminal (C8/C12) carbon atoms. The longest bond is at 2.683(2) or 2.649(6) Å for 51 and 52, respectively, the Zr–B bond, but it is still significantly shorter than in related complexes such as [(η6-C5H5B–CH3)2ZrCl2] (2.845(5)/2.783(4) Å).60 Consequently, the slip-distortion found in the half-open trozircenes (vide supra) is recognized here.
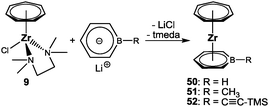 |
| | Scheme 12 Synthesis of boratatrozircenes. | |
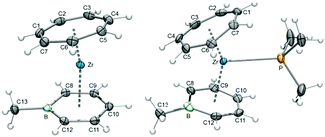 |
| | Fig. 21 ORTEP diagrams of 51 (left) and its PMe3 adduct 53 (right). | |
In the η6-coordination mode of a boratabenzene ligand, the Lewis acidity of boron is suppressed, because all orbitals are involved in bond formation. In the context of the stability of [(η7-C7H7)Zr(η5-Pdl)] species, an empty orbital at boron could be provided upon η6–η5 hapticity interconversion to an η5-pentadienyl-type coordination mode. We tried to promote such a rearrangement by adding either PMe3 or DMAP to 51 (Scheme 13). In agreement with NMR spectroscopic investigations and X-ray diffraction studies (Fig. 21), only Zr–L bond formation was observed, while the Zr–B bond remained intact. Even excess of PMe3 or pyridine did not result in B–L bond formation, leading to the conclusion that the formation of a B–P or B–N bond could not compensate for breaking the Zr–B bond.59
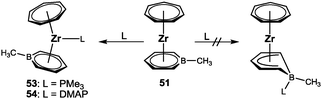 |
| | Scheme 13 Formation of the adducts 53 and 54. | |
Detour to a cycloheptatrienyl zirconium imidazolin-2-iminato complex with pogo stick geometry
So far, [(η7-C7H7)ZrCl(tmeda)] (9) has appeared to have no limitations or disadvantages; the reactions always turned out the way they were expected to. The initial problems were encountered when we targeted cycloheptatrienyl zirconium imidazolin-2-iminato complexes of the general formula [(η7-C7H7)Zr(ImRN)]. Although the reaction of [(η7-C7H7)ZrCl(tmeda)] (9) with Li(ImDippN) (ImDippN = 1,3-bis(2,6-diisopropylphenyl)imidazolin-2-iminato) gave with [(η7-C7H7)Zr(ImDippN)(tmeda)] (55) a well-defined product, which was also confirmed by X-ray diffraction analysis, it became clear that tmeda was only lost in the former reactions, because the electron-demand of the resulting complexes was satisfied.49 Our goal still remained to obtain a base-free cycloheptatrienyl zirconium imidazolin-2-iminato complex, because such a system would be in an isolobal relationship to Mountford's cyclooctatetraenyl titanium imido complexes (Scheme 14).61
![Isolobal relationship of [(η8-C8H8)Ti(NR)] and [(η7-C7H7)Zr(ImRN)]; the numbers represent formal charges.](/image/article/2013/CS/c2cs35321k/c2cs35321k-s14.gif) |
| | Scheme 14 Isolobal relationship of [(η8-C8H8)Ti(NR)] and [(η7-C7H7)Zr(ImRN)]; the numbers represent formal charges. | |
The successful approach was grounded on a novel reactivity pattern in the area of the bis(trimethylsilyl)allyl ligand.47,48 As discussed earlier, replacement of THF in [(η7-C7H7)Zr{C3H3(TMS)2}(thf)] (35) is possible and leads to stable adducts (vide supra).50 However, this was not the case when the imidazolin-2-imine ImDippNH was offered as a ligand. Here, the adduct 56 was formed only as an intermediate, followed by a presumably intramolecular deprotonation reaction affording the monoanionic imidazolin-2-iminato ligand and neutral 1,3-bis(trimethylsilyl)propene as by-products (Scheme 15). Accordingly, the desired complex [(η7-C7H7)Zr(ImDippN)] (57) is also stable in the absence of tmeda, but could not be synthesized directly from 9.49
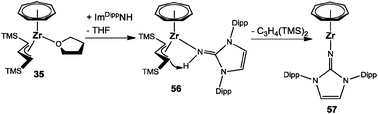 |
| | Scheme 15 Reaction sequence leading to 57. | |
The molecular structure of 57 exhibits a rare one-legged piano stool (“pogo stick”) geometry as opposed to [(η8-C8H8)Zr(NR)] complexes, where exclusive dimer formation was observed (Fig. 22).62 We suspect that the combination of bulky Dipp substituents and a close approach of the wide cycloheptatrienyl ligand (Zr–Chtcent = 1.661 Å) is crucial for the stability of the monomer in the present case. Remarkable about 57 is also the extremely short Zr–N bond distance of 1.997(2) Å (2.003(2) Å for the second independent molecule in the asymmetric unit), which can be attributed to the capability of the imidazolin-2-iminato ligand to act as a 2σ,4π-electron donor, resulting from the efficient stabilization of a positive charge within the imidazoline ring.63 Furthermore, as revealed by theoretical studies, the bonding situation in 57 resembles that of [(η8-C8H8)Ti(NR)] as well as of [(η7-C7H7)Zr(η5-C5H5)], which led to the conclusion that imidazolin-2-iminato ligands “can be regarded as monoanionic imido-type ligands or as monodentate analogues of the ubiquitous cyclopentadienyl ligand”.49
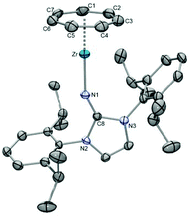 |
| | Fig. 22 ORTEP diagram of 57. | |
In the context of the relationship of imido and imidazolin-2-iminato ligands, experiments were performed to test whether 57 also features a typical imdo-type reactivity.64 The reaction with isocyanides resulted in terminal coordination and isolation of [(η7-C7H7)Zr(ImDippN)(CNR)] (58: R = o-Xy; 59 = tBu) rather than in the formation of single or multiple coupling products (Scheme 16); these adducts were even stable for prolonged time at higher temperatures. In contrast, the addition of 2,6-dimethylphenyl isocyanate (OCN-o-Xy) gave 60 with a four-membered metallacycle. The N,N′-ureato(1-) moiety arises from a regioselective [2+2] cycloaddition of the NC and the Zr–N bond and is not followed by a cycloreversion reaction as observed in some related cases involving imido ligands.62,64 The molecular structure (Fig. 23) indicates a stronger Zr–N4 than the Zr–N1 bond, because the corresponding Zr–N distance is significantly shorter, 2.2059(16) vs. 2.3177(15) Å. A comparable reactivity could not be observed with either SCN-o-Xy, CO2 or CS2, because only complex mixtures of products were formed.
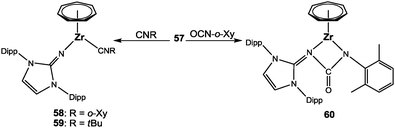 |
| | Scheme 16 Synthesis of 58–60. | |
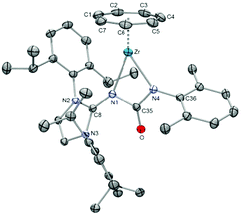 |
| | Fig. 23 ORTEP diagram of 60. | |
The reactivity of the Zr–N bond in 57 was also used to initiate the ring-opening polymerization (ROP) of ε-caprolactone (ε-CL). This catalytic process most likely occurs via the generally accepted coordination–insertion mechanism and involves the attack of the ImDippN ligand at the carbonyl carbon atom.65 The performance of 57 as catalyst is only moderate under the chosen experimental conditions (0.5 or 1 mol% catalyst, 30 or 60 °C, THF as solvent, 4 h), because the polydispersity indices range between 1.43 and 1.81, indicating no or at least delayed participation of some catalyst molecules in the catalytic process. Finally, NMR studies based on low cat:ε-CL ratios showed the presence of –CH2OH end groups, which is in agreement with the formation of linear rather than cyclic polymers.
Approaching cycloheptatrienyl zirconium multi-decker complexes
Throughout the aforementioned examples, the seven-membered ring always acted as an innocent spectator ligand that certainly plays an important, if not crucial role in the stabilization of these (sandwich) complexes, but it has not disclosed any unprecedented features on its own. One perspective, which would certainly add something new to the area of organotransition metal chemistry, is the use of the cycloheptatrienyl ligand for the construction of multi-decker complexes. In view of only very few examples with the C7H7 ligand bridging two metals, this appears to be a challenging task. In fact, antifacially coordinated Cht ligands are only found in a series of uranium and neodymium complexes, where an exceptional μ-η7:η7-coordination mode is present.66 In stark contrast, exclusively synfacial coordination has been known for transition metal complexes to date.67 For the observation of the elusive trans coordination, three requirements have to be fulfilled. (1) The second metal must have a high electron demand, which is synonymous with a high Lewis acidity. (2) No other ligands are available, which preferentially bind to the second metal. (3) One side of the ring is blocked.
Access to the first transition metal complex with an antifacial coordination of the C7H7 ligand was achieved by an unexpected pathway. Inspiration came originally from Siemeling's iron pogo stick complex, [(η5-C5Me5)Fe{N(TMS)2}], which bears a bulky bis(trimethylsilyl)amido ligand.68 In analogy, a reaction of [(η7-C7H7)ZrCl(tmeda)] (9) and Na{N(TMS)2} in THF was performed, yielding the structurally characterized complex [(η7-C7H7)Zr{N(TMS)2}(thf)] (61, Scheme 17).69 This is not the desired pogo stick, but according to NMR studies, THF coordination is rather weak. Subsequent sublimation under dynamic vacuum resulted in the loss of THF and the formation of [(η7-C7H7)Zr{N(TMS)2}]n (62). The insolubility of 62 in aromatic or aliphatic solvents indicates the presence of a multinuclear or polymeric arrangement instead of the expected one-legged piano stool geometry. Confirmation was provided by an X-ray diffraction study, which established the unusual molecular structure of 62 as a polymer of slipped multi-decker complexes (Fig. 24). They originate from trans coordinated seven-membered rings in an unprecedented μ-η7:η2-coordination mode. While the Zr–C bond lengths to the η7-bound metal are at 2.394(4)–2.369(4) Å in the expected range, comparatively long bonds of 2.642(4) and 2.645(4) Å are developed to the second metal. The unique structural motif can be rationalized by a combination of electron deficiency of the metal upon THF loss and the steric prevention of dimer formation via the electron lone pair on nitrogen.69
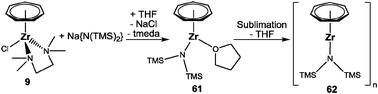 |
| | Scheme 17 Synthesis of 61 and 62. | |
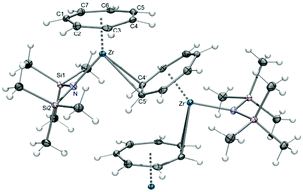 |
| | Fig. 24 ORTEP diagram of polymeric 62. | |
Complex 62 can be utilized as starting material (Scheme 18): in simple NMR tube reactions, addition of THF or one equivalent of PMe3 to 62 gave 61 and [(η7-C7H7)Zr{N(TMS)2}(PMe3)] (63), respectively. Of certainly higher interest are the reactions, where the basic amido ligand comes into play. Thus, reaction of triphenylsilanol (Ph3SiOH) with 62 afforded [(η7-C7H7)Zr(OSiPh3)]2 (64), while the reaction with 2,4,6-tri(tert-butyl)phenol gave [(η7-C7H7)Zr(O-2,4,6-tBu3C6H2)] (65). The dimeric structure of 64 with μ-OSiPh3 ligands was unambiguously elucidated by X-ray diffraction analysis,69 but the nuclearity of 65 still remains unclear. Both examples strongly suggest that 62 could become another versatile starting material for the introduction of monoanionic ligands into the cycloheptatrienyl zirconium coordination sphere by acid–base reactions. In contrast to the use of 9, this approach avoids the presence of tmeda and, as exemplified for the synthesis of 65, even coordinating solvents. Whenever electron deficient and/or coordinatively unsaturated complexes are aimed for, this can become of essential importance.
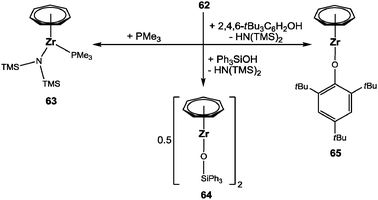 |
| | Scheme 18 Reactivity of 62. | |
Conclusions
Although [(η7-C7H7)ZrCl(tmeda)] is already known for more than two decades,26 its potential to serve as an efficient starting material remained unrecognized until our initial report on the synthesis and characterization of half-open trozircenes.27 This new approach is undoubtedly a great advancement in the still fairly unexplored chemistry of the seven-membered C7H7 ring,12,17 because a plethora of (sandwich) complexes involving the Cht ligand can now be addressed, which are inaccessible by previous methods. While this has already led to new aspects, it still leaves numerous unexploited areas open for future investigations. Among them, some appear particularly worthwhile.
First of all, the library of [(η7-C7H7)Zr(η5-Cp)] complexes is currently extended in collaboration with Sitzmann. The focus is on unknown cyclopentadienyl ligands bearing more than one type of aliphatic substituent and the assessment of their steric bulk based on cone angle measurements. In view of a recent report,70 the corresponding trozircenes appear to be ideal precursors for atomic layer deposition, because the steric bulk should increase the thermal and air stability, but still provide reasonable low sublimation temperatures.
As already mentioned above, the tmeda-free complex [(η7-C7H7)Zr{N(TMS)2}] (62) should allow for the incorporation of several ligands via acid–base reactions. Additionally, mild deprotonation of 62 with an acid in a suitable aromatic solvent could yield cationic, 16-electron cycloheptatrienyl zirconium arene complexes of the type [(η7-C7H7)Zr(η6-arene)]+; they can also be targeted by chloride abstraction in 9 with a silver salt (Scheme 19). The obvious subsequent reaction with such cationic species in hand would be the reduction to neutral, paramagnetic Zr(+III) cycloheptatrienyl complexes, which should offer unusual physical properties and interesting reactivity. As an alternative to the synthesis of “real” trivalent zirconium complexes, non-innocent ligands such as 2,2′-bypyridine could be employed, which might allow for the observation of reactivity associated with low-valent species. Certainly, the use of redox-active ligands would complicate the formal oxidation state assignment in Cht complexes even more, but this would entail some interesting physical and theoretical studies.
![Two possible approaches to [(η7-C7H7)Zr(η6-C6H6)]+ complexes.](/image/article/2013/CS/c2cs35321k/c2cs35321k-s19.gif) |
| | Scheme 19 Two possible approaches to [(η7-C7H7)Zr(η6-C6H6)]+ complexes. | |
Most rewarding, but probably also most challenging, is a continuation and detailed exploration of zirconium multi-decker complexes with bridging C7H7 ligands. Such examples still remain extremely rare, because reliable approaches have not yet been developed, but especially the remarkable μ-η7:η2 coordination mode found in complex 62 (Fig. 24) tempts to envisage related systems with μ-η7:η7-C7H7 ligands. In this context, it is worth trying to react the bis(cycloheptatrienyl)uranium complex, [(η7-C7H7)2U]−,28 with 9, because a salt metathesis reaction could result in [(η7-C7H7)U(μ-η7:η7-C7H7)Zr(η7-C7H7)] (Scheme 20).
As a final note, [(η7-C7H7)ZrCl(tmeda)] is clearly a potent candidate for more intriguing discoveries along these lines and it will thereby help to keep the renewed interest in group 4 transition metal sandwich complexes fresh.11
Acknowledgements
Many of these studies were jointly performed with a number of national and international collaborators. We are grateful to Richard D. Ernst (University of Utah), who played a crucial role in launching this project, Marc D. Walter (Technische Universität Braunschweig), Helmut Sitzmann (Technische Universität Kaiserslautern), Yaofeng Chen (State Key Laboratory of Organometallic Chemistry, Shanghai), Manfred Scheer (Universität Regensburg), Zuowei Xie (Chinese University of Hong Kong) and Moris S. Eisen (Israel Institute of Technology). Their dedication to this project as well as that of additional contributors, whose names are listed throughout the references, is highly appreciated. This work was generously supported by the German Academic Exchange Service (DAAD), the Bayer Science & Education Foundation, the Fonds der Chemischen Industrie through fellowships awarded to AG and, in part, by the Deutsche Forschungsgemeinschaft (DFG).
Notes and references
- H. Werner, Angew. Chem., Int. Ed., 2012, 51, 6052 CrossRef CAS.
- A. K. Fischer and G. Wilkinson, J. Inorg. Nucl. Chem., 1956, 2, 149 CrossRef CAS.
- H. H. Brintzinger and J. E. Bercaw, J. Am. Chem. Soc., 1970, 92, 6182 CrossRef CAS.
- S. I. Troyanov, H. Antropiusová and K. Mach, J. Organomet. Chem., 1992, 427, 49 CrossRef CAS ; and references cited therein.
- P. B. Hitchcock, F. M. Kerton and G. A. Lawless, J. Am. Chem. Soc., 1998, 120, 10264 CrossRef CAS.
- J.-Z. Liu and R. D. Ernst, J. Am. Chem. Soc., 1982, 104, 3737 CrossRef CAS.
- R. W. Gedridge, A. M. Arif and R. D. Ernst, J. Organomet. Chem., 1995, 501, 95 CrossRef CAS.
- H. O. van Oven and H. J. de Liefde Meijer, J. Organomet. Chem., 1970, 23, 159 CrossRef CAS ; and references cited therein.
- P. A. Kroon and R. B. Helmholdt, J. Organomet. Chem., 1970, 25, 451 CrossRef CAS.
- J. D. Zeinstra and J. L. de Boer, J. Organomet. Chem., 1973, 54, 207 CrossRef CAS.
- P. J. Chirik, Organometallics, 2010, 29, 1500 CrossRef CAS.
- M. L. H. Green and D. K. P. Ng, Chem. Rev., 1995, 95, 439 CrossRef CAS ; and references cited therein.
- H. J. Dauben Jr. and L. R. Honnen, J. Am. Chem. Soc., 1958, 80, 5570 CrossRef.
- H. Braunschweig, M. Friedrich, K. Radacki and J. Wolf, Organometallics, 2012, 31, 3027 CrossRef CAS ; and references cited therein.
- Trovacene chemistry part 18: C. Elschenbroich, F. Lu, O. Burghaus, K. Harms and J. Pebler, Z. Anorg. Allg. Chem., 2011, 637, 1750 CrossRef CAS ; and references cited therein.
- W. Noh and G. S. Girolami, Inorg. Chem., 2008, 47, 10682 CrossRef CAS ; and references cited therein.
- M. Tamm, Chem. Commun., 2008, 3089 RSC ; and references cited therein.
- P. Nguyen, P. Gómez-Elipe and I. Manners, Chem. Rev., 1999, 99, 1515 CrossRef CAS ; and references cited therein.
- H. Braunschweig and T. Kupfer, Acc. Chem. Res., 2010, 43, 455 CrossRef CAS ; and references cited therein.
- A. C. Tagne Kuate, C. G. Daniliuc, P. G. Jones and M. Tamm, Eur. J. Inorg. Chem., 2012, 1727 CrossRef CAS ; and references cited therein.
- S. K. Mohapatra, S. Büschel, C. G. Daniliuc, P. G. Jones and M. Tamm, J. Am. Chem. Soc., 2009, 131, 17014 CrossRef CAS.
- A. C. Tagne Kuate, S. K. Mohapatra, C. G. Daniliuc, P. G. Jones and M. Tamm, J. Organomet. Chem., 2012, 696, 4281 CrossRef CAS.
- S. Büschel, A.-K. Jungton, T. Bannenberg, S. Randoll, C. G. Hrib, P. G. Jones and M. Tamm, Chem.–Eur. J., 2009, 15, 2176 CrossRef.
- S. Büschel, T. Bannenberg, C. G. Hrib, A. Glöckner, P. G. Jones and M. Tamm, J. Organomet. Chem., 2009, 694, 1244 CrossRef.
- The trivial name trozircene was derived from tropylium, zirconium and cyclopentadienyl; see for instance ref. 19. A prefix such as “half-open”, “phospha” or “borata” is applied to indicate the presence of a different η5-ligand than the conventional cyclopentadienyl ligand.
- G. M. Diamond, M. L. H. Green, P. Mountford and N. M. Walker, J. Chem. Soc., Dalton Trans., 1992, 2259 RSC ; and references cited therein.
- A. Glöckner, T. Bannenberg, M. Tamm, A. M. Arif and R. D. Ernst, Organometallics, 2009, 28, 5866 CrossRef.
- T. Arliguie, M. Lance, M. Nierlich, J. Vigner and M. Ephritikhine, J. Chem. Soc., Chem. Commun., 1995, 183 RSC.
- H. Wang, Y. Xie, R. B. King and H. F. Schaefer, Eur. J. Inorg. Chem., 2008, 3698 CrossRef CAS.
- J. T. Miller and C. W. Dekock, J. Organomet. Chem., 1981, 216, 39 CrossRef CAS ; and references cited therein.
- Unpublished results.
- H. W. Bosch, H.-U. Hund, D. Nietlispach and A. Salzer, Organometallics, 1992, 11, 2087 CrossRef CAS.
- A. Glöckner, Ò. Àrias, T. Bannenberg, C. G. Daniliuc, P. G. Jones and M. Tamm, Dalton Trans., 2011, 40, 11511 RSC.
- R. D. Ernst, Chem. Rev., 1988, 88, 1255 CrossRef CAS ; and references cited therein.
- L. Stahl and R. D. Ernst, Adv. Organomet. Chem., 2007, 55, 137 CrossRef ; and references cited therein.
- J. D. Zeinstra and W. C. Nieuwpoort, Inorg. Chim. Acta, 1978, 30, 103–117 CrossRef CAS ; and references cited therein.
- J. C. Green, N. Kaltsoyannis, K. H. Sze and M. MacDonald, J. Am. Chem. Soc., 1994, 116, 1994 CrossRef CAS.
- G. Menconi and N. Kaltsoyannis, Organometallics, 2005, 24, 1189 CrossRef CAS.
- K. A. Lyssenko, M. Y. Antipin and S. Y. Ketkov, Russ. Chem. Bull., 2001, 50, 130 CrossRef CAS.
- R. J. Baker, T. Bannenberg, A. Kunst, S. Randoll and M. Tamm, Inorg. Chim. Acta, 2006, 359, 4797 CrossRef CAS.
- A. Glöckner, A. M. Arif, R. D. Ernst, T. Bannenberg, C. G. Daniliuc, P. G. Jones and M. Tamm, Inorg. Chim. Acta, 2010, 364, 23 CrossRef.
- A. Glöckner, M. Tamm, A. M. Arif and R. D. Ernst, Organometallics, 2009, 28, 7041 CrossRef.
- A. Glöckner, H. Bauer, M. Maekawa, T. Bannenberg, C. G. Daniliuc, P. G. Jones, Y. Sun, H. Sitzmann, M. Tamm and M. D. Walter, Dalton Trans., 2012, 41, 6614 RSC ; and references cited therein.
- C. A. Tolman, Chem. Rev., 1977, 77, 313 CrossRef CAS.
- P. C. Möhring and N. J. Coville, Coord. Chem. Rev., 2006, 250, 18 CrossRef ; and references cited therein.
- U. Rosenthal, V. V. Burlakov, M. A. Bach and T. Beweries, Chem. Soc. Rev., 2007, 36, 719 RSC ; and references cited therein.
- S. C. Chmely and T. P. Hanusa, Eur. J. Inorg. Chem., 2010, 1321 CrossRef CAS ; and references cited therein.
- S. A. Solomon and R. A. Layfield, Dalton Trans., 2010, 39, 2469 RSC ; and references cited therein.
- A. Glöckner, T. Bannenberg, C. G. Daniliuc, P. G. Jones and M. Tamm, Inorg. Chem., 2012, 51, 4368 CrossRef.
-
A. Glöckner, S. Kronig, T. Bannenberg, C. G. Daniliuc, P. G. Jones and M. Tamm, submitted.
- S. Büschel, C. G. Daniliuc, P. G. Jones and M. Tamm, Organometallics, 2010, 29, 671 CrossRef.
-
K. B. Dillon, F. Mathey and J. F. Nixon, Phosphorus: The Carbon Copy, John Wiley & Sons Ltd., Sussex, 1998; and references cited therein Search PubMed.
- P. Le Floch, Coord. Chem. Rev., 2006, 250, 627 CrossRef CAS ; and references cited therein.
- Y. J. Ahn, R. J. Rubio, T. K. Hollis, F. S. Tham and B. Donnadieu, Organometallics, 2006, 25, 1079 CrossRef CAS ; and references cited therein.
- A. Glöckner, T. Bannenberg, S. Büschel, C. G. Daniliuc, P. G. Jones and M. Tamm, Chem.–Eur. J., 2011, 17, 6118 CrossRef.
- S. Bellemin-Laponnaz, M. M.-C. Lo, T. H. Peterson, J. M. Allen and G. C. Fu, Organometallics, 2001, 20, 3453 CrossRef CAS ; and references cited therein.
- N. M. Kostić and R. F. Fenske, Organometallics, 1983, 2, 1008 CrossRef ; and references cited therein.
- G. C. Fu, Adv. Organomet. Chem., 2001, 47, 101 CrossRef CAS ; and references cited therein.
- A. Glöckner, P. Cui, Y. Chen, C. G. Daniliuc, P. G. Jones and M. Tamm, New J. Chem., 2012, 36, 1392 RSC.
- G. E. Herberich, U. Englert and A. Schmitz, Organometallics, 1997, 16, 3751 CrossRef CAS.
- S. C. Dunn, N. Hazari, N. M. Jones, A. G. Moody, A. J. Blake, A. R. Cowley, J. C. Green and P. Mountford, Chem.–Eur. J., 2005, 11, 2111 CrossRef CAS ; and references cited therein.
- S. C. Dunn, N. Hazari, A. R. Cowley, J. C. Green and P. Mountford, Organometallics, 2006, 25, 1755 CrossRef CAS ; and references cited therein.
- A. G. Trambitas, T. K. Panda and M. Tamm, Z. Anorg. Allg. Chem., 2010, 636, 2156 CrossRef CAS ; and references cited therein.
- N. Hazari and P. Mountford, Acc. Chem. Res., 2005, 38, 839 CrossRef CAS ; and references cited therein.
- A. Arbaoui and C. Redshaw, Polym. Chem., 2010, 1, 801 RSC ; and references cited therein.
- T. Arliguie, M. Lance, M. Nierlich and M. Ephritikhine, J. Chem. Soc., Dalton Trans., 1997, 2501 RSC ; and references cited therein.
- V. Beck and D. O'Hare, J. Organomet. Chem., 2004, 689, 3920 CrossRef CAS ; and references cited therein.
- U. Siemeling, U. Vorfeld, B. Neumann and H.-G. Stammler, Organometallics, 1998, 17, 483 CrossRef CAS.
- A. Glöckner, C. G. Daniliuc, M. Freytag, P. G. Jones and M. Tamm, Chem. Commun., 2012, 48, 6598 RSC.
- J. Niinistö, T. Hatanpää, M. Kariniemi, M. Mäntymäki, L. Costelle, K. Mizohata, K. Kukli, M. Ritala and M. Leskelä, Chem. Mater., 2012, 24, 2002 CrossRef.
|
| This journal is © The Royal Society of Chemistry 2013 |
Click here to see how this site uses Cookies. View our privacy policy here.