Selective aerobic oxidation of activated alkanes with MOFs and their use for epoxidation of olefins with oxygen in a tandem reaction
I.
Luz
,
A.
León
,
M.
Boronat
,
F. X.
Llabrés i Xamena
* and
A.
Corma
*
Instituto de Tecnología Química UPV-CSIC, Universidad Politécnica de Valencia, Consejo Superior de Investigaciones Científicas, Avda. de los Naranjos s/n, 46022 Valencia, Spain. E-mail: fllabres@itq.upv.es; acorma@itq.upv.es; Fax: +34 963877809
First published on 30th August 2012
Abstract
MOFs with Cu2+ centers linked to four nitrogen atoms from azaheterocyclic compounds, i.e., pyrimidine [Cu(2-pymo)2] and imidazole [Cu(im)2], are active catalysts for aerobic oxidation of activated alkanes, such as tetralin, cumene and ethylbenzene. Differences in activity among the two MOFs appear to be related to differences in their ability to decompose the hydroperoxide and to coordinate to the resulting radical ˙OH species. Copper ions in [Cu(im)2] can coordinate by expanding their coordination sphere from 4 to 5 in a reversible way, while in the case of [Cu(2-pymo)2] it results in a displacement of one of the pyrimidine ligands. The MOFs can be used in combination with a silylated Ti-MCM-41 to catalyze the epoxidation of olefins with oxygen by means of a tandem reaction in which the MOF produces cumene hydroperoxide, which is used by Ti-MCM-41 to epoxidize the olefin.
1. Introduction
There is much interest in the design of hybrid materials for selective and multi-step catalytic processes.1 In this field, Metal–Organic Frameworks (MOFs) can play an important role due to the possibility to control and fine tune the crystalline structure, porosity, chemical environment and functionalities of the material. Compared to purely inorganic porous materials, MOFs offer in general higher flexibility for the design of the catalytic center,2–4 which makes them interesting materials for heterogeneous catalysis, provided that mild reaction conditions are required. It is possible to design MOF-based heterogeneous catalysts in which the active sites are located either at the metallic sites or at the organic linkers, or even associated to catalytic species bonded or entrapped inside the pore system of the MOF.5,6 Moreover, this large flexibility can readily lead to the design of multi-functional MOF catalysts7,8 by combining two or more types of active centers in the same material, such as metal–metal, metal–acid, metal–base or acid–base functionalities.We have recently reported9 that a copper-containing MOF, [Cu(2-pymo)2] (2-pymo = 2-oxypyrimidinolate10), could be of interest as heterogeneous catalyst for the liquid-phase oxidation of tetralin (T–H) using air as oxidant. Under the reaction conditions studied, a relatively high T–H conversion (ca. 52%) was attained without affecting the crystalline structure of the solid, which allowed to reuse the MOF without important loss of activity.9 However, this Cu-MOF showed some limitations. The most important was the high level of tetralinhydroperoxide (T–OOH) accumulated at the beginning of the reaction (up to 24 mol% yield) and a low selectivity to the ketone, α-tetralone (T![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O). We found that this inconvenience can be avoided by physically mixing [Cu(2-pymo)2] with another MOF, i.e., cobalt benzimidazole ZIF-9,11 that decomposes the hydroperoxides.
O). We found that this inconvenience can be avoided by physically mixing [Cu(2-pymo)2] with another MOF, i.e., cobalt benzimidazole ZIF-9,11 that decomposes the hydroperoxides.
In the present work, we have extended our studies on the aerobic oxidation of tetralin to a copper-containing MOF, [Cu(im)2] (im = imidazolate12), which has a chemical composition and crystalline structure related to that of [Cu(2-pymo)2]. Recently, we have successfully used both materials for coupling reactions; viz. 1,3-dipolar cycloadditions of azides to alkynes,13 and three-component coupling of aldehydes, amines and terminal alkynes.14 In the present work, we have found that [Cu(im)2] has a better catalytic performance as compared to [Cu(2-pymo)2] for tetralin oxidation, in terms of activity, maximum T–H conversion and selectivity to the ketone. At the same time, [Cu(im)2] produces less T–OOH accumulation in the reaction medium than [Cu(2-pymo)2] under the same conditions, avoiding the use of a second MOF to deal with the hydroperoxides. Therefore, [Cu(im)2] largely overcomes the limitations of the previously reported [Cu(2-pymo)2] as a catalyst for aerobic tetralin oxidation, without any need to prepare physical mixtures with a second catalyst. Besides tetralin, we have also studied the aerobic oxidation of other substrates; viz., cumene and ethylbenzene, which let us make an evaluation of the potential of both [Cu(2-pymo)2] and [Cu(im)2] as catalysts for the oxidation of benzilic paraffins. Since both MOFs were active for such oxidations we have attempted a cascade reaction. Then, we will also show that, when [Cu(2-pymo)2] is combined with a second solid catalyst (silylated Ti-MCM-41), the in situ generated hydroperoxide can be used to transform an olefin into the corresponding epoxide with good selectivity and acceptable yield. A comparison will be made between the performance of this mixed catalytic system under different working setups (i.e., one-pot and two-pot setups).
2. Materials and methods
2.1. Preparation of the Cu-MOFs
The Cu-MOFs were prepared according to the corresponding procedures reported in the original references for [Cu(2-pymo)2]10 and [Cu(im)2].12 X-ray diffraction (Phillips X'Pert, Cu Kα radiation) was used to confirm the expected crystalline structure of the materials.2.2. Silylated Ti-MCM-41
Silylated Ti-MCM-41 was prepared following the original procedure.15 First, Ti-MCM-41 was obtained from a gel having the following molar composition: SiO2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :0.015 Ti(OEt)4:0.26 CTABr:0.26 TMAOH:24.3 H2O where CTABr is cetyltrimethylammonium bromide and TMAOH is tetramethylammonium hydroxide. The silica source, Aerosil-200, was obtained from Degussa. The crystallization was performed at 100 °C for 48 h in Teflon lined stainless steel autoclaves. The occluded surfactant was completely removed following a two step extraction procedure. Ti-MCM-41 was then fully silylated with hexamethyldisilazane (HMDS) as the silylating agent. The silylation was carried out at 120 °C with a solution of HMDS in toluene under inert atmosphere. The MCM-41 structure was preserved after silylation and the surface area of the silylated sample was close to 1000 m2 g−1.
:0.015 Ti(OEt)4:0.26 CTABr:0.26 TMAOH:24.3 H2O where CTABr is cetyltrimethylammonium bromide and TMAOH is tetramethylammonium hydroxide. The silica source, Aerosil-200, was obtained from Degussa. The crystallization was performed at 100 °C for 48 h in Teflon lined stainless steel autoclaves. The occluded surfactant was completely removed following a two step extraction procedure. Ti-MCM-41 was then fully silylated with hexamethyldisilazane (HMDS) as the silylating agent. The silylation was carried out at 120 °C with a solution of HMDS in toluene under inert atmosphere. The MCM-41 structure was preserved after silylation and the surface area of the silylated sample was close to 1000 m2 g−1.
2.3. Computational details
Calculations were carried out by means of the Gaussian03 program package16 using the density functional B3PW91 method17,18 and the standard 6-311G(d,p) basis set.19 The Cu active sites in [Cu(im)2] and [Cu(2-pymo)2] MOFs were modeled by means of two clusters of atoms consisting of a central Cu2+ cation surrounded by either four imidazole or four 2-hydroxypyrimidine molecules. Each of the four organic ligands in the resulting models was saturated with a proton placed in the direction of the next Cu2+ cation in the real MOF material, so that each model bears a net positive charge of +2, and the geometry of the two cluster models was fully optimized without any restriction. For the complexes formed by interaction of the Cu2+ sites with CM–OOH, with CM–O˙ radical and with a ˙OH radical, two types of geometry optimizations were performed: (i) a restricted optimization, in which the positions of the four terminal hydrogen atoms saturating the organic ligands were kept fixed to model the partial flexibility of the MOF material, and (ii) a full optimization in which the positions of all atoms in the system were allowed to relax without any restriction. Calculations were performed with different spin states of the central copper atom for all models, and the singlet state was found the most stable in all cases. Finally, thermal corrected Gibbs free energies at 298 K were obtained from vibrational frequency calculations at the same B3PW91/6-311G(d,p) level of theory.2.4. Catalytic reactions
3. Results and discussion
3.1. Liquid phase oxidation of tetralin
Oxidation of tetralin to the corresponding ketone, α-tetralone, is interesting for the preparation of a number of chemicals (insecticides, agrochemicals and drugs) and for the production of oxygenated diesel molecules.21 This reaction can be performed using air as the oxidant and in the presence of a suitable oxidation catalyst, yielding a mixture of tetralinhydroperoxide (T–OOH), α-tetralol (T–OH) and α-tetralone (TO), as depicted in Scheme 1:
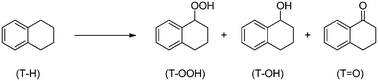 | ||
| Scheme 1 Tetralin (T–H) oxidation reaction, leading to α-tetralinhydroperoxide (T–OOH), α-tetralol (T–OH) and α-tetralone (TO). | ||
As we have shown in a previous work,9 the copper-containing MOF [Cu(2-pymo)2] is an active and reusable catalyst for the liquid phase oxidation of T–H using air as the oxidant. A maximum T–H conversion of about 52% was achieved after 48 h of reaction at 90 °C, with a T–H/metal molar ratio of 2000. Conversely, when the reaction was carried out in the presence of [Cu(im)2], keeping all the other reaction conditions unchanged, the maximum amount of T–H converted increased up to 68%, and this value was reached after only 22 h of reaction. A comparison of the two catalysts for the oxidation of T–H can be seen in Fig. 1 and Table 1. [Cu(im)2] also showed a higher activity than [Cu(2-pymo)2], with turnover frequencies (TOF) calculated from the initial reaction rates from Fig. 1 being 19000 and 4000 h−1 for [Cu(im)2] and [Cu(2-pymo)2], respectively. Thus, for instance, after 8 h of reaction, the conversion of T–H over [Cu(2-pymo)2] was only 18%, while the conversion over [Cu(im)2] already reached 55%. Blank experiments performed in the absence of any MOF catalyst (i.e., autothermal oxidation) gave very poor conversions, attaining a maximum T–H conversion of 1.3 mol% after 24 h (see Table 1), T–OOH being the only product formed.
| Tetralin (T–H) | Conv. 8 h | Conv. max | TO/T–OH |
[T–OOH]max |
|---|---|---|---|---|
| T–H: tetralin; T–OOH: tetralinhydroperoxide; TO: α-tetralone; T–OH: α-tetralol; CM: cumene; CM–OOH: cumene hydroperoxide (2-phenyl-2-propylhydroperoxide); CM–OH: cumene alcohol (2-phenyl-2-propanol); EB: ethylbenzene; AP: acetophenone. |
||||
| [Cu(im)2] | 55% | 68% (22 h) | 3.5 | 9% (1 h) |
| [Cu(2-pymo)2] | 18% | 52% (48 h) | 2.7 | 24% (18 h) |
| Blank: | 4 h: 0.2% T–OOH; 24 h: 1.3% T–OOH | |||
| Cumene (CM) | Conv. Max | Select. CM–OH | [CM–OOH]max |
|---|---|---|---|
| [Cu(im)2] | 99% (23 h) | 74% | 2 (1.5 h) |
| [Cu(2-pymo)2] | 87% (23 h) | 64% | 24 (8 h) |
| Blank: | 4 h: 0.5% CM–OOH; 24 h: 21% CM–OOH + 3% AP | ||
| Ethylbenzene (EB) | Conv. 5 h | Conv. max | Select. AP |
|---|---|---|---|
| [Cu(im)2] | 23% | 29% (40 h) | 86% |
| [Cu(2-pymo)2] | 6% | 25% (40 h) | 87% |
| Blank: | 4 h: No conversion; 24 h: 2% EB–OH + 0.5% EB–OOH | ||
![Conversion of T–H over [Cu(im)2] and [Cu(2-pymo)2] (part a). Time conversion of T–H and time evolution of products over [Cu(im)2] and [Cu(2-pymo)2] is also shown in parts (b) and (c), respectively.](/image/article/2013/CY/c2cy20449e/c2cy20449e-f1.gif) | ||
| Fig. 1 Conversion of T–H over [Cu(im)2] and [Cu(2-pymo)2] (part a). Time conversion of T–H and time evolution of products over [Cu(im)2] and [Cu(2-pymo)2] is also shown in parts (b) and (c), respectively. | ||
Another important parameter of the oxidation reaction is the final selectivity to the ketone, which is usually expressed as the molar ratio of α-tetralone to α-tetralol, TO/T–OH. As it can be seen in Table 1, also from this point of view the performance of [Cu(im)2] is better than [Cu(2-pymo)2]: the values of TO/T–OH calculated at maximum conversion were 3.5 and 2.7 for [Cu(im)2] and [Cu)2-pymo)2], respectively. Finally, as we mentioned above, the use of [Cu(2-pymo)2] as catalyst for T–H oxidation showed an undesired accumulation of hydroperoxides in the reaction medium, which reaches up to 24% of the initial amount of T–H after 18 h. The presence of large quantities of the hydroperoxide during the reaction can be a safety hazard, so must be avoided. On the contrary, when [Cu(im)2] was used as catalyst, the concentration of T–OOH was kept much lower throughout the reaction, reaching a maximum concentration of only 9 mol% at short reaction time (1 h) and dropping below 1–2 mol% shortly afterwards (see Fig. 1b).
In summary, as we have shown in Fig. 1 and Table 1, the use of [Cu(im)2] as catalyst for the liquid phase aerobic oxidation of T–H presents a number of benefits with respect to [Cu(2-pymo)2], which makes it a very interesting material for this reaction. We also observed that both catalysts were stable under the experimental conditions used, since the materials recovered after the catalytic reaction showed XRD patterns practically undistinguishable from those of the fresh materials. The occurrence of copper leaching from the framework was also ruled out by a hot filtration test and by chemical analysis of the filtrate after removing the solid catalyst at the end of the reaction. Stability and leaching tests were already reported and discussed in detail for [Cu(2-pymo)2] in our previous work,9 so they are not reproduced again here, the results obtained with [Cu(im)2] being completely analogous.
Encouraged by the good results obtained with [Cu(im)2] for T–H oxidation, we extended our studies to other substrates: cumene and ethylbenzene. We wanted on the one hand to determine the general applicability of these copper-containing MOFs as catalysts for paraffin oxidation. And if the result were positive, we thought of designing a multistep catalytic process in which the cumene hydroperoxide formed could further react, as oxidizing agent, for the synthesis of epoxides from olefins.
3.2. Liquid phase oxidation of cumene
Oxidation of cumene (CM) with air yields cumene hydroperoxide (CM–OOH) in the first place, which is then converted into cumene alcohol (CM–OH), usually accompanied by other byproducts (mainly acetophenone, AP) coming from non-selective decomposition side reactions.22Fig. 2 and Table 1 show a comparison of the results obtained for the liquid phase aerobic oxidation of cumene (80 °C, 0.5 mol% Cu) using either [Cu(2-pymo)2] or [Cu(im)2] as catalyst.![Conversion of CM over [Cu(im)2] and [Cu(2-pymo)2] (part a). Time conversion of CM and time evolution of products over [Cu(im)2] and [Cu(2-pymo)2] is also shown in parts (b) and (c), respectively.](/image/article/2013/CY/c2cy20449e/c2cy20449e-f2.gif) | ||
| Fig. 2 Conversion of CM over [Cu(im)2] and [Cu(2-pymo)2] (part a). Time conversion of CM and time evolution of products over [Cu(im)2] and [Cu(2-pymo)2] is also shown in parts (b) and (c), respectively. | ||
The results obtained for cumene oxidation are qualitatively very similar to those obtained for the oxidation of T–H, which demonstrates that both Cu-MOFs are able to catalyze this reaction. As occurred before for T–H, the imidazolate material [Cu(im)2] also shows a better oxidation performance as compared to [Cu(2-pymo)2], although the differences observed between the two catalysts are less pronounced than in the oxidation of T–H. Complete CM conversion was achieved after 23 h over [Cu(im)2], while some lower conversion (i.e., 87%) was obtained with [Cu(2-pymo)2]. The selectivity towards CM–OH was also higher in the case of [Cu(im)2] (74%, versus 64% obtained for [Cu(2-pymo)2]). In both cases, at the end of the reaction the sole product observed besides CM–OH was acetophenone, which accounted for the rest of CM converted (25% and 23%, respectively). It can also be observed in Fig. 2 that the amount of accumulated CM–OOH is higher (and lasts longer) in the case of [Cu(2-pymo)2] (up to 24% CM–OOH after 8 h, see Table 1). However, the activity of both materials is practically the same at short reaction times (up to 2 h), as evidenced by the superposition of both curves in Fig. 2a. From the slopes of the time-conversion curves at short reaction times, a TOF of about 3500 h−1 was calculated for both copper catalysts. A blank experiment in the absence of catalyst yielded 21% CM–OOH and 3% AP after 24 h (see Table 1).
3.3. Ethylbenzene oxidation
Oxidation of ethylbenze is expected to be more difficult than in the case of tetralin or cumene, since the carbon atom to be oxidized is less activated. In the case of ethylbenzene, oxygen is added onto a secondary carbon atom, while in cumene the preferred position for the attack is on a tertiary carbon atom, which allows a major stabilization of the radical species formed. In tetralin the addition of oxygen is onto a secondary carbon atom, but in this case, it belongs to a six-member ring, which adds an extra activation as compared to ethylbenzene.Although the conversion of ethylbenzene was considerably lower than that observed for cumene under similar conditions (at 80 °C, 0.5 mol% Cu), both [Cu(2-pymo)2] and [Cu(im)2] demonstrated to be active catalysts for this reaction, as shown in Fig. 3. In this case, [Cu(im)2] showed a better performance at short reaction time, although at the end of the reaction the two materials gave similar results: 29% (25%) conversion after 40 h and 86% (87%) selectivity to acetophenone for [Cu(im)2] and for [Cu(2-pymo)2], respectively.
![Conversion of EB over [Cu(im)2] and [Cu(2-pymo)2] (part a). Time conversion of EB and time evolution of products over [Cu(im)2] and [Cu(2-pymo)2] is also shown in parts (b) and (c), respectively.](/image/article/2013/CY/c2cy20449e/c2cy20449e-f3.gif) | ||
| Fig. 3 Conversion of EB over [Cu(im)2] and [Cu(2-pymo)2] (part a). Time conversion of EB and time evolution of products over [Cu(im)2] and [Cu(2-pymo)2] is also shown in parts (b) and (c), respectively. | ||
If we compare the kinetic data obtained for EB oxidation (Fig. 3) with the data obtained for TH and CM oxidation (Fig. 1 and 2), clear differences can be observed. On the one hand, accumulation of hydroperoxide in the reaction medium does not occur during EB oxidation, not even when [Cu(2-pymo)2] was used as catalyst. This is not surprising, since the hydroperoxide derived from ethylbenzene is the least stable among all the substrates studied in this work. Thus, when 1-phenylhydroperoxide (EB–OOH) is generated, it is immediately converted into other products (i.e., acetophenone and 1-phenylethanol). On the other hand, we also observed clear induction periods of about 2–3 h, in which EB is not converted, irrespective of the catalyst used. Both observations indicate that in the EB oxidation reaction, formation of the hydroperoxide species is difficult for both Cu-MOF catalysts. However, once this primary species is formed, it is converted to reaction products more effectively over [Cu(im)2] than over [Cu(2-pymo)2]. A blank experiment in the absence of catalyst yielded only 2% EB–OH and 0.5% EB–OOH after 24 h (see Table 1).
3.4. Origin of the different reactivities of [Cu(2-pymo)2] and [Cu(im)2]
In light of the results presented above, and which are summarized in Table 1, it is evident that [Cu(im)2] is a better catalyst than [Cu(2-pymo)2] for the oxidation of TH, CM and EB. In general, we have observed that both formation of the primary hydroperoxide species and its conversion into final products is faster for the imidazolate compound. Both compounds feature Cu2+ ions coordinated to four N atoms from different diazaheteorcyclic compounds (imidazole and pyrimidine), thus leading to CuN4 centers. However, Cu2+ ions are in square planar coordination in [Cu(2-pymo)2], while they are in a highly distorted coordination in [Cu(im)2], with trans N–Cu–N angles of 140° and 155°. In a first approximation, one can expect that these structural differences could account for the different reactivity observed for both materials. However, it is difficult for us to explain intuitively the structure–composition–reactivity correlation for the two Cu2+ MOFs studied. Then, and in order to get more insights into the structural and electronic properties of the two compounds, we have carried out first principle DFT calculations on MOF model clusters, as described in detail in the Experimental section. The models used consisted of a central Cu2+ ion surrounded by either four imidazole (im) or four 2-hydroxypyrimidine (2-pymo) molecules. Each of the four organic ligands in the cluster models was saturated with a proton placed in the direction of the next Cu2+ ion in the real MOF material, so that each model bears a net positive charge of +2. The geometry of the two resulting clusters was fully optimized without restrictions.To shed some light on the different catalytic behavior of the two copper-containing MOFs [Cu(im)2] and [Cu(2-pymo)2], we have theoretically investigated using DFT the interaction of these two materials with cumene-hydroperoxide (CM–OOH) to form an adsorption complex [Cu2+–HOO–CM], and its subsequent dissociation into a hydroxyl radical that remains adsorbed on the copper center [Cu2+–OH] and a free cumyl (˙O–CM) radical. The fully optimized structures (see Computational details) resulting from CM-OOH adsorption on the two Cu2+ active site models are depicted in Fig. 4. CM-OOH adsorption was found to be energetically favorable on the two materials studied, with calculated free energy values of −6 kcal mol−1 for [Cu(im)2] and −2 kcal mol−1 for [Cu(2-pymo)2], irrespectively of the type of geometry optimization performed (full or restricted). The optimized Cu2+–O distances found were 2.47 and 2.30 Å in [Cu(im)2] and [Cu(2-pymo)2] materials, respectively, also indicating a certain degree of interaction. It is worth noting that the initial geometry of the active center in both MOFs is not significantly distorted in any case as a consequence of the interaction with the hydroperoxide. However, after cumene hydroperoxide dissociation, substantial differences were found in the resulting [Cu2+-OH] complexes formed in the two materials. On the one hand, Cu2+ ions in [Cu(im)2] were found to expand their coordination sphere from 4 to 5 upon binding of a ˙OH radical. On the contrary, the interaction of the ˙OH radical with the Cu2+ centre in [Cu(2-pymo)2] implies the de-coordination of one of the four 2-pymo ligands. Note that this ligand displacement would not necessary imply the collapse of the crystalline structure of the MOF. Actually, the Cu2+ centers would still remain connected to the framework through three out of the four initial 2-pymo ligands. Once the catalytic cycle is finished and the product desorbs from the active site, the 2-pymo ligand that has been displaced can coordinate again to the Cu2+ site to recover the initial catalytic centre, as shown in Scheme 2. A similar ligand displacement and re-coordination cycle has been demonstrated to occur in a series of zinc(II) benzoate coordination polymers during transesterification reactions.23
![Optimized structures of the complexes formed by interaction of: (a) cumene-hydroperoxide with [Cu(im)2] (b) cumene-hydroperoxide with [Cu(2-pymo)2] (c) hydroxyl radical with [Cu(im)2] and (d) cumene-hydroperoxyl radical and [Cu(im)2]. Carbon, nitrogen, oxygen, hydrogen and copper atoms are grey, blue, red, white and yellow, respectively.](/image/article/2013/CY/c2cy20449e/c2cy20449e-f4.gif) | ||
| Fig. 4 Optimized structures of the complexes formed by interaction of: (a) cumene-hydroperoxide with [Cu(im)2] (b) cumene-hydroperoxide with [Cu(2-pymo)2] (c) hydroxyl radical with [Cu(im)2] and (d) cumene-hydroperoxyl radical and [Cu(im)2]. Carbon, nitrogen, oxygen, hydrogen and copper atoms are grey, blue, red, white and yellow, respectively. | ||
![Ligand displacement and re-coordination of 2-hydroxypyrimidine in [Cu(2-pymo)2] upon coordination/release of a radical species.](/image/article/2013/CY/c2cy20449e/c2cy20449e-s2.gif) | ||
| Scheme 2 Ligand displacement and re-coordination of 2-hydroxypyrimidine in [Cu(2-pymo)2] upon coordination/release of a radical species. | ||
In conclusion, the results obtained from first principle calculations indicate that [Cu(im)2] has a more adaptable crystalline framework than [Cu(2-pymo)2], which allows the copper sites to expand their coordination sphere from 4 to 5 upon interaction with ˙OH radical species. On the contrary, binding of the same radical to [Cu(2-pymo)2] produces the displacement of one of the 2-pymo ligands from the coordination sphere around the central Cu site. These differences could account for the higher activity of [Cu(im)2] to decompose the hydroperoxide into final products, and at the same time could explain the experimentally observed accumulation of the CM–OOH intermediate in the oxidation of CM catalyzed by [Cu(2-pymo)2]. Even if this preliminary computational study does not include calculation of the transition states corresponding to the two Cu-MOFs, it is not unreasonable to expect that a higher energy would be required in the case of [Cu(2-pymo)2] to break a Cu–pyrimidine bond that in the case of [Cu(im)2] in which only a rearrangement of the ligands is required to accommodate the ˙OH radical.
Nevertheless, in order to demonstrate that the two MOFs have indeed different abilities for decomposing the hydroperoxide, we have designed an additional experiment. Thus, when we contacted the two MOFs with cumene hydroperoxide, it was observed that [Cu(im)2] decomposes the hydroperoxide significantly faster than [Cu(2-pymo)2] under identical conditions (0.01 mmol Cu, 0.9 ml CM, 0.1 ml CM–OOH, 80 °C under a N2 atmosphere). After 1 hour, [Cu(im)2] decomposed 55 mol% of the initial CM–OOH, while only 31 mol% was decomposed over [Cu(2-pymo)2].
3.5. Tandem cumene oxidation and 1-octene epoxidation using a combination of catalysts: one-pot versus two-pot setups
As we have mentioned above, one of the drawbacks of [Cu(2-pymo)2] as oxidation catalyst is that a considerable amount of hydroperoxide is accumulated in the reaction medium, especially in the case of cumene and tetralin oxidation. This limitation can be turned into an advantage if we are able to use the hydroperoxide generated in situ during the oxidation as a reagent for a second reaction. This becomes even more interesting if one can couple the two reactions in a one-pot process, thus leading to a tandem catalytic process. Taking as a source of inspiration the Sumitomo process24,25 for olefin epoxidation using cumene hydroperoxide, we have combined [Cu(2-pymo)2] with a state-of-the-art epoxidation catalyst (silylated Ti-MCM-4115) to perform the tandem process depicted in Scheme 3. The experimental setup is shown in Fig. 5a. Thus, [Cu(2-pymo)2] is used in the first reaction to oxidize CM into CM–OOH using air as the oxidant (reaction R1). Then, silylated Ti-MCM-41 can use the generated CM–OOH to oxidize an olefin (1-octene) in reaction R2, producing 1-octene oxide and one equivalent of CM–OH.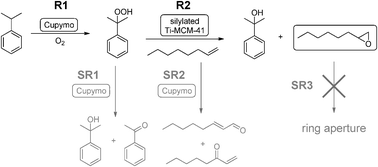 | ||
| Scheme 3 | ||
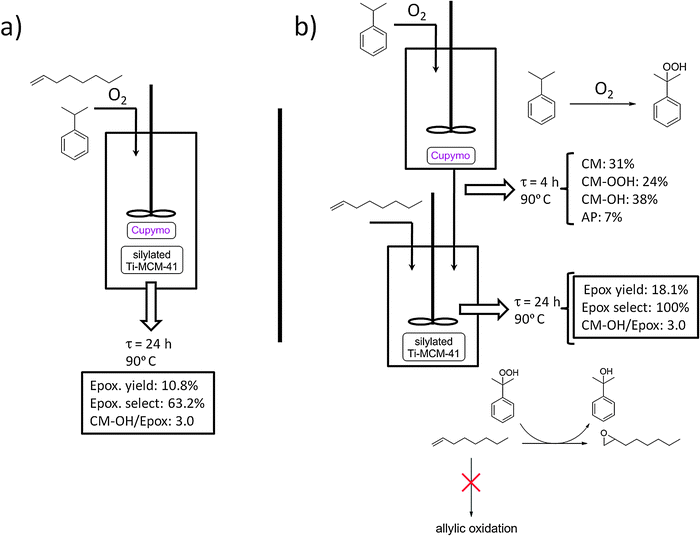 | ||
| Fig. 5 Schematic representation of the one-pot (a) and two-pot setups (b) adopted for the tandem cumene oxidation/1-octene epoxidation reaction. CM = cumene; CM–OOH = cumene hydroperoxide; AP = acetophenone; Epox = 1-octene oxide. | ||
However, this simple reaction scheme is complicated by the occurrence of competing unwanted side reactions, which decrease the overall selectivity to 1-octene oxide by either yielding secondary products of 1-octene (SR2 and/or SR3), or by spuriously consuming CM–OOH without transferring the oxygen to 1-octene (SR1). Side reaction SR1 consists in the decomposition of the CM–OOH into CM–OH and AP. This reaction is catalyzed by [Cu(2-pymo)2], as we have already demonstrated in the previous section, and it can also take place thermically (non-catalytic decomposition). SR2 consists in the direct allylic oxidation/isomerization of 1-octene, to produce a mixture of oxidized products. SR2 can also be catalyzed by copper or an uncatalyzed thermal reaction. Finally SR3, consisting in the ring opening of the epoxide, is another reaction that in principle could decrease the overall yield of 1-octene oxide.
After a preliminary screening of different conditions, we found a satisfactory experimental setup, as detailed in the experimental section. Among others, the following parameters were considered: (i) [Cu(2-pymo)2] to silylated Ti-MCM-41 ratio; (ii) cumene to 1-octene ratio; (iii) temperature of the reaction; (iv) order of addition of the reagents; and (v) oxygen pressure. To evaluate the relevance of the different competing side reactions, we carried out the oxidation process at two different temperatures, viz. 333 and 363 K. A summary of the results obtained is shown in Table 2.
| Temp (K) | Conv./Time (mol%/h) | Yield epox (mol%) | Yield oxdn. (mol%) | Select. epox (%) | CM–OH/epox |
|---|---|---|---|---|---|
| epox = 1-octene oxide; oxdn = products coming from the allylic oxidation of 1-octene. | |||||
| 333 | 7.1 (48) | 4.5 | 2.6 | 63.4 | 1.1 |
| 363 | 17.1 (24) | 10.8 | 6.3 | 63.2 | 3.0 |
When the reaction was performed at 333 K, an overall conversion of 1-octene of 7.1 mol% was achieved after 48 h, producing 1-octene oxide with a selectivity of 63.4%, together with other products coming from the allylic oxidation of 1-octene. In a separate experiment we have observed that pure [Cu(2-pymo)2] can indeed catalyze the allylic oxidation of 1-octene using air as the oxidant, yielding mainly the products indicated in Scheme 3 (12% maximum conversion of 1-octene achieved after 24 h at 363 K). Note that these oxidation products can also be formed through a non-catalyzed thermal reaction. It is important to stress that in our system we did not detect any traces of products coming from the ring aperture of the formed epoxide (SR3), which is largely suppressed by the hydrophobic character of the silylated Ti-MCM-41 catalyst used and the absence of water in the reaction medium.15
When the reaction temperature was increased to 363 K, the overall reaction rate dramatically increased: 17.1% 1-octene conversion after 24 h was observed, which represents an almost 5-fold increase of the reaction rate. Interestingly, the selectivity to 1-octene oxide was practically the same (63.2%), with a similar distribution of secondary products than at lower temperature.
Another important parameter of the tandem reaction is given by the CM-OH/epoxide ratio, which reveals the extent to which the competing side reaction SR1 is taking place. Indeed, if all the CM-OOH formed in R1 is used to produce 1-octene oxide following R2, the final CM-OH/epoxide ratio should be equal to 1. Any excess amount of CM-OH with respect to 1-octene oxide (as well as the presence of acetophenone in the products mixture) indicates that CM-OOH has been spuriously decomposed without producing the desired epoxide. This is an important factor to take into account, since in the envisaged reaction scheme the CM-OH produced has to be recovered and reconverted into CM (through a dehydration and hydrogenation process) for recycling. If we compare our results obtained at 333 and 363 K, we can see that at the lower temperature, although the reaction rate is considerably low, the process is highly selective concerning the utilization of the CM-OOH formed (CM-OH/epoxide = 1.1). On the contrary, upon increasing the temperature of the reaction, the alternative SR1 side reaction becomes the main reaction pathway for CM-OOH decomposition (CM-OH/epoxide = 3.0). Thus, an increase of the reaction temperature has negative effects on the amount of CM-OH waste generated per mol of final product.
One evident limitation of this one-pot setup is the relatively low selectivity to the epoxide (63.2% at 363 K), which is due, as mentioned above, to the occurrence of the competing side-reaction SR2: allylic oxidation of 1-octene catalyzed by [Cu(2-pymo)2]. The only way of suppressing this reaction, thus increasing the selectivity to the epoxide product, is to avoid the contact between 1-octene and the copper-MOF by keeping them in separate reactors. In other words, it becomes necessary to adopt a two-pot setup, such as that shown in Fig. 5b. In such a reaction setup, [Cu(2-pymo)2] is allowed to interact with CM under an O2 atmosphere at 363 K for 4 h, to produce CM-OOH in 24 mol% yield (see Fig. 5b). Then, the copper-catalyst is removed by filtration and the liquid filtrate is fed into a second reactor, containing silylated Ti-MCM-41, and 1-octene is added at this point. Under these conditions, 1-octene was selectively converted into the corresponding epoxide, yielding 18.1 mol% after 24 h at 363 K. In this case, no traces of products coming from either the allylic oxidation of 1-octene, or from the ring-opening of the epoxide, were detected. A final CM-OH/epoxide ratio of 3.0 was obtained. Note that most of this CM-OH was already present in the filtrate coming from the first reactor (38 mol%, see Fig. 5b). Indeed, CM-OOH is used to oxidize the olefin in the second reactor with a high selectivity (24 mol% CM-OOH produces 18.1 mol% of 1-octene oxide), while direct decomposition of the hydroperoxide occurs only to a minor extent in the second reactor. This is largely prevented by the absence of the copper catalyst at this stage (which would catalyze the side reaction SR1) and due to the relatively low temperature (which minimizes the autocatalytic thermal decomposition of CM-OOH).
4. Conclusion
In this work, we have shown that copper-containing MOFs in which Cu2+ centers are linked to 4 nitrogen atoms from azaheterocyclic compounds (i.e., pyrimidine and imidazole) have interesting potential as heterogeneous catalysts for aerobic liquid phase oxidation of activated paraffins. We have shown this for two materials, [Cu(im)2] and [Cu(2-pymo)2], and for three different substrates of increasing demands: tetralin, cumene and ethylbenzene. Our results indicate that [Cu(im)2] has in general better performance than [Cu(2-pymo)2], which results in higher alkane conversion, higher selectivity and low accumulation of alkylhydroperoxides in the reaction medium. According to DFT calculations, the differences between the two catalysts could be related to the different ability of the two MOFs to decompose the hydroperoxide and to coordinate to the resulting radical ˙OH species. Copper ions in [Cu(im)2] can coordinate to these radicals by expanding their coordination sphere from 4 to 5. On the contrary, in the case of [Cu(2-pymo)2], coordination of a radical species to the Cu2+ ions results in the displacement of one of the pyrimidine ligands. This different behavior of the two MOFs also explains why the ROOH intermediate is accumulated in the reaction medium when [Cu(2-pymo)2] is used as catalyst, and not in the case of [Cu(im)2].We have also shown that the hydroperoxide accumulated in the reaction medium when [Cu(2-pymo)2] used as catalyst can be used as oxidant in a parallel reaction. To illustrate this, we have combined [Cu(2-pymo)2] with a good epoxidation catalyst (silylated Ti-MCM-41) to carry out a tandem process consisting in cumene oxidation and 1-octene epoxidation. We have studied this reaction using either a one-pot or a two-pot setup. A main drawback of the one-pot setup is that [Cu(2-pymo)2] can also catalyze the allylic oxidation of 1-octene, thus decreasing the net epoxide yield. To overcome this problem, it is necessary to avoid the contact between the copper MOF and the olefin. Thus, it becomes necessary to work in two batch reactors.
Acknowledgements
Financial support by Ministerio de Educación y Ciencia e Innovación (Project MIYCIN, CSD2009-00050; PROGRAMA CONSOLIDER. INGENIO 2009), Generalidad Valenciana (GV PROMETEO/2008/130) and CSIC (Proyectos Intramurales Especiales 201080I020) is gratefully acknowledged. CSIC and Fundación Bancaja are gratefully acknowledged for a research contract to I.L. AL thanks the ITQ for a postgraduate scholarship.References
- A. Corma, U. Diaz, T. Garcia, G. Sastre and A. Velty, J. Am. Chem. Soc., 2010, 132, 15011–15021 CrossRef CAS.
- A. Corma, H. Garcia and F. X. Llabrés i Xamena, Chem. Rev., 2010, 110, 4606–4655 CrossRef CAS.
- D. Farrusseng, S. Aguado and C. Pinel, Angew. Chem., Int. Ed., 2009, 48, 7502–7513 CrossRef CAS.
- J. Y. Lee, O. K. Farha, J. Roberts, K. A. Scheidt, S. T. Nguyen and J. T. Hupp, Chem. Soc. Rev., 2009, 38, 1450–1459 RSC.
- M. Meilikhov, K. Yusenko, D. Esken, S. Turner, G. Van Tendeloo and R. A. Fischer, Eur. J. Inorg. Chem., 2010, 3701–3714 CrossRef CAS.
- J. Juan-Alcaniz, J. Gascon and F. Kapteijn, J. Mater. Chem., 2012, 22, 10102–10118 RSC.
- Y. Pan, B. Yuan, Y. Li and D. He, Chem. Commun., 2010, 46, 2280–2282 RSC.
- F. G. Cirujano, F. X. Llabrés i Xamena and A. Corma, Dalton Trans., 2012, 41, 4249–4254 RSC.
- F. X. Llabrés i Xamena, O. Casanova, R. Galiasso Tailleur, H. Garcia and A. Corma, J. Catal., 2008, 255, 220–227 CrossRef.
- L. C. Tabares, J. A. R. Navarro and J. M. Salas, J. Am. Chem. Soc., 2001, 123, 383–387 CrossRef CAS.
- K. S. Park, Z. Ni, A. P. Cote, J. Y. Choi, R. D. Huang, F. J. Uribe-Romo, H. K. Chae, M. O'Keeffe and O. M. Yaghi, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 10186–10191 CrossRef CAS.
- N. Masciocchi, S. Bruni, E. Cariati, F. Cariati, S. Galli and A. Sironi, Inorg. Chem., 2001, 40, 5897–5905 CrossRef CAS.
- I. Luz, F. X. Llabrés i Xamena and A. Corma, J. Catal., 2010, 276, 134–140 CrossRef CAS.
- I. Luz, F. X. Llabrés i Xamena and A. Corma, J. Catal., 2012, 285, 285–291 CrossRef CAS.
- A. Corma, M. E. Domine, J. Gaona, J. L. Jorda, M. T. Navarro, J. Perez-Pariente, F. Rey, B. McCulloch, L. Nemeth and J. Tsuji, Chem. Commun., 1998, 2211–2212 RSC.
- M. J. Frisch, G. W. Trucks and H. B. Schlegel, Gaussian 03, Revision B.04, Gaussian, Inc., Pittsburgh, PA, Editon edn, 2003 Search PubMed.
- J. P. Perdew and Y. Wang, Phys. Rev. B: Condens. Matter Mater. Phys., 1992, 45, 13244–13249 CrossRef.
- A. D. Becke, J. Chem. Phys., 1993, 98, 5648–5652 CrossRef CAS.
- P. C. Hariharan and J. A. Pople, Theor. Chim. Acta, 1973, 28, 213–222 CrossRef CAS.
- M. M. Fares, M. El-Khateeb and K. J. Asali, J. Inorg. Organomet. Polym., 2003, 13, 143–155 CrossRef CAS.
- G. Y. Tian, D. H. Xia and F. T. Zhan, Energy Fuels, 2004, 18, 49–53 CrossRef CAS.
- C. Aprile, A. Corma, M. E. Domine, H. Garcia and C. Mitchell, J. Catal., 2009, 264, 44–53 CrossRef CAS.
- H. Kwak, S. H. Lee, S. H. Kim, Y. M. Lee, B. K. Park, Y. J. Lee, J. Y. Jun, C. Kim, S.-J. Kim and Y. Kim, Polyhedron, 2009, 28, 553–561 CrossRef CAS.
- J. Tsuji, J. Yamamoto, A. Corma and F. Rey, US Patent, 6211388, 1998 Search PubMed.
- J. Tsuji, J. Yamamoto, M. Ishino and N. Oku, Sumitomo Kagaku (Osaka, Jpn.), 2006, 1, 4–10 Search PubMed.
| This journal is © The Royal Society of Chemistry 2013 |