 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Oxo-carboxylato-molybdenum(VI) complexes possessing dithiolene ligands related to the active site of type II DMSOR family molybdoenzymes†
Hideki Sugimoto*a, Masanori Satoa, Logan J. Gilesb, Kaori Asanoc, Takeyuki Suzukic, Martin L. Kirk*b and Shinobu Itoh*a
aDepartment of Material and Life Science, Graduate School of Engineering, Osaka University, 2-1 Yamadaoka, Suita, Osaka 565-0871, Japan. E-mail: sugimoto@mls.eng.osaka-u.ac.jp; shinobu@mls.eng.osaka-u.ac.jp
bDepartment of Chemistry and Chemical Biology, The University of New Mexico, MSC03 2060, 1 University of New Mexico, Albuquerque, New Mexico 87131-0001, USA. E-mail: mkirk@unm.edu
cComprehensive Analysis Centre, The Institute of Scientific and Industrial Research (ISIR), Osaka University, 8-1 Mihogaoka, Ibaraki, Osaka 567-0057, Japan
First published on 28th August 2013
Abstract
Spectroscopic and kinetic studies indicate that oxo-carboxylato-molybdenum(VI) bis-dithiolene complexes, (MoVIO(p-X-OBz)L2), have been generated at low temperature as active site structural models for the type II class of pyranopterin molybdenum DMSOR family enzymes. A DFT analysis of low energy charge transfer bands shows that these complexes possess a Mo–Sdithiolene π-bonding interaction between the Mo(dxy) redox active molecular orbital and a cis S(pz) donor orbital located on one of the dithiolene ligands.
The vast majority of pyranopterin molybdenum enzymes catalyse oxygen atom transfer reactions between the substrate and solvent water coupled with proton and electron transfer.1 The dimethyl sulfoxide reductase (DMSOR) family of pyranopterin molybdenum enzymes is unique in that they possess two pyranopterin ene-1,2-dithiolate ligands bound to the Mo ion.1 The active site Mo centre is the locus of the oxygen atom transfer reactivity and can adopt desoxomolybdenum(IV) and monooxomolybdenum(VI) structures during the catalytic cycle.1,2 Phylogenic analysis and protein sequence alignments of the metal-binding regions have been used to further subdivide DMSOR family enzymes into three types: I, II and III (Fig. 1).3 Type I enzymes possess a Cys or SeCys residue that coordinates to the molybdenum centre,4 while type II and type III enzymes have a metal center coordinated by an Asp and a Ser residue, respectively.2,5
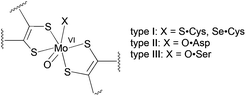 | ||
| Fig. 1 The active site structures of oxidized type I, II and III enzymes. | ||
Extensive studies of bis(ene-1,2-dithiolate)molybdenum complexes as active site structural models have been carried out by Holm and co-workers.6–8 Specifically, these researchers have synthesised and structurally characterised a number of desoxomolybdenum(IV) complexes employing 1,2-dimethylethylene-1,2-dithiolate (S2C2Me2) together with RS−, RSe−, RCOO−, and RO− ligands (X)6 that are structurally related to the molybdenum(IV) active site structures of DMSOR type I, II and III enzymes.7 However, the corresponding monooxomolybdenum(VI) complexes, [MoVIO(L)(S2C2Me2)2]−, have proved too unstable for full characterisation due to an auto-redox reaction between the MoVI ion and the monodentate ligand L that results in the formation of the five-coordinate [MoVO(S2C2Me2)2]− complex.7,8 The only crystallographically characterised example of a [MoVIO(L)(ene-1,2-dithiolate)2]− complex is [MoVIO(OSiiPr3)(S2C2(COOMe)2)2]− (MoVIO(OSiiPr3)(LCOOMe)2, S2C2(COOMe)2 = 1,2-dicarbomethoxyethylene-1,2-dithiolate), where the six-coordinate structure appears to be stabilised by the presence of electron-withdrawing methoxycarbonyl (–COOMe) groups on the dithiolene.9MoVIO(OSiiPr3)(LCOOMe)2 best represents a synthetic analogue of the DMSOR type III enzyme active sites. Recently, we prepared and characterised oxosulfido- and oxoselenido-molybdenum(VI) complexes at low temperature as active site models for the xanthine oxidase family of pyranopterin molybdenum enzymes.10 Herein, we report the successful application of low temperature techniques to generate and characterise new bis(ene-1,2-dithiolate)oxocarboxylatomolybdenum(VI) complexes, which can be regarded as structural models for oxidised members of type II DMSOR family enzymes. Additionally, we provide an initial electronic structure description of these bis(ene-1,2-dithiolate)oxocarboxylatemolybdenum(VI) complexes using DFT calculations. The designation and abbreviation of the complex structures are given in Chart S1.†
Initial attempts to prepare the oxocarboxylatomolybdenum(VI) complex MoVIO(p-H-OBz)L2 (L = cyclohexene-1,2-dithiolate (S2C2C4H8)) utilised an oxo-transfer reaction from the tertiary amine oxide (Me3NO) to the benzoatomolybdenum(IV) complex, MoIV(p-H-OBz)L2, at room temperature under an inert Ar atmosphere. However, the grey coloured oxomolybdenum(V) complex [MoVO(S2C2C4H8)2]− (MoVOL2) was obtained instead of the anticipated oxocarboxylatomolybdenum(VI) complex MoVIO(p-H-OBz)L2. The formation of MoVO(ene-1,2-dithiolate)2 has also been reported in the reaction of [MoIV(X)(S2C2Me2)2]− with tertiary amine oxides or sulfoxides.8 We attempted to observe the formation of (MoVIO(p-H-OBz)L2 at low temperatures, but the reaction between MoIV(p-H-OBz)L2 and tertiary amine oxides does not proceed to a significant extent below −40 °C. Next, the reaction of the five-coordinate oxomolybdenum(VI) complex, MoVIOL2 (synthesis in ESI†),11 with the benzoate anion, Et4N(p-H-OBz), was examined in C2H5CN under an Ar atmosphere. At room temperature, this reaction also yielded the oxomolybdenum(V) complex (MoVOL2) (Fig. S1†), but at low temperature (−60 °C) the reaction yielded a deep-green EPR silent product. Fig. 2 shows the observed spectral changes upon addition of p-H-OBz− to MoVIOL2 in C2H5CN at −60 °C. Here, the absorption band at 395 nm due to MoVIOL2 decreases with the concomitant appearance of new absorption bands at 807 nm (band 1) and 597 nm (band 2). Tight isosbestic points are observed at 336, 367 and 445 nm. Titration plots clearly indicate that the stoichiometry of MoVIOL to p-H-OBz− is 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 (Fig. 2, inset). The final spectrum is very similar to that of [MoVIO(OSiiPr3)(S2C2(COOMe)2)2]−,9 consistent with the formation of a six-coordinate oxocarboxylatomolybdenum(VI) complex, [MoVIO(p-H-OBz)(S2C2C4H8)2]−, (MoVIO(p-H-OBz)L2). MoVIO(p-OMe-OBz)L2 and MoVIO(p-Cl-OBz)L2 possess similar spectral features to MoVIO(p-H-OBz)L2 and were generated in a similar manner using Et4N(p-OMe-OBz) and Et4N(p-Cl-OBz), respectively (Fig. S2†). The λmax for band 1 shifts to lower energy as the substituent X changes from electron withdrawing to electron donating (X = Cl, 793 nm; X = H, 807 nm; X = OMe, 814 nm). This strongly supports the direct coordination of the benzoate ligand to the molybdenum(VI) centre. When warmed to room temperature, the UV-vis and EPR spectra converted to those of MoVOL2.
:1 (Fig. 2, inset). The final spectrum is very similar to that of [MoVIO(OSiiPr3)(S2C2(COOMe)2)2]−,9 consistent with the formation of a six-coordinate oxocarboxylatomolybdenum(VI) complex, [MoVIO(p-H-OBz)(S2C2C4H8)2]−, (MoVIO(p-H-OBz)L2). MoVIO(p-OMe-OBz)L2 and MoVIO(p-Cl-OBz)L2 possess similar spectral features to MoVIO(p-H-OBz)L2 and were generated in a similar manner using Et4N(p-OMe-OBz) and Et4N(p-Cl-OBz), respectively (Fig. S2†). The λmax for band 1 shifts to lower energy as the substituent X changes from electron withdrawing to electron donating (X = Cl, 793 nm; X = H, 807 nm; X = OMe, 814 nm). This strongly supports the direct coordination of the benzoate ligand to the molybdenum(VI) centre. When warmed to room temperature, the UV-vis and EPR spectra converted to those of MoVOL2.
![Spectral changes observed upon addition of 1/5, 2/5, 3/5, 4/5, and 5/5 equiv. of Et4N(p-H-OBz) to MoVIOL2 (0.2 mM) in C2H5CN at −60 °C; (inset) plots of the absorbance at 395 (●) and 807 (■) nm against the molar ratio of [p-H-OBz−]/[MoVIOL2].](/image/article/2013/DT/c3dt51485d/c3dt51485d-f2.gif) | ||
| Fig. 2 Spectral changes observed upon addition of 1/5, 2/5, 3/5, 4/5, and 5/5 equiv. of Et4N(p-H-OBz) to MoVIOL2 (0.2 mM) in C2H5CN at −60 °C; (inset) plots of the absorbance at 395 (●) and 807 (■) nm against the molar ratio of [p-H-OBz−]/[MoVIOL2]. | ||
Cyclic voltammograms of 0.1 M nBu4NPF6–C2H5CN solutions containing MoVIOL2 and Et4N(p-X-OBz) (X = Cl, H, OMe) in a 1:1 ratio were measured at −60 °C and yielded one irreversible reduction wave below −1 V vs. SCE. The reduction peak potential is observed to shift in a negative direction (Cl, Epc = −1.08 V; H, Epc = −1.16; OMe, Epc = −1.18 V vs. SCE) as the pKa value of the p-substituted benzoic acid increases (3.99 for X = Cl, 4.20 for X = H and 4.50 for X = OMe). This result provides support for the coordination of the p-X-OBz anion to the Mo center of MoVIOL2 since the electron-donating substituent increases the basicity of the benzoate anion and enhances its ability to coordinate to MoVI. The CSI-mass spectrum of a C2H5CN solution containing MoVIOL2 and 1 equiv. of Et4N(p-OMe-OBz) showed a peak cluster attributable to [Mo(p-OMe-OBz)(S2C2C4H8)2]− at m/z = 537 at −60 °C (Fig. S3†). Since the UV-vis spectrum of the C2H5CN solution of the product shown in Fig. 2 is different from that of [MoIV(p-OMe-OBz)(S2C2C4H8)2]− synthesised separately from [MoIV(S2C2C4H8)2] and p-OMe-OBz−, this peak cluster is likely to be a fragment of MoVIO(p-OMe-OBz)L2. Therefore, we conclude that MoVIO(p-H-OBz)L2 and its para substituted derivatives are formed at low temperature by coordination of the benzoates to the molybdenum(VI) centre of MoVIOL2.
In order to obtain information about the mechanism of MoVIO(p-X-OBz)L2 formation, a low temperature kinetic study was performed. The spectral changes observed upon addition of the benzoate ligand to MoVIOL2 were observed to be biphasic. The data show a rapid disappearance of the MoVIOL2 395 nm band and the appearance of a new absorption band at 360 nm due to intermediate A that converts to MoVIO(p-X-OBz)L2 with characteristic absorption bands at 597 and 807 nm (Fig. S4a and 4b†). Although the time course of the first step was too fast to be followed accurately, the conversion of intermediate A to MoVIO(p-X-OBz)L2 obeys first-order kinetics. It should be noted that the observed first-order rate constant, kobs, was independent of the concentration of Et4N(p-H-OBz) (Fig. S5†). The formation of MoVIO(p-Cl-OBz)L2 and MoVIO(p-OMe-OBz)L2 displayed kinetic behavior similar to MoVIO(p-H-OBz)L2 with kobs increasing as the electron-withdrawing character of X increases: 65.8 × 10−3 s−1 for X = Cl, 7.2 × 10−3 s−1 for X = H and 1.4 × 10−3 s−1 for X = OMe. These observations suggest that the first step is the association (coordination) of the benzoate anion with the Mo center of MoVIOL2, giving intermediate A, and the second step is the intramolecular rearrangement of intermediate A to the product (Scheme 1). DFT calculations support an idealised C2v structure with respect to the two S2C2C4H8 ligands for intermediate A that subsequently rearranges via a Ray–Dutt type twist12 to form a more stable product with a distorted octahedral geometry (Fig. 3).
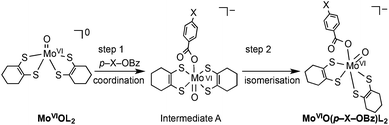 | ||
| Scheme 1 Proposed mechanism for formation of MoVIO(p-X-OBz)L. | ||
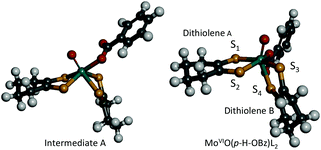 | ||
| Fig. 3 Computed structures of intermediate A (left) and MoVIO(p-H-OBz)L2 (right). | ||
The DFT optimised structure of MoVIO(p-H-OBz)L2 possesses a distorted octahedral coordination environment that is similar to the related MoVIO(OSiiPr3)(LCOOMe)2 (vide supra; Fig. 3), which has been previously characterised by X-ray crystallography.9 The computations indicate a slightly larger S1–S2–S3–S4 dithiolene dihedral angle for MoVIO(p-H-OBz)L2 of 126° compared with a 108° dihedral angle for structurally characterised MoVIO(OSiiPr3)(LCOOMe)2. As was observed for MoVIO(OSiiPr3)(LCOOMe)2, the Mo–S4 bond distance (2.55 Å) is elongated when compared to the three Mo–S bonds (mean 2.42 Å) as a result of a strong trans influence due to the Mo![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) O bond. Geometry optimisations indicate that MoVIO(p-Cl-OBz)L2 and MoVIO(p-OMe-OBz)L2 possess S2–S1–S3–S4 dihedral angles, and Mo–S, MoOoxo and Mo–OOBz bond distances, which are nearly identical to those of MoVIO(p-H-OBz)L2. The computed structures for the MoVIO(p-X-OBz)L2 complexes display ∼172° Ooxo–Mo–S3–C dihedral angles, allowing for strong Mo dxy–Sdithiolene π-bonding involving a single S3 donor on dithiolene B. This is supported by bonding calculations that show a LUMO wavefunction which possesses a strong Mo–Sdithiolene π* bonding interaction between the Mo(dxy) orbital and the cis S3(pz) donor orbital located on dithiolene (B) (Table 1). This Mo–S π* bonding description is similar to what we observed in the related complex MoVIO(OSiiPr3)(LCOOMe)2,9 and this derives from the ∼180° Ooxo–Mo–S–C dihedral angle involving the cis S of dithiolene B.
O bond. Geometry optimisations indicate that MoVIO(p-Cl-OBz)L2 and MoVIO(p-OMe-OBz)L2 possess S2–S1–S3–S4 dihedral angles, and Mo–S, MoOoxo and Mo–OOBz bond distances, which are nearly identical to those of MoVIO(p-H-OBz)L2. The computed structures for the MoVIO(p-X-OBz)L2 complexes display ∼172° Ooxo–Mo–S3–C dihedral angles, allowing for strong Mo dxy–Sdithiolene π-bonding involving a single S3 donor on dithiolene B. This is supported by bonding calculations that show a LUMO wavefunction which possesses a strong Mo–Sdithiolene π* bonding interaction between the Mo(dxy) orbital and the cis S3(pz) donor orbital located on dithiolene (B) (Table 1). This Mo–S π* bonding description is similar to what we observed in the related complex MoVIO(OSiiPr3)(LCOOMe)2,9 and this derives from the ∼180° Ooxo–Mo–S–C dihedral angle involving the cis S of dithiolene B.
The electronic absorption spectra of the MoVIO(p-X-OBz)L2 complexes display well-resolved bands at ∼800 nm and ∼600 nm (band 1: ∼12500 cm−1, ε ∼ 1375 M−1 cm−1; and band 2: 16700 cm−1, ε ∼ 1600 M−1 cm−1, respectively). Using a combination of time-dependent DFT computations and our previous band assignments for MoVIO(OSiiPr3)(LCOOMe)2,9 we assigned band 1 as dominantly deriving from a HOMO → LUMO one-electron promotion with appreciable dithiolene A → (Mo dxy + dithiolene B) charge transfer character. The dominant contributor to band 2 is a HOMO − 1 → LUMO (Mo–S3dithiolene π → Mo–S3dithiolene π*) one-electron promotion with dithiolene A → Mo(dxy) charge transfer character. The computations also suggest a variable HOMO → LUMO + 2 contribution with benzoate acceptor character that leads to intensity differences in band 2.
Conclusions
New bis(ene-1,2-dithiolato)oxocarboxylatomolybdenum(VI) complexes have been synthesized as active site analogues of type II DMSOR family enzymes. Low temperature kinetic and spectroscopic analyses, in conjunction with DFT calculations, have been used to understand their formation and electronic structure. Provided DMSORox possesses a hexacoordinate geometry; these results suggest that the ancillary Oasp ligand likely functions to fine tune the redox potential of the Mo ion. Interestingly, a hexacoordinate geometry also allows for a specific pyranopterin dithiolene to couple the active site into long-range superexchange pathways for electron transfer regeneration of the catalytically relevant Mo(IV) site.This work was partly supported by grants (no. 23350027, 24108725 and 2410915 to H.S. and no. 22105007 to S.I.). M.L.K. acknowledges the National Institutes of Health (GM 057378) for financial support. The authors also thank Dr Kei Ohkubo and Prof. Shunichi Fukuzumi of Osaka University for their help in collecting the EPR spectrum.
Notes and references
- R. Hille, Chem. Rev., 1996, 96, 2757 CrossRef CAS PubMed; S. J. N. Burgmayer, Prog. Inorg. Chem., 2004, 52, 491 CrossRef; M. J. Romao, Dalton Trans., 2009, 4053 RSC; R. Hille, Dalton Trans., 2013, 42, 3029 RSC.
- H. Schindelin, C. Kisker, J. Hilton, K. V. Rajagopalan and D. C. Rees, Science, 1996, 272, 1615 Search PubMed; H.-K. Li, C. Temple, K. V. Rajagopalan and H. Schindelin, J. Am. Chem. Soc., 2000, 122, 7673 CrossRef CAS.
- C. A. McDevitt, P. Hugenholtz, G. R. Hanson and A. G. McEwan, Mol. Microbiol., 2002, 44, 1575 CrossRef CAS; A. G. McEwan, J. P. Ridge and C. A. McDevitt, Geomicrobiol. J., 2002, 19, 3 CrossRef.
- J. C. Boyington, V. N. Gladyshev, S. V. Khangulov, T. C. Stadtman and P. D. Sun, Science, 1997, 275, 1305 CrossRef CAS; J. M. Dias, M. E. Than, A. Humm, R. Huber, G. P. Bourenkov, H. D. Bartunik, S. Bursakov, J. Calvete, J. Calderia, C. Carneiro, J. J. G. Moura, I. Moura and M. J. Romao, Structure, 1999, 7, 65 CrossRef; M. Jormakka, S. Tornroth, B. Byrne and S. Iwata, Science, 2002, 295, 1863 CrossRef PubMed.
- M. G. Bertero, R. A. Rothery, M. Palak, C. Hou, D. Lim, F. Blasco, J. H. Weiner and N. C. J. Strynadka, Nat. Struct. Biol., 2003, 10, 681 CrossRef CAS PubMed; M. Jormakka, D. Richardson, B. Byrne and S. Iwata, Structures, 2004, 12, 95 CrossRef PubMed.
- J. H. Enemark, J. J. A. Cooney, J.-J. Wang and R. H. Holm, Chem. Rev., 2004, 104, 1175 CrossRef CAS PubMed; J. McMaster, J. M. Tunney and C. D. Garner, Prog. Inorg. Chem., 2004, 52, 539 CrossRef; H. Sugimoto and H. Tsukube, Chem. Soc. Rev., 2008, 37, 2609 RSC; F. J. Hine, A. J. Taylor and C. D. Garner, Coord. Chem. Rev., 2010, 254, 1570 CrossRef PubMed; C. Schulzke, Eur. J. Inorg. Chem., 2011, 1189 CrossRef.
- R. H. Holm, E. I. Solomon, A. Majumdar and A. Tenderholt, Coord. Chem. Rev., 2011, 255, 993 CrossRef CAS PubMed.
- B. S. Lim and R. H. Holm, J. Am. Chem. Soc., 2001, 123, 1920 CrossRef CAS PubMed.
- H. Sugimoto, S. Tatemoto, K. Suyama, H. Miyake, R. P. Mtei, S. Itoh and M. L. Kirk, Inorg. Chem., 2010, 49, 5368 CrossRef CAS PubMed.
- H. Sugimoto, S. Tatemoto, K. Toyota, M. Kubo, T. Ogura and S. Itoh, Chem. Commun., 2013, 49, 4358 RSC.
- H. Sugimoto, M. Harihara, M. Shiro, K. Sugimoto, K. Tanaka, H. Miyake and H. Tsukube, Inorg. Chem., 2005, 44, 6386 CrossRef CAS PubMed.
- P. Ray and N. K. Dutt, J. Indian Chem. Soc., 1943, 20, 81 CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental details for the preparation, low temperature measurements and DFT calculations (Tables S1–S2, Fig. S1–S8). See DOI: 10.1039/c3dt51485d |
| This journal is © The Royal Society of Chemistry 2013 |