 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Reactivity of Ir(III) carbonyl complexes with water: alternative by-product formation pathways in catalytic methanol carbonylation†‡
Paul I. P.
Elliott§
a,
Susanne
Haak
a,
Anthony J. H. M.
Meijer
a,
Glenn J.
Sunley
b and
Anthony
Haynes
*a
aDepartment of Chemistry, University of Sheffield, Sheffield, S3 7HF, UK. E-mail: a.haynes@sheffield.ac.uk
bBP Chemicals Limited, Hull Research and Technology Centre, Saltend, Hull, HU12 8DS, UK
First published on 17th September 2013
Abstract
The reactions of water with a number of iridium(III) complexes relevant to the mechanism for catalytic methanol carbonylation are reported. The iridium acetyl, [Ir(CO)2I3(COMe)]−, reacts with water under mild conditions to release CO2 and CH4, rather than the expected acetic acid. Isotopic labeling and kinetic experiments are consistent with a mechanism involving nucleophilic attack by water on a terminal CO ligand of [Ir(CO)2I3(COMe)]− to give an (undetected) hydroxycarbonyl species. Subsequent decarboxylation and elimination of methane gives [Ir(CO)2I2]−. Similar reactions with water are observed for [Ir(CO)2I3Me]−, [Ir(CO)2(NCMe)I2(COMe)] and [Ir(CO)3I2Me] with the neutral complexes exhibiting markedly higher rates. The results demonstrate that CO2 formation during methanol carbonylation is not restricted to the conventional water gas shift mechanism mediated by [Ir(CO)2I4]− or [Ir(CO)3I3], but can arise directly from key organo-iridium(III) intermediates in the carbonylation cycle. An alternative pathway for methane formation not involving the intermediacy of H2 is also suggested. A mechanism is proposed for the conversion MeOH + CO → CO2 + CH4, which may account for the similar rates of formation of the two gaseous by-products during iridium-catalysed methanol carbonylation.
Introduction
The carbonylation of methanol to acetic acid represents one of the most successful industrial scale applications of organometallic catalysis by transition metal complexes and has been primarily achieved using group 9 metals in combination with iodide co-catalysts.1–12 After the initial introduction by BASF of a cobalt-based process, higher activity and selectivity under milder conditions was identified by Monsanto for rhodium and iridium-based catalysts.13 The rhodium/iodide catalysed process was commercialised by Monsanto and has been operated, along with related variants, for more than 40 years. In 1995, BP Chemicals commercialised a promoted iridium/iodide catalysed methanol carbonylation process, Cativa™, which now operates at a number of sites worldwide.14,15 The Cativa™ process has a number of benefits compared to the rhodium-based process, including high activity and catalyst stability at low water concentrations. The iridium-based catalyst also gives reduced levels of liquid by-products and improved yield based on CO.For both the rhodium- and iridium-based processes, however, a significant side reaction is the water-gas-shift (WGS) reaction (eqn (1)).1,16 A mechanism proposed by Forster17 for the Ir-catalysed WGS reaction is shown in Scheme 1, involving anionic and neutral cycles. Oxidation of [Ir(CO)2I2]− or [Ir(CO)3I] by HI leads to a hydride complex that can react with a second equivalent of HI to release H2. Carbon dioxide then results from nucleophilic attack by water on an anionic or neutral Ir(III) iodocarbonyl complex, with reduction of the iridium back to Ir(I).
| H2O + CO → CO2 + H2 | (1) |
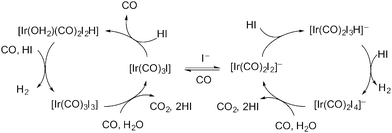 | ||
| Scheme 1 Proposed cycles for the iridium/iodide catalysed WGS reaction. | ||
The hydrogen that is formed in the WGS reaction can participate in other side reactions such as methane formation via the formal hydrogenolysis of methanol (eqn (2)). When coupled with the WGS reaction this results in the net conversion of methanol and CO into methane and CO2 (eqn (3)).18 Data reported previously show that CO2 and CH4 are formed at comparable rates (ca. 1% of the carbonylation rate) during Ru-promoted Ir-catalysed methanol carbonylation.14
| MeOH + H2 → CH4 + H2O | (2) |
| MeOH + CO → CO2 + CH4 | (3) |
We have shown previously that methane can be formed from the iridium methyl complex, [Ir(CO)2I3Me]−, by reaction with H2 (eqn (4)) or on heating in carboxylic acid solvents, presumably by protonolysis of the methyl ligand (eqn (5)).19
| [Ir(CO)2I3Me]− + H2 → [Ir(CO)2I3H]− + CH4 | (4) |
| [Ir(CO)2I3Me]− + RCO2H → [Ir(CO)2I3(O2CR)]− + CH4 | (5) |
Since [Ir(CO)2I3Me]− has been identified as the resting state for the iridium catalyst,14,17,20 its reactions with H2 (from the WGS reaction) or acetic acid (the major component of the reaction medium) can be considered plausible pathways for the formation of methane during catalytic carbonylation. In this paper we present results that suggest an alternative mechanism for formation of methane and CO2 from iridium species that participate in the carbonylation cycle. These reactions involve nucleophilic attack by water on a carbonyl ligand of an iridium methyl or acetyl complex, and occur without the intermediacy of H2.
Results and discussion
Reactivity of [Ir(CO)2I3(COMe)]− with water
Mechanistic cycles for iridium-catalysed methanol carbonylation generally depict the Ir(III) acetyl complex [Ir(CO)2I3(COMe)]− reacting with water to eliminate acetic acid (eqn (6)), either directly or via initial reductive elimination of acetyl iodide and subsequent hydrolysis.| [Ir(CO)2I3(COMe)]− + H2O → [Ir(CO)2I2]− + MeCO2H + HI | (6) |
The initial intention of the present study was to investigate the kinetics of this product-forming step of the carbonylation cycle. The isolation and structural characterization of both cis,fac and trans,mer isomers of [Ir(CO)2I3(COMe)]− have been reported previously.20–23 When the reaction of the cis,fac isomer with water was monitored spectroscopically under mild conditions (MeCN, 42 °C) an unexpected outcome resulted. In a typical series of IR spectra (Fig. 1) the decay of the reactant ν(CO) absorptions at 2110, 2062 and 1658 cm−1 is accompanied by the growth of new bands at 2046 and 1968 cm−1, assigned to [Ir(CO)2I2]−, consistent with the expected reaction (eqn (6)). However the IR spectra did not indicate the formation of any acetic acid in the region of 1750 cm−1. Instead, inspection of the region between 2200 and 2500 cm−1 revealed the appearance of an intense new band at 2342 cm−1 characteristic of the formation of CO2. The formation of CO2 in this reaction is indicative of nucleophilic attack by water on coordinated CO. Furthermore, since a dicarbonyl species is formed and no organic acyl product is detected, the observations are consistent with concomitant decarbonylation of the acetyl ligand according to the reaction stoichiometry shown in eqn (7). The analogous reaction of trans,mer-[Ir(CO)2I3(COMe)]− with water under the same conditions was also found to result in CO2 formation.
| [Ir(CO)2I3(COMe)]− + H2O → [Ir(CO)2I2]− + CO2 + CH4 + HI | (7) |
![IR spectra recorded during the reaction of cis,fac-[Ir(CO)2I3(COMe)]− (as its Ph4As+ salt) with H2O (0.56 mol dm−3) in MeCN at 42 °C. The region around 2300 cm−1 is masked due to the strong solvent ν(CN) band.](/image/article/2013/DT/c3dt52092g/c3dt52092g-f1.gif) | ||
| Fig. 1 IR spectra recorded during the reaction of cis,fac-[Ir(CO)2I3(COMe)]− (as its Ph4As+ salt) with H2O (0.56 mol dm−3) in MeCN at 42 °C. The region around 2300 cm−1 is masked due to the strong solvent ν(CN) band. | ||
Further evidence is provided by the detection of methane. In an NMR tube experiment, a solution of cis,fac-[Ir(CO)2I3(COMe)]− in d3-MeCN containing water (1% v/v) was heated to 35 °C for 2 days, resulting in the appearance of a 1H resonance at δ 0.19, assigned to dissolved methane. When the experiment was repeated with the 13C-labelled analogue, [Ir(CO)2I3(CO13CH3)]−, a doublet (JCH = 125.5 Hz) was observed at the same chemical shift corresponding to the formation of 13CH4.
The results presented above do not distinguish whether the CO2 is derived from one of the terminal CO ligands or the acetyl carbonyl of [Ir(CO)2I3(COMe)]−. In principle, nucleophilic attack by water on a CO ligand could occur either before or after the decarbonylation step. We therefore undertook isotopic labeling experiments to elucidate the mechanism further. We first sought to prove the participation of added water in the formation of CO2 using ca. 10% 18O enriched water. The experiment was performed using THF as solvent to avoid interference from the strong solvent ν(CN) absorption encountered with acetonitrile. A spectrum recorded during this reaction (Fig. 2) shows the band for C16O2 at 2337 cm−1 as well as a weaker band at 2324 cm−1 corresponding to C16O18O. The formation of this isotopologue provides direct evidence of the involvement of added water in the formation of CO2.
![IR absorptions for CO2 formed in the reaction of cis,fac-[Ir(CO)2I3(COMe)]− with water (10.3% 18O labelled) in THF at 42 °C showing bands for C16O2 and C16O18O.](/image/article/2013/DT/c3dt52092g/c3dt52092g-f2.gif) | ||
| Fig. 2 IR absorptions for CO2 formed in the reaction of cis,fac-[Ir(CO)2I3(COMe)]− with water (10.3% 18O labelled) in THF at 42 °C showing bands for C16O2 and C16O18O. | ||
To distinguish whether a terminal or acyl carbonyl is the origin of the observed CO2, a 13C-labelled sample of cis,fac-[Ir(CO)2I3(COMe)]− was prepared. Initially the methyl complex [Ir(*CO)2I3Me]− was synthesised with ca. 65% 13C enrichment of the CO ligands. Carbonylation of this complex using non-enriched CO (10 bar) resulted in the acetyl complex [Ir(CO)2I3(*COMe)]− in which ca. 30–35% 13C label was retained in the acetyl carbonyl position but the label was largely lost from the terminal carbonyl sites (ca. 5% as judged from the IR spectrum). A series of IR spectra recorded during the reaction of this labelled compound with water in THF is shown in Fig. 3. It is clear that the CO2 formed is largely unlabelled, with only a weak band due to 13CO2. This matches the level of 13C enrichment in the terminal CO ligands of the reactant and proves that the CO2 is derived from a terminal CO ligand of [Ir(CO)2I3(*COMe)]−. Consistent with this, the IR spectra indicate formation of a ca. 2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 mixture of [Ir(12CO)2I2]− (2036, 1958 cm−1) and [Ir(12CO)(13CO)I2]− (2020, 1930 cm−1), indicating that the 13C label from the acetyl group is retained as a terminal CO ligand in the product complex. The outcome of the labeling experiments is summarized in Scheme 2.
:1 mixture of [Ir(12CO)2I2]− (2036, 1958 cm−1) and [Ir(12CO)(13CO)I2]− (2020, 1930 cm−1), indicating that the 13C label from the acetyl group is retained as a terminal CO ligand in the product complex. The outcome of the labeling experiments is summarized in Scheme 2.
![Series of IR spectra during the reaction of cis,fac-[Ir(CO)2I3(*COMe)]− with water (0.56 mol dm−3) in THF at 42 °C.](/image/article/2013/DT/c3dt52092g/c3dt52092g-f3.gif) | ||
| Fig. 3 Series of IR spectra during the reaction of cis,fac-[Ir(CO)2I3(*COMe)]− with water (0.56 mol dm−3) in THF at 42 °C. | ||
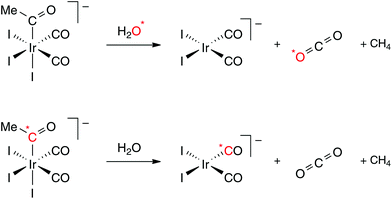 | ||
| Scheme 2 Isotopic labelling experiments. | ||
Kinetics
The kinetic behaviour of the reaction of cis,fac-[Ir(CO)2I3(COMe)]− with water was assessed from plots of IR absorbance vs. time. Under pseudo-first order conditions (excess water), the decay of the 2110 cm−1 band for the reactant was fitted approximately by an exponential curve, although there was some deviation from ideal behaviour. This might arise from the change in acidity of the reaction solution with time, due to the release of HI according to eqn (6). A somewhat better fit to first order behaviour was found for the growth of the band due to CO2 at 2342 cm−1. Values of a pseudo first order rate constant, kobs, were therefore obtained by analysis of this absorption to determine the effect of water concentration. A plot of kobsvs. [H2O] is reasonably linear (Fig. 4) with an intercept close to the origin, and the slope gives an estimated second order rate constant of 1.2(±0.1) × 10−4 dm3 mol−1 s−1 at 42 °C.24 The approximate second order kinetics are consistent with a bimolecular reaction between the iridium complex and water. A detailed kinetic study was not undertaken for the reaction of water with trans,mer-[Ir(CO)2I3(COMe)]−, but the rate for this isomer was found to be marginally slower than the cis,fac complex under the same conditions.![Plot of kobsvs. [H2O] for reaction of cis,fac-[Ir(CO)2I3(COMe)]− with water (MeCN, 42 °C).](/image/article/2013/DT/c3dt52092g/c3dt52092g-f4.gif) | ||
| Fig. 4 Plot of kobsvs. [H2O] for reaction of cis,fac-[Ir(CO)2I3(COMe)]− with water (MeCN, 42 °C). | ||
Since the observed reaction products implicate loss of an iodide ligand, we tested the effect of iodide salt on the rate. Addition of Bu4NI caused a modest promotional effect (e.g. by a factor of ca. 2 at 0.15 M Bu4NI). The results are therefore consistent with a direct nucleophilic attack by water on a CO ligand of the anionic reactant complex, since a mechanism involving initial iodide dissociation would result in rate inhibition by iodide salt.
Reactivity of other anionic Ir(III) complexes
In situ IR spectroscopy has identified [Ir(CO)2I3Me]− as the dominant catalyst species during catalytic methanol carbonylation and [Ir(CO)2I4]− can also accumulate as a resting state in the competing WGS reaction.14,17,20 The reactions of both of these anionic complexes with water have been studied under comparable conditions to those used for [Ir(CO)2I3(COMe)]− (42 °C in MeCN or THF). In each case CO2 formation was apparent from the growth of an IR band at ca. 2340 cm−1. Reactions using 13CO enriched [Ir(CO)2I3Me]− or [Ir(CO)2I4]− gave 12CO2 and 13CO2 in the expected ratio based on the level of isotopic enrichment of the metal complex. The IR spectra again indicate formation of [Ir(CO)2I2]−, although the maximum possible yield of this dicarbonyl is 50% in the absence of added CO (assuming conversion of one CO ligand per reactant complex to CO2). The reactions shown in eqn (8) and (9) include a non-carbonyl iridium(I) side-product, “[IrI2]−” to account for this. In the presence of added CO this would be expected to be converted into [Ir(CO)2I2]−.| [Ir(CO)2I3Me]− + H2O → 1/2[Ir(CO)2I2]− + 1/2“[IrI2]−” + CO2 + CH4 + HI | (8) |
| [Ir(CO)2I4]− + H2O → 1/2[Ir(CO)2I2]− + 1/2“[IrI2]−” + CO2 + 2HI | (9) |
The reactions with water observed for the series of complexes [Ir(CO)2I3R]− (R = I, Me, COMe), demonstrate that CO2 formation during Ir-catalysed methanol carbonylation is not restricted to the conventional WGS cycle involving attack by water on [Ir(CO)2I4]− or [Ir(CO)3I3]. Susceptibility to nucleophilic attack by water clearly extends to other Ir(III) complexes and can be coupled with methane formation when R = Me or COMe.
Reactivity of neutral Ir(III) complexes
Mechanistic investigations of iridium-catalysed methanol carbonylation have established the participation of neutral as well as anionic complexes.17,20,25,26 We therefore extended our study to neutral Ir(III) acetyl and methyl complexes, which are expected to be more electrophilic than the anionic species. Abstraction of iodide from cis,fac-[Ir(CO)2I3(COMe)]− in acetonitrile was accomplished by reaction with a small excess of InI3, as previously reported for the analogous methyl complex, cis,fac-[Ir(CO)2I3Me]−.20 The IR spectrum of the product in MeCN showed ν(CO) bands at 2136, 2094 and 1701 cm−1. The shift of each absorption to high frequency with respect to the anionic precursor is consistent with loss of an iodide ligand to give a solvated neutral complex, [Ir(CO)2(NCMe)I2(COMe)], by analogy with the previously reported [Ir(CO)2(NCMe)I2Me].20,27 Addition of water to this solution resulted in formation of CO2 and absorptions at 2077 and 2006 cm−1 that are assigned to [Ir(CO)2I(NCMe)]27 along with a very small amount of [Ir(CO)2I2]−. Hence, the results are analogous to those for the anionic system, and accord with the reaction in eqn (10). A detailed kinetic study was not undertaken, but a pseudo-first order rate constant of 1.04 × 10−3 s−1 was measured at 34 °C, which is an order of magnitude greater than that for [Ir(CO)2I3(COMe)]− under the same conditions.| [Ir(CO)2(NCMe)I2(COMe)] + H2O → [Ir(CO)2(NCMe)I] + CO2 + CH4 + HI | (10) |
Another significant species in the catalytic carbonylation mechanism is the methyl complex [Ir(CO)3I2Me], formed upon substitution of an iodide ligand in [Ir(CO)2I3Me]− by CO. We have previously characterised this tricarbonyl as the fac,cis isomer using in situ high pressure IR and NMR spectroscopy.26 This neutral species undergoes migratory CO insertion much more readily than the anion, explaining the promotional effect on the carbonylation process of certain species that bind iodide.20
In the present investigation, high pressure IR spectroscopy was used to probe the reactivity of [Ir(CO)3I2Me] towards water. The tricarbonyl was generated from the dimer, [Ir(CO)2I2Me]2 under 20 bar CO in CH2Cl2–THF (3:1) and identified by its ν(CO) bands at 2156, 2116 and 2098 cm−1. In the presence of water, at ambient temperature, these absorptions decayed and new bands grew at 2072 and 2042 cm−1, corresponding to the Ir(I) tricarbonyl, [Ir(CO)3I]. The eventual IR spectrum was more complex and also indicated formation of the anionic species, [Ir(CO)2I3Me]− and [Ir(CO)2I2]− (vide infra). GC-MS analysis of the headspace of the IR cell at the end of the reaction demonstrated the presence of methane and CO2 but no organic acetyl products (e.g. acetic acid) were detected. Isotopic labeling experiments confirmed that the iridium complex is the source of both gaseous products; CD3H was generated from the CD3-labelled complex and a significant quantity of 13CO2 resulted from the 13CO-enriched complex, despite the large excess of natural abundance CO used. The observations are therefore consistent with eqn (11). Since HI is formed during the reaction, this can act as a source of iodide that can be scavenged by either [Ir(CO)3I2Me] or [Ir(CO)3I] to give the anions [Ir(CO)2I3Me]− or [Ir(CO)2I2]−, as observed by IR spectroscopy.
| [Ir(CO)3I2Me] + H2O + CO → [Ir(CO)3I] + CO2 + CH4 + HI | (11) |
Kinetic data were obtained by analyzing the exponential decay of the high frequency ν(CO) band of [Ir(CO)3I2Me] at 2157 cm−1 to give values of a pseudo-first order rate constant, kobs. A plot of kobsvs. [H2O] (Fig. 5) showed an approximate linear dependence on water concentration. The non-zero intercept may arise from contributions to the measured kobs values from side reactions such as those noted above involving HI. Kinetic measurements were also made for reactions using D2O and reveal a small kinetic isotope effect, kH/kD of ca. 1.3. This is similar to the value reported previously for the reaction of [TpIr(CO)2] with water (Tp = HBpz3, pz = pyrazolyl).28 The reactivity of [Ir(CO)3I2Me] is more than an order of magnitude higher than that reported above for [Ir(CO)2I3(COMe)]−,29 exemplifying the more facile nucleophilic attack by water on the carbonyl ligand of a neutral complex relative to an anion.
![Plot of kobsvs. [H2O] for the reaction of [Ir(CO)3I2Me] with water in 3 : 1 CH2Cl2–THF, 20 bar CO at 23 °C.](/image/article/2013/DT/c3dt52092g/c3dt52092g-f5.gif) | ||
| Fig. 5 Plot of kobsvs. [H2O] for the reaction of [Ir(CO)3I2Me] with water in 3:1 CH2Cl2–THF, 20 bar CO at 23 °C. | ||
Mechanism
The results demonstrate that a range of Ir(III) complexes of relevance to catalytic methanol carbonylation react with water under relatively mild conditions to liberate CO2. For complexes that also contain an alkyl or acetyl ligand, formation of methane is also observed, resulting from (formal) proton transfer from water to a methyl moiety. Scheme 3 shows some alternative pathways for the reaction of [Ir(CO)2I3(COMe)]− with water. Initial nucleophilic attack by water on coordinated CO to form a hydroxycarbonyl complex B could occur with concerted loss of HI or in a stepwise manner via a di-anionic intermediate A. The absence of rate inhibition on addition of iodide salt suggests that initial substitution of an iodide ligand by water is not required, although an alternative mechanism in which water coordinates by substitution of a CO ligand, followed by intramolecular nucleophilic attack, might be considered. Iridium(III) hydroxycarbonyl species have been reported previously, e.g. [IrCl2(CO2H)(CO)L2] (L = PMe2Ph, AsMe2Ph),30,31 [TpIr(CO)(CO2H)H] (Tp = HBpz3− or HB(3,5-Me2pz)3−)28,32,33 and [TpmIr(CO)(CO2H)H]+ (Tpm = HC(3,5-Me2pz)3).34 The proposed reactive intermediate B has a vacant site that could mediate decarboxylation via β-H transfer to give a hydride, C, followed by migration of methyl from CO to the Ir center and reductive elimination of methane from the hydrido methyl complex D. Alternatively, B might undergo migratory de-insertion to give E, which could eliminate CO2 and methane in a stepwise fashion (via hydride D) or in a concerted mechanism involving intramolecular proton transfer from the hydroxycarbonyl ligand to methyl. Similar mechanisms can be envisaged starting from the Ir methyl complexes [Ir(CO)2I3Me]− and [Ir(CO)3I2Me]. For example nucleophilic attack by water on a carbonyl ligand of [Ir(CO)3I2Me] would lead to the hydroxycarbonyl intermediate E in Scheme 3.![Possible mechanisms for reaction of cis,fac-[Ir(CO)2I3(COMe)]− with water.](/image/article/2013/DT/c3dt52092g/c3dt52092g-s3.gif) | ||
| Scheme 3 Possible mechanisms for reaction of cis,fac-[Ir(CO)2I3(COMe)]− with water. | ||
Preliminary DFT calculations on intermediate E resulted in optimized structures with two alternative conformations of the hydroxycarbonyl ligand. The lowest energy conformation has a hydrogen-bonding interaction between the CO2H group and a cis iodide ligand as illustrated in Fig. 6a. An isomer of E with methyl and hydroxycarbonyl ligands mutually trans was calculated to be ca. 47 kJ mol−1 higher in energy. A transition state for H-transfer to the methyl ligand was located (Fig. 6b) with a calculated ΔG‡298 (relative to the lowest energy conformer of E) of 118 kJ mol−1 (in the gas phase) or 109 kJ mol−1 (using the PCM solvation model with acetonitrile as solvent). The transition state exhibits lengthening of both the Ir–CH3 and Ir–CO2H bonds (by ca. 0.22 and 0.10 Å respectively) compared to complex E. Calculations were also carried out for a neutral 5-coordinate analogue of E with the iodide ligand trans to CO2H omitted from the model. A similar transition state for H-transfer to methyl was located, with somewhat lower ΔG‡298 values of 83 kJ mol−1 (gas phase) or 92 kJ mol−1 (PCM, MeCN).35 Since the calculated activation barriers appear a little high to be consistent with the facile elimination of CO2 and CH4 observed experimentally, it is possible that more specific solvation effects that are not accounted for by the PCM method play a role. Further computational studies of these systems are in progress.
![DFT optimized structures for (a) complex E, [Ir(CO)2(CO2H)I2Me]− and (b) transition state for H transfer to the methyl ligand.](/image/article/2013/DT/c3dt52092g/c3dt52092g-f6.gif) | ||
| Fig. 6 DFT optimized structures for (a) complex E, [Ir(CO)2(CO2H)I2Me]− and (b) transition state for H transfer to the methyl ligand. | ||
Relation to catalytic carbonylation and water gas shift reactions
Conventionally the methanol carbonylation and WGS reactions are depicted as occurring via coupled catalytic cycles in which Ir(I) reacts with either methyl iodide, to initiate the carbonylation cycle, or with HI to initiate the WGS cycle.12,17,22 Carbon dioxide then results from nucleophilic attack of water on an Ir(III) iodocarbonyl, [Ir(CO)2I4]− or [Ir(CO)3I3]. The present study has demonstrated that CO2 formation can also occur by reaction of water with Ir(III) species that are part of the methanol carbonylation cycle, i.e. [Ir(CO)2I3R]− (R = Me, COMe) and [Ir(CO)3I2Me]). Scheme 4 shows a proposed catalytic cycle for the net conversion of methanol and carbon monoxide to methane and CO2 (eqn (3)). Oxidative addition of methyl iodide to [Ir(CO)2I2]− proceeds in the same manner as for the methanol carbonylation cycle. The resulting Ir(III) methyl species then reacts with water and CO to release CO2 and methane and regenerate [Ir(CO)2I2]− and HI.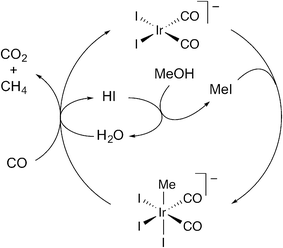 | ||
| Scheme 4 Proposed cycle for combined CO2/CH4 formation under catalytic carbonylation conditions. | ||
The proposed cycle shown in Scheme 4 provides a mechanism for combined CO2/CH4 formation (eqn (3)) without the intermediacy of H2 that would be generated by a conventional WGS reaction. Analogous cycles can be drawn in which water reacts with any of the Ir(III) methyl or acetyl complexes that participate in the methanol carbonylation mechanism. Indeed, under a high pressure of CO, the reaction of [Ir(CO)2I3Me]− with water shown in Scheme 4 might actually involve initial substitution of an iodide ligand by CO to give the more electrophilic neutral tricarbonyl, [Ir(CO)3I2Me]. Likewise, CO2 formation could occur from an acetyl species (as shown in Scheme 3) after migratory CO insertion has occurred. Previously reported data from a pilot plant unit operating under steady state conditions show that the rates of CO2 and methane formation are very similar in Ir/Ru catalysed reactions, at ca. 1% of the carbonylation rate.14 The close correspondence of CO2 and CH4 formation rates is consistent with a mechanism such as that shown in Scheme 4.
Since our results indicate that these CO2-forming reactions occur readily under mild conditions, an obvious question is why high selectivity to acetic acid is achieved in iridium-catalysed methanol carbonylation. The formation of CO2 and CH4 (eqn (3)) is favored thermodynamically with respect to acetic acid (ΔG298 = −57.7 kJ mol−1 for MeCO2H → CH4 + CO2)36 so the selectivity is governed by kinetic considerations. Under process conditions, the key steps that facilitate turnover in the carbonylation cycle must compete effectively with the CO2-forming reactions. Hence, the rates of migratory CO insertion (for Ir-methyl species) and elimination of acetyl iodide/acetic acid (for Ir-acetyl species) must exceed that of nucleophilic attack by water on coordinated CO. The different conditions used for the model reactions reported here and the catalytic process are presumably crucial in determining the outcomes. For two competing reactions, the one with lower activation energy will dominate at lower temperature but the Arrhenius relationship shows that the rate of a reaction with higher activation energy will increase more as the temperature is raised, and so it can become dominant at higher temperature. Since the temperature difference between the catalytic and model reactions here is ∼150 °C, the relative reaction rates can be expected to be markedly different in the two systems. The reaction medium may also be important. In particular, the catalytic process operates under acidic conditions (MeCO2H–HI–H2O) that will influence the behaviour of hydroxycarbonyl species. Under acidic conditions, hydroxycarbonyl complexes are known to undergo dehydroxylation to reform a terminal carbonyl ligand whereas decarboxylation is often promoted by basic conditions.28,31,33,37,38 Hence, CO2 forming reactions may be inhibited in the reaction medium used for catalytic carbonylation.
The overall rate of CO2 formation will result from a combination of the possible pathways and our data do not demonstrate whether one particular iridium species is the principal source of CO2. However, the neutral tricarbonyl, [IrMe(CO)3I2] has been shown to be particularly reactive towards water and is also thought to be the dominant species in which migratory CO insertion occurs. Therefore it may be speculated that these two reactions of [IrMe(CO)3I2] form a branching point in the mechanisms of acetic acid and CO2/CH4 formation and therefore determine selectivity. Analogous reactions of iridium acetyl species might also be responsible for the formation of other by-products like acetaldehyde, a precursor (via hydrogenation and carbonylation steps) to propionic acid that is reported to be present at levels of 290–1150 ppm in the acetic acid product in pilot plant studies.14 Thus, nucleophilic attack by water on a terminal carbonyl ligand followed by proton transfer to acetyl would give a route to CO2/MeCHO formation. Although [Ir(CO)2I3(COMe)]− reacts with water to release CO2 and CH4 (Scheme 2) the high CO pressure during catalytic carbonylation may inhibit the decarbonylation step proposed in Scheme 3 and allow acetaldehyde elimination from species such as A, or B.
Conclusions
This study has identified a pathway for the co-formation of CO2 and CH4 from iridium methyl and acetyl complexes that participate in iridium/iodide catalysed methanol carbonylation. Isotopic labeling experiments demonstrate that, on reaction with water under mild conditions, a terminal CO ligand of [Ir(CO)2I3(COMe)]− is liberated as CO2 and decarbonylation of the acetyl moiety leads to loss of the methyl fragment as CH4. A mechanism involving nucleophilic attack by water on coordinated CO to form a hydroxycarbonyl ligand is implicated, followed by decarboxylation and methane elimination in stepwise or concerted fashion. Analogous reactions occur for [Ir(CO)2I3Me]−, [Ir(CO)2(NCMe)I2(COMe)] and [Ir(CO)3I2Me], with the neutral species being an order of magnitude more reactive than the anions. A particularly notable reaction is that of [Ir(CO)3I2Me] which has been shown previously to undergo facile migratory CO insertion. The relative rates of methyl migration and attack by water on this complex may be significant in determining catalytic selectivity. The results show that the formation of CO2 and CH4 can occur in a coupled fashion, possibly from an intermediate such as E (Scheme 3), consistent with the similar rates of formation of these two gaseous by-products in catalytic reactions. Preliminary DFT calculations identified a transition state for direct H transfer from hydroxycarbonyl to methyl in E, resulting in concerted elimination of CO2 and CH4.The observed reactions provide an alternative mechanism for methane formation in addition to the protonolysis and hydrogenolysis of iridium methyl complexes for which evidence has been presented previously. The new mechanism is closely related to that of the WGS reaction but does not involve the intermediacy of H2. It is likely that the gaseous by-products arise from multiple pathways during catalytic carbonylation and the relative contributions of the different routes likely depends on the process conditions employed. Nonetheless, the results presented in this paper provide further mechanistic insight into an important industrial process.
Experimental
Solvents were purified by distillation or using a column purification system. Methyl iodide (Sigma-Aldrich) was distilled over calcium hydride and stored at 5 °C in a foil-wrapped Schlenk tube under nitrogen and over mercury. Other reagents were used as supplied: Ph4AsCl (Sigma-Aldrich), IrCl3·nH2O (PMO Pty Ltd); carbon monoxide (BOC CP grade); 13C-enriched carbon monoxide (99% 13C, Euriso-top). Standard Schlenk techniques and glassware were used for preparative reactions. The iridium compounds Ph4As[Ir(CO)2I2], Ph4As[Ir(CO)2I3Me], Ph4As[Ir(CO)2I3(COMe)], [IrMe(CO)2I2]2 (and their 13C labelled analogues) were synthesised using published methods.20,22,391H NMR spectra were recorded on a Bruker AC250 instrument in pulsed Fourier transform mode, fitted with a Bruker B ACS-60 sample changer, and using solvent as internal reference. GC and GC-MS measurements were made using Perkin-Elmer Autosystem XL and TurboMass instruments respectively (capillary column PE-1, 60 m × 0.320 mm, df = 5.00 μm; temperature program 0 °C for 5 min, raise 10° min−1 up to 200 °C, 200 °C for 5 min).Solution-phase infrared spectra were recorded on a Mattson Genesis FTIR spectrometer controlled by WinFirst software using a CaF2 liquid cell (0.5 mm pathlength). For kinetic experiments, the cell was maintained at the desired temperature by use of a thermostatted water jacket and circulating water bath. In a typical experiment, a solution of water at the appropriate concentration in MeCN or THF was prepared in a 5 cm3 graduated flask. A portion of this solution was transferred to the IR cell to record a background spectrum. The reaction was then initiated by dissolving the iridium compound (ca. 5 mg) in 1 cm3 of the water-containing solution. After thorough mixing, a portion of the resulting solution was transferred to fill the pre-equilibrated IR cell. IR spectra were recorded at programmed time-intervals under computer control. Absorbance vs. time data were extracted for analysis using Kaleidagraph curve-fitting software. Observed rate constants are tabulated in the ESI.‡
Reactions of [Ir(CO)3I2Me] with water were monitored under CO pressure using a cylindrical internal reflectance (CIR) cell comprising an autoclave (Parr) modified (by SpectraTech) to accommodate a crystalline silicon CIR rod as described by Moser.40,41 Spectra were recorded using a Perkin-Elmer 1710 FTIR spectrometer fitted with an MCT detector. In a typical experiment, [Ir(CO)2I2Me]2 (160 mg) was dissolved in a 3:1 mixture of CH2Cl2–THF (8 cm3) containing the required concentration of water. The resulting solution was transferred to the CIR cell (maintained at 23 °C) that was then sealed and purged three times with CO. The cell was then pressurized to 20 bar CO with continuous stirring to generate [Ir(CO)3I2Me] (2156, 2116 and 2098 cm−1) as described previously.20,26 IR spectra were recorded at intervals under these conditions to monitor the evolution of ν(CO) absorptions with time. After completion of the reaction a sample of the cell head-space was collected by venting into a glass tube. A 300 μl sample was analysed by GC-MS (split mode 1/20, 0 °C). Experiments using [Ir(CO)2I2(CD3)]2 and [Ir(13CO)2I2Me]2 were conducted using the same procedure.
Density functional theory (DFT) calculations were performed using the Gaussian 09 program package,42 compiled using the Portland compiler (version 8.0-2) on an EMT64 architecture using Gaussian-supplied versions of BLAS and ATLAS.43,44 All calculations employed the B3LYP functional45 with Stuttgart/Dresden pseudopotentials46,47 on iridium and iodine and the 6-311G** basis set on all other atoms.48,49 This basis set/functional combination has resulted in (semi-) quantitative agreement with experiment in previous work.50–52 Geometry optimizations were performed using the default settings. Frequency calculations confirmed the absence of imaginary frequencies for minimum energy structures and the presence of a single imaginary frequency for transition state structures. Visual inspection showed this frequency to correspond to the reaction coordinate for H-transfer. Medium effects were modeled using the PCM method,53,54 with acetonitrile as solvent. Cartesian coordinates and relative energies of optimised structures and are provided in the ESI.‡
Acknowledgements
We thank the EPSRC and BP Chemicals Limited for funding this research.References
- D. Forster, Adv. Organomet. Chem., 1979, 17, 255–267 CrossRef CAS.
- T. W. Dekleva and D. Forster, Adv. Catal., 1986, 34, 81–130 CAS.
- M. J. Howard, M. D. Jones, M. S. Roberts and S. A. Taylor, Catal. Today, 1993, 18, 325–354 CrossRef.
- P. M. Maitlis, A. Haynes, G. J. Sunley and M. J. Howard, J. Chem. Soc., Dalton Trans., 1996, 2187–2196 RSC.
- N. Yoneda, S. Kusano, M. Yasui, P. Pujado and S. Wilcher, Appl. Catal., A, 2001, 221, 253–265 CrossRef CAS.
- P. Torrence, in Applied Homogeneous Catalysis with Organometallic Compounds, ed. B. Cornils and W. A. Herrmann, Wiley-VCH, Weinheim, 2nd edn, 2002, vol. 1, pp. 104–136 Search PubMed.
- P. Maitlis and A. Haynes, in Metal-Catalysis in Industrial Organic Processes, ed. P. Maitlis and G. P. Chiusoli, RSC, 2006, pp. 114–162 Search PubMed.
- G. E. Morris, in Mechanisms in Homogeneous Catalysis; a Spectroscopic Approach, ed. B. T. Heaton, Wiley-VCH, Weinheim, 2005, pp. 195–230 Search PubMed.
- A. Haynes, in Catalytic Carbonylation Reactions, ed. M. Beller, Springer, Berlin, 2006, vol. 18, pp. 179–205 Search PubMed.
- A. Haynes, in Comprehensive Organometallic Chemistry III, ed. R. H. Crabtree and D. M. P. Mingos, Elsevier, 2006, vol. 7, pp. 427–444 Search PubMed.
- P. Kalck and P. Serp, in Iridium Complexes in Organic Synthesis, ed. L. A. Oro and C. Claver, Wiley-VCH, Weinheim, 2009, pp. 195–209 Search PubMed.
- A. Haynes, Adv. Catal., 2010, 53, 1–45 CAS.
- F. E. Paulik and J. F. Roth, Chem. Commun., 1968, 1578 RSC.
- G. J. Sunley and D. J. Watson, Catal. Today, 2000, 58, 293–307 CrossRef CAS.
- J. H. Jones, Platinum Met. Rev., 2000, 44, 94–105 CAS.
- E. C. Baker, D. E. Hendriksen and R. Eisenberg, J. Am. Chem. Soc., 1980, 102, 1020–1027 CrossRef CAS.
- D. Forster, J. Chem. Soc., Dalton Trans., 1979, 1639–1645 RSC.
- D. Forster and T. C. Singleton, J. Mol. Catal., 1982, 17, 299–314 CrossRef CAS.
- T. Ghaffar, J. P. H. Charmant, G. J. Sunley, G. E. Morris, A. Haynes and P. M. Maitlis, Inorg. Chem. Commun., 2000, 3, 11–12 CrossRef CAS.
- A. Haynes, P. M. Maitlis, G. E. Morris, G. J. Sunley, H. Adams, P. W. Badger, C. M. Bowers, D. B. Cook, P. I. P. Elliott, T. Ghaffar, H. Green, T. R. Griffin, M. Payne, J. M. Pearson, M. J. Taylor, P. W. Vickers and R. J. Watt, J. Am. Chem. Soc., 2004, 126, 2847–2861 CrossRef CAS PubMed.
- M. Volpe, G. Wu, A. Iretskii and P. C. Ford, Inorg. Chem., 2006, 45, 1861–1870 CrossRef CAS PubMed.
- S. Gautron, N. Lassauque, C. Le Berre, L. Azam, R. Giordano, P. Serp, G. Laurenczy, J.-C. Daran, C. Duhayon, D. Thiébaut and P. Kalck, Organometallics, 2006, 25, 5894–5905 CrossRef CAS.
- A. Haynes, A. J. H. M. Meijer, J. R. Lyons and H. Adams, Inorg. Chem., 2009, 48, 28–35 CrossRef CAS PubMed.
- In view of the approximate pseudo-first order kinetic behaviour observed, we do not assign any mechanistic significance to the non-zero intercept in the absence of further evidence.
- J. M. Pearson, A. Haynes, G. E. Morris, G. J. Sunley and P. M. Maitlis, J. Chem. Soc., Chem. Commun., 1995, 1045–1046 RSC.
- T. Ghaffar, H. Adams, P. M. Maitlis, G. J. Sunley, M. J. Baker and A. Haynes, Chem. Commun., 1998, 1023–1024 RSC.
- A. Haynes, P. M. Maitlis, I. A. Stanbridge, S. Haak, J. M. Pearson, H. Adams and N. A. Bailey, Inorg. Chim. Acta, 2004, 357, 3027–3037 CrossRef CAS.
- P. I. P. Elliott, C. E. Haslam, S. E. Spey and A. Haynes, Inorg. Chem., 2006, 45, 6269–6275 CrossRef CAS PubMed.
- For example, for [Ir(CO)3I2Me] (in 3:1 CH2Cl2–THF, 1.04 M H2O, 23 °C) kobs = 1.55 × 10−3 s−1 compared with 3.64 × 10−5 s−1 for [Ir(CO)2I3(COMe)]− (in MeCN, 0.56 M H2O, 25 °C).
- A. J. Deeming and B. L. Shaw, J. Chem. Soc. A, 1969, 443–446 RSC.
- A. J. Deeming and G. P. Proud, J. Organomet. Chem., 1986, 301, 385–390 CrossRef CAS.
- E. Gutierrez-Puebla, A. Monge, M. C. Nicasio, P. J. Perez, M. L. Poveda, L. Rey, C. Ruiz and E. Carmona, Inorg. Chem., 1998, 37, 4538–4546 CrossRef CAS PubMed.
- M. J. Fernandez, M. J. Rodriguez and L. A. Oro, J. Organomet. Chem., 1992, 438, 337–342 CrossRef CAS.
- I. I. Padilla-Martinéz, M. L. Poveda and E. Carmona, Organometallics, 2002, 21, 93–104 CrossRef.
- For comparison, a transition state was also located for reductive elimination of methane from intermediate D, giving a ΔG‡298 of 88 kJ mol−1 (gas phase).
- NIST Chemistry WebBook, NIST Standard Reference Database Number 69, ed. P. J. Linstrom and W. G. Mallard, National Institute of Standards and Technology, Gaithersburg, MD, http://webbook.nist.gov, (retrieved June 13, 2013) Search PubMed.
- K. Bowman, A. J. Deeming and G. P. Proud, J. Chem. Soc., Dalton Trans., 1985, 857–860 RSC.
- M. A. Bennett, G. B. Robertson, A. Rokicki and W. A. Wickramasinghe, J. Am. Chem. Soc., 1988, 110, 7098–7105 CrossRef CAS.
- S. Gautron, R. Giordano, C. Le Berre, J. Jaud, J.-C. Daran, P. Serp and P. Kalck, Inorg. Chem., 2003, 42, 5523–5530 CrossRef CAS PubMed.
- W. R. Moser, J. E. Cnossen, A. W. Wang and S. A. Krouse, J. Catal., 1985, 95, 21–32 CrossRef CAS.
- W. R. Moser, B. J. Marshik-Guerts and S. J. Okrasinski, J. Mol. Catal. A: Chem., 1999, 143, 57–69 CrossRef CAS.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. A. Montgomery, Jr., J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, N. J. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, Ö. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski and D. J. Fox, Gaussian 09, Revision C.01, Gaussian, Inc., Wallingford, CT, 2009 Search PubMed.
- R. C. Whaley and A. Petitet, Software: Pract. Exper., 2005, 35, 101–121 CrossRef.
- R. C. Whaley, A. Petitet and J. J. Dongarra, Parallel Comput., 2001, 27, 3–35 CrossRef.
- A. D. Becke, J. Chem. Phys., 1993, 98, 5648–5652 CrossRef CAS.
- X. Cao and M. Dolg, J. Chem. Phys., 2001, 115, 7348–7355 CrossRef CAS.
- A. Nicklass, M. Dolg, H. Stoll and H. Preuss, J. Chem. Phys., 1995, 102, 8942–8952 CrossRef CAS.
- A. D. McLean and G. S. Chandler, J. Chem. Phys., 1980, 72, 5639–5648 CrossRef CAS.
- R. Krishnan, J. S. Binkley, R. Seeger and J. A. Pople, J. Chem. Phys., 1980, 72, 650–654 CrossRef CAS.
- S. P. Foxon, C. Green, M. G. Walker, A. Wragg, H. Adams, J. A. Weinstein, S. C. Parker, A. J. H. M. Meijer and T. J. A, Inorg. Chem., 2012, 51, 463–471 CrossRef CAS PubMed.
- H. Ahmad, A. J. H. M. Meijer and J. A. Thomas, Chem.–Asian J., 2011, 6, 2339–2351 CrossRef CAS PubMed.
- A. B. Wragg, S. Derossi, T. L. Easun, M. W. George, X.-Z. Sun, F. Hartl, A. H. Shelton, A. J. H. M. Meijer and M. D. Ward, Dalton Trans., 2012, 41, 10354–10371 RSC.
- B. Mennucci and J. Tomassi, J. Chem. Phys., 1997, 106, 5151–5158 CrossRef CAS.
- M. Cossi, V. Barone, B. Mennucci and J. Tomasi, Chem. Phys. Lett., 1998, 286, 253–260 CrossRef CAS.
Footnotes |
| † Dedicated to Professor David Cole-Hamilton on the occasion of his retirement and for his outstanding contribution to transition metal catalysis. |
| ‡ Electronic supplementary information (ESI) available. See DOI: 10.1039/c3dt52092g |
| § Current address: Department of Chemical & Biological Sciences, University of Huddersfield, Huddersfield, UK. |
| This journal is © The Royal Society of Chemistry 2013 |