DOI:
10.1039/C2GC36542A
(Paper)
Green Chem., 2013,
15, 236-244
Selective reductions using visible light photocatalysts of supported gold nanoparticles†
Received
30th September 2012
, Accepted 13th November 2012
First published on 13th November 2012
Abstract
Photocatalytic synthesis using visible light is a desirable chemical process because of its potential to utilize sunlight. Supported gold nanoparticles (Au-NPs) were found to be efficient photocatalysts and the effects of the supports were identified including CeO2, TiO2, ZrO2, Al2O3, and zeolite Y. In particular Au/CeO2 exhibited the high catalytic activity to reduce nitro-aromatics to azo compounds, hydrogenate azobenzene to hydroazobenzene, reduce ketones to alcohols, and deoxygenate epoxides to alkenes at ambient temperatures, under irradiation of visible light (or simulated sunlight). The reactive efficiency depends on two primary factors: one is the light adsorption of catalysts and the other is the driving ability of catalysts corresponding to the reactants. The light absorption by Au-NPs is due to the surface plasmon resonance effect or interband electron transition; this is related to the reduction ability of the photocatalysts. Irradiation with shorter wavelengths can excite the conduction electrons in Au-NPs to higher energy levels and, as a result, induce reduction with more negative reduction potentials. It is known that when irradiated with light the Au-NPs can abstract hydrogen from isopropanol forming Au–H species on the Au-NP surface. Hence, we proposed that the active Au–H species will react with the N![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O, NN, CO double bonds or epoxide bonds, which are weakened by the interaction with the excited electrons in the Au-NPs, and yield the final reductive products. The reacting power of the Au–H species depends on the energy of the excited electrons in Au-NPs: the higher the electronic energy, the stronger the reduction ability of the Au–H species. This finding demonstrates that we can tune the reduction ability of the photocatalysts by manipulating the irradiation wavelength.
O, NN, CO double bonds or epoxide bonds, which are weakened by the interaction with the excited electrons in the Au-NPs, and yield the final reductive products. The reacting power of the Au–H species depends on the energy of the excited electrons in Au-NPs: the higher the electronic energy, the stronger the reduction ability of the Au–H species. This finding demonstrates that we can tune the reduction ability of the photocatalysts by manipulating the irradiation wavelength.
Introduction
The photocatalytic process has been extensively applied to degrade environmental pollutants, develop new solar cells, and conduct homogeneous reactions of photochemistry.1 The process has salient advantages: it is generally conducted at ambient temperatures with the potential to be driven by sunlight—an inexhaustible and clean energy source. If the photocatalytic process is applied to synthesize fine organic chemicals, it can bring about significant benefits such as achieving better selectivity to the desired product via a green approach. Moreover, the substances that are unstable at high temperatures may be synthesized through this unique light-excited progress. However, the application of the sunlight-driven photocatalytic process to synthesize fine organic chemicals only achieved limited progress.2 Titania (TiO2) and silica-supported titania are active for photocatalytic oxidation of light alkanes and alkenes under ultraviolet (UV) irradiation at ambient temperatures.2 UV light accounts for only about 4% of the solar energy received by the earth while visible light (wavelength 400–700 nm) accounts for 43%.2d The efficiency of the photocatalytic synthesis becomes a primary concern. Various methods have been developed to produce visible light photocatalysts. For example, some research groups reported the synthesis of a series of aldehydes from the corresponding alcohols by photocatalytic oxidation with dye-sensitized TiO2 or molecular oxygen on TiO2 under visible light.3 The dye-sensitized photocatalysts can absorb visible light and this process achieved high conversion and product selectivity. Nonetheless, this approach is not applicable to catalyze reduction reactions. Given that reduction is a major process for the synthesis of organic fine chemicals, it will be of great interest if photocatalytic reduction processes can be applied to synthesize fine chemicals under visible light. However, the synthesis of organic compounds via visible light photocatalytic reduction is even more challenging than that via visible-light photocatalytic oxidation. Currently there are a few reports on this topic, which focus on the reduction of nitro-aromatics, such as the photoreduction of nitrobenzene, using photocatalysts of cadmium sulphide or modified TiO2, and the main products are aniline or imines.4 The core issue regarding photocatalytic reduction is to devise efficient photocatalysts that can catalyze the reduction reactions with superior product selectivity under visible light irradiation. If we can find photocatalysts that are able to absorb visible light and activate chemical reactions, photocatalytic processes driven by sunlight can be developed.
Since the 1980s Au-NPs have been recognized as a fascinating material that shows great potential in sustainable chemical synthesis.5 In the photocatalytic process on gold science, we found that the supported Au-NPs exhibited superior performance in the photoredox catalysis of fine organic compounds under both visible and UV irradiation.6 It highlights that sustainable sunlight resources can be used to drive some chemical process with high efficiency. Au-NPs absorb visible light mainly due to the surface plasmon resonance (SPR) effect.7 The SPR effect is the collective oscillation of conduction electrons in the NPs, which resonate with the electromagnetic field of the incident light, and this resonance gives rise to significant field enhancement of the local electromagnetic fields near surfaces of the Au-NPs. The SPR absorption for typical spherical Au-NPs is generally observed between 520 and 550 nm,6,7 and the conducting electrons at the NP surfaces gain energy from the absorption. The energetic electrons may strongly interact with the molecules on the Au-NP surface and even migrate to the molecules absorbed on the support6,7b and will relax back to their thermal equilibrium states and release heat to the Au-NP lattice, resulting in rapid heating of the Au-NPs.7c Together, the two effects can induce chemical reactions of molecules on the Au-NPs. The mechanism of the photocatalysis with Au-NPs is distinctly different from that of semiconductor photocatalysts.2,6 This poses an opportunity and question that it may be possible to use the supported Au-NPs to catalyze reduction reactions under visible and UV light. If successful, we can drive important reactions using sunlight, gain insight into the interaction between light, Au-NPs and reactant molecules and understand the mechanism of photocatalytic reductions, which are valuable for designing new photocatalytic processes.
In the present study, gold nanoparticles (Au-NPs) supported on CeO2, TiO2, ZrO2, Al2O3, and zeolite Y were prepared and the effects of the supports were identified. In particular we investigated four typical reduction processes on supported Au-NP photocatalysts under visible light (or simulated sunlight): reduction of nitro-aromatics to azo compounds, hydrogenation of azobenzene to hydroazobenzene, reduction of ketones to alcohols, and deoxygenation of epoxides to alkenes. These reduction reactions are important in organic synthesis and biological chemistry.8–10 For example, in the vitamin K cycle the recycling of the oxidized vitamin K back to the reduced form is a key step currently relying on enzyme catalysis,8 which involves deoxygenation of epoxides. The reduction of ketones to alcohols is a significant transformation in organic synthesis and the chemical industry.9 Azo compounds are widely used as dyes in the polymer, textile and food industries as well as in the production of medicines to treat inflammation and a type of arthritis.10 The supported Au-NPs catalysts exhibited excellent activity for these photocatalytic syntheses. Moreover, the reduction potentials of the four reductions vary from −0.8 V to −2.4 V.1,11 Hence, it is possible to examine the impact of the irradiation wavelength on the ability of the supported Au-NPs catalysts to drive the reductions with different reduction potential. Indeed, we found that the irradiation of shorter wavelength can drive the reduction of more negative reduction potential. Since the irradiation wavelength is related to the energy of the electrons excited by the light, this finding means that the higher the energy of the excited electrons in the photocatalysts, the stronger the reduction ability of the catalyst. Thus a tentative mechanism of the photochemical reduction can be proposed.
Results and discussion
1. Effects of Au-NPs on different supports
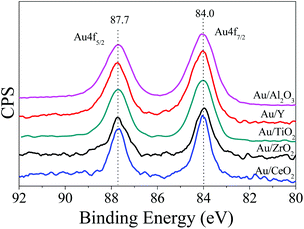
The size and dispersion of the Au-NPs should influence the photocatalytic performance, which can be readily tuned by adjusting the ratios of sodium borohydride to HAuCl4 and HAuCl4 to supports.12Fig. 1 shows the typical transmission electron microscopy (TEM) images of the Au-NPs on the various supports including CeO2, TiO2, ZrO2, Al2O3, and zeolite Y (namely Au/CeO2, Au/TiO2, Au/ZrO2, Au/Al2O3, and Au/Y). The Au-NPs are well dispersed and have narrow size distributions. The sizes of the Au-NPs on CeO2, TiO2, and ZrO2 are estimated to be in the range of 5 nm to 8 nm while those on Al2O3 are smaller (about 3 nm) and those on zeolite Y are bigger (about 10 nm). The X-ray photoelectron spectroscopy (XPS) spectra of the photocatalysts are shown in Fig. 2, Au 4f spectra of all Au-NPs photocatalysts exhibit similar patterns. The binding energies of Au 4f7/2 and Au 4f5/2 electrons are 84.0 and 87.7 eV, respectively. It is consistent with the reports on gold metal, which indicates that Au-NPs on the supports exist in the metallic state.12
 |
| | Fig. 1 TEM images of Au-NPs on different supports. The length of the scale bars in the images is 10 nm. | |
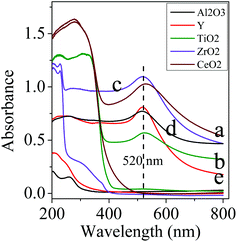
UV/Vis spectra of Au-NPs on different supports as well as supports themselves were collected and shown in Fig. 3. An absorption band at about 520 nm is observed in the UV/Vis spectra of the photocatalysts, which is the characteristic SPR absorption by Au-NPs. The light absorption of the Au-NPs in the photocatalysts varies with the change of the supports, such as CeO2, TiO2, ZrO2, Al2O3, and zeolite Y, while the supports exhibit little absorption of light with wavelengths longer than 400 nm (Fig. 3). From the UV/Vis spectra, the TiO2 support can absorb most light with wavelengths less than 400 nm, and CeO2 can absorb light with wavelengths less than 500 nm. On the other hand, the Au/CeO2 catalyst shows a little shift to the long-wavelength direction, and has higher intensity relative to Au/TiO2, indicating a stronger SPR effect of Au/CeO2.13 Even though Au-NPs on different supports have similar size (Fig. 1), the strong light absorption of Au/CeO2 may be attributed to the strong interface action between Au-NPs and CeO2.13 The light absorption is important to the photocatalytic performance, but the catalysis activity is also dependent on other factors, particularly the interface action between the Au-NPs and supports as the photocatalytic reactions take place on the Au-NP surface with the aid of supports. Some researchers propose electron transitions between Au-NPs and the support (TiO2), which is not perfect to clarify the functions of supports, particularly in the photocatalytic process. Au/CeO2 has a stronger interaction between Au-NPs and CeO2 and has a stable structure with Au-NPs incorporated into CeO2, which has been identified previously.14 Thus in a reactive system the aggregation of Au-NPs can be avoided and intensive light absorption is maintained. Au-NPs were also loaded on TiO2 and ZrO2 and we found that for reduction of styrene oxide the selectivity to the product styrene is moderately lower than that of the Au/CeO2 (Table 1). Nevertheless, all the supports themselves show trivial activities in these photocatalytic reductions. It appears that the support materials have a limited impact on the conversion of photocatalytic reductions. In view of the strong linkage between Au-NPs and CeO2, the Au-NPs on the CeO2 support were selected to be an efficient and stable photocatalyst.
| Catalysts |
Reaction time (h) |
Visible light |
No light |
| Conversion (%) |
Selectivity (%) |
Conversion (%) |
Selectivity (%) |
| Au/CeO2 |
16 |
19.7 |
88 |
4.4 |
72 |
| Au/ZrO2 |
16 |
17.4 |
72 |
7.8 |
75 |
| Au/TiO2 |
16 |
22.5 |
73 |
7.8 |
75 |
| Au/Al2O3 |
16 |
23.9 |
60 |
5.8 |
51 |
| Au/Y |
16 |
4.5 |
62 |
1.6 |
55 |
2. Effects of gold loads for the reduction of styrene oxide
The photocatalytic efficiency of supported Au-NPs depends on two primary aspects: the light absorption of catalysts to the emission light and the driving capability of catalysts with regard to the reactants. In a certain reactive system, for instance, the reduction of styrene oxide, light absorption of catalysts plays a significant role in the enhancement of reactive activity due to the contribution of light absorption of Au-NPs. Fig. 4A exhibits the UV/Vis diffuse reflectance spectra of Au/CeO2 with various amounts of gold loads. It appears that the light absorption of Au/CeO2 at about 520 nm increases with the increasing loadings of Au-NPs. The results of the photocatalytic reduction of styrene oxide under visible light irradiation are shown in Fig. 4B. High conversions are achieved for this reaction and the photocatalytic efficiency is significantly enhanced with the increase of gold loading. Meanwhile, the corresponding conversions in the dark are found to maintain a minimal increase. For instance, when the gold loading increased from 1.5% to 3%, the conversion under visible light increased from 12% to 20% while the conversion in the dark had negligible improvement. This indicates that the light absorption of catalysts is a crucial factor to enhance the photocatalytic efficiency. To minimize the influence of the reduction reaction in the dark, 3% Au/CeO2 was selected to be an exemplar photocatalyst for the study of four types of reductions.
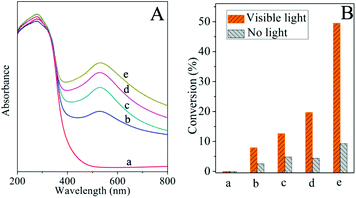 |
| | Fig. 4 A. UV/Vis diffuse reflectance spectra of Au/CeO2. B. Catalytic conversions of Au/CeO2 for the reduction of styrene oxide with various amounts of gold loads: (a) 0 (CeO2); (b) 0.5%; (c) 1.5%; (d) 3%; (e) 5%. | |
3. Performance of Au/CeO2 for the four photocatalytic reductions
The results of the four photocatalytic processes on the photocatalyst of Au/CeO2 under visible light irradiation are summarized in Table 2. High conversion rates were achieved for these reactions. Typically, 43.5% of nitrobenzene was reduced to azobenzene in 2 h with a selectivity of 96%. 40% of azobenzene was reduced to hydroazobenzene in 6 h with a selectivity of 78%. 31% of acetophenone transformed into benzyl ethanol with a selectivity of 99%. 20% of styrene oxide was converted to styrene in 16 h with a high selectivity of about 88%. These reactions were also conducted in the dark with other experimental conditions unchanged—very low conversions of the reactants were observed. This indicates that the reduction reactions can be driven by visible light irradiation and may follow different mechanisms from those of thermal processes and photocatalytic reductions using other catalysts under UV irradiation.4,8–10 Take the reduction of nitrobenzene as an example (entry 1), the SPR effect of Au-NPs plays a key role in assisting activation of the NO bonds, which is distinct from the case under UV light irradiation.6a Thus, the product is mainly azoxybenzene rather than aniline and the mechanism of this process will be discussed later.
Table 2 Performance of 3 wt% Au/CeO2 for the four photocatalytic reductions
| Entry |
Reagents |
Products |
Time (h) |
Temp. (°C) |
Visible light |
No light |
| Conv. (%) |
Sel. (%) |
Conv. (%) |
Sel. (%) |
| 1 |
|

|
2 |
30 |
43.5 |
96 |
8.1 |
>99 |
| 2 |

|

|
6 |
30 |
40 |
78 |
10 |
79 |
| 3 |

|

|
24 |
30 |
31 |
>99 |
1 |
>99 |
| 4 |

|

|
16 |
25 |
20 |
88 |
4.4 |
72 |
4. Effects of light wavelength
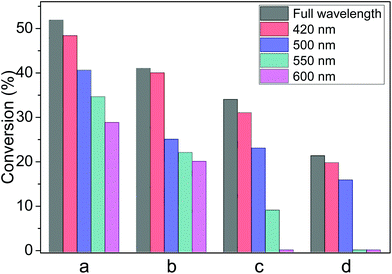
The wavelength of the irradiation has influence on the photocatalytic process.15 In photocatalytic experiments, glass filters were employed to block the irradiation below a certain wavelength (e.g. <550 nm), this wavelength is called the cut-off wavelength. With the higher cut-off wavelength, i.e. the light source focuses on longer wavelength, all the reactions declined gradually even under other identical experimental conditions. Therefore, it can be deduced that the SPR effect has a primary influence on these photocatalytic reductions due to the strong absorption around 520 nm (Fig. 3). When the light with wavelength longer than 550 nm was used—by filtering out wavelength shorter than 550 nm—the reduction of nitrobenzene still proceeded even though the conversion (35%) decreased (Fig. 5a). This was similarly observed in the reduction of azobenzene of which the conversion decreased to 22% as well as the hydrogenation of acetophenone of 9% (Fig. 5b and 5c). However, the reduction of styrene oxide could not be activated under such irradiation at the cut-off wavelength of 550 nm, shown in Fig. 5d. Furthermore, when the cut-off wavelength of the light source was around 600 nm, the reduction of azobenzene still proceeded (20% of conversion), but acetophenone and styrene oxide could not be reduced.
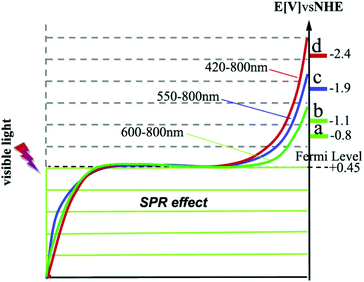
We recognized a relationship between the cut-off wavelength of the irradiation and the reduction potentials of the reactants. The reduction potentials of the four reactants are −0.8 eV (nitrobenzene), −1.1 eV (azobenzene), −1.9 eV (acetophenone), and −2.4 eV (styrene oxide).1,11 Correlating these reduction potentials with the conversions under the irradiation of different cut-off wavelengths, it can be seen that a shorter cut-off wavelength is required to reduce a compound with a more negative reduction potential. Conduction electrons in the Au-NPs gain energy from the light absorption and their distribution over the energy levels shifts to higher levels in the 6sp band.6,7 As shown in Scheme 1, the cut-off wavelength determines the energy of the excited electrons as well as their distribution over the high energy levels.6 The shorter the wavelength is, the higher the energy of the excited electrons is (Scheme 1). The electrons excited by the light with wavelength longer than 600 nm are able to induce the reduction of nitrobenzene (reduction potential −0.8 eV) and azobenzene (reduction potential −1.1 eV), but cannot induce the reduction of more negative reduction potentials. It is rational from the view of thermodynamics that reductions with more negative reductive potentials could not proceed because the reactant molecules could not gain the photo-excited electrons at lower energy levels. This may imply that the reactant molecules in the reduction with more negative reduction potentials can only gain the excited electrons of Au-NPs at the higher energy levels (Scheme 1), which are excited by light of shorter wavelength. The wavelength of the light determines the reduction power of the photocatalysts. This finding highlights important opportunities that the conversion and product selectivity of the reduction reactions can be optimized and the by-products may be avoided by manipulating the irradiation wavelength. As the SPR absorption of the Au-NPs is very weak in the range longer than 650 nm (Fig. 3), the conversion of all the four reactants is negligible. This confirms again that the SPR absorption of Au-NPs is crucial for the catalytic activity.
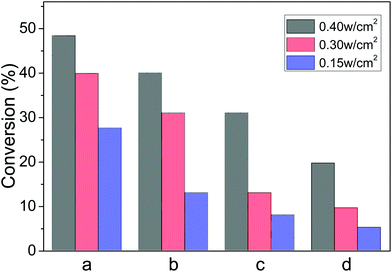
5. Effects of light intensity
In an attempt to clarify the influence of light irradiation intensity, the irradiation intensity was reduced from 0.40 to 0.30 and 0.15 W cm−2, while other experimental conditions were unchanged. We found that the conversion of nitrobenzene decreased from 48% to 40% and then to 28%, correspondingly. Similar changes were observed for the reduction of the styrene oxide, azoaromatics and the ketones (Fig. 6). Such a tendency reveals that these photocatalytic reductions are dependent on the light intensity, as a higher intensity can excite more energetic electrons in Au-NPs and enhance the interaction with reactants absorbed on the Au-NPs under visible light irradiation. These results further confirm that the reduction reactions were driven by visible light.
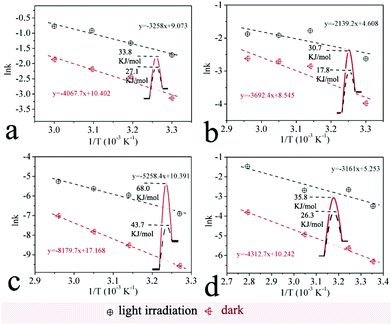
6. Apparent activation energy of photocatalysis and thermal reactions
The contribution of visible light irradiation to reducing the apparent activation energies of the reductions is also investigated. These reduction reactions were conducted both under irradiation (photocatalytic process) and in the dark (thermal process) at various temperatures to examine the reduction kinetics. According to the first-order reaction rate equation, the values of the rate constants were calculated. Furthermore, the Arrhenius equation was applied to derive the apparent activation energies of the reductions via photocatalytic and thermal processes.6d For one reduction reaction, the difference between the activation energies of the two processes indicates the contribution of light irradiation to reducing the apparent activation energy. The results of four reactions are shown in Fig. 7. For azobenzene, the activation energy is 17.8 kJ mol−1 for the photocatalytic reduction and 30.7 kJ mol−1 for the thermal reduction. The difference between the two activation energies (12.9 kJ mol−1) reveals the contribution of light irradiation (Fig. 7b). It indicates that the light irradiation plays a major role in activating the reduction reaction.
7. Photocatalytic reductions with simulated sunlight
Photocatalysis with simulated sunlight was conducted under the same experimental conditions but using a different light source (model: xenon lamp, 150 W, Farrand Optical Co.). The results with simulated sunlight are roughly the same corresponding to those with the visible light, which further confirm that light absorption and the reaction system are two primary aspects of the photocatalytic efficiency. As light intensity and the catalytic system are unchanged, the minimal deviations in conversions may be attributed to the differences of wavelength ranges in the light sources. These reduction reactions exhibited the great potential to excite the photocatalytic reactions utilizing sunlight (Table 3).
Table 3 Photocatalytic reduction reactions conducted with simulated light
8. Mechanistic discussions
The fact that light irradiation substantially reduces the apparent activation energy of the reduction process also suggests that the photocatalytic process follows a different mechanism from that of the thermal process (Scheme 2). Generally, isopropanol is a hydrogen donor in the studied reduction reactions. Under visible light irradiation, the Au-NPs are able to abstract hydrogen from isopropanol forming the transient Au–H species.6,13b The active Au–H species attack the double bonds (NO, CO, NN) or the epoxide bond leading to the hydrogenation or deoxygenation, in which the hydrogen of Au–H species is consumed and the final reductive products form without releasing hydrogen molecules. In a control experiment, an efficient hydrogen-abstracting reagent, 2,2′,6,6′-tetramethylpiperidine N-oxyl (TEMPO)—which can abstract hydrogen from the surface of gold and other metals to form hydroxylamine—was added into the reaction system while the other experimental conditions were unchanged.16 No final reductive products were detected. This confirms the important role of the surface Au–H species in the reductions. The SPR effect also enhances the local electromagnetic fields near rough surfaces of the Au-NPs and this could assist activation of the double bonds or the epoxide bond when the reactant molecules are adsorbed on the surface of Au-NPs. The ability of the Au–H species to react with the double bonds (NO, CO, NN) or the epoxide bond should depend also on the energy of the excited electrons in Au-NPs, which is higher when the irradiation wavelength is shorter as discussed previously. The Au–H species can release hydrogen to reactant molecules. Moreover, higher conversions were generally achieved for reactants with an aromatic ring than those without the ring. It is believed that the aromatic reactants have stronger affinity to the Au-NP surface.
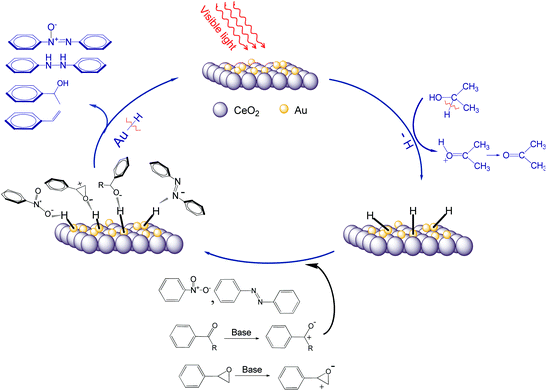 |
| | Scheme 2 Schematic mechanisms of the reaction pathways in the photocatalytic reductions with supported Au-NPs. | |
Conclusions
In summary, this study demonstrated that the four reductions, reduction of nitro-aromatics to azo compounds, hydrogenation of azobenzene to hydroazobenzene, reduction of ketones to alcohols, and deoxygenation of epoxides to alkenes, can be efficiently catalyzed by the Au/CeO2 photocatalyst under visible light irradiation or simulated sunlight. The reduction power of the photocatalysts can be optimized by manipulating the irradiation wavelength. Irradiation with shorter wavelength can induce the more difficult reduction with more negative reduction potentials. The efficiency of the photocatalytic reductions increased with the increasing irradiation intensity under ambient conditions. The kinetic study showed that the apparent activation energy can be significantly reduced under visible light irradiation compared with that in the thermal-driven processes. The photocatalytic reductions on Au/CeO2 followed a mechanism distinctly different from that on the semiconductor photocatalysts and gave an insight into the formation of Au–H species and interaction with the reactants. Such photocatalytic syntheses are sustainable and clean as these production processes can utilize sunlight for chemical synthesis.
Experimental
Raw materials
Chemicals of chloroauric acid (99.999%, HAuCl4), L-lysine (98.0%), sodium borohydride (99.99%, NaBH4), hydrochloric acid (37%, HCl), nitrobenzene (99.0%), potassium hydroxide (99.0%, KOH), isopropanol (99.5%), acetophenone (99.0%), azobenzene (98.0%) and styrene oxide (97.0%) were purchased from Sigma-Aldrich. The inert gas of argon was from the BOC (99.99%). The supports of cerium(IV) oxide (nanopowder, 99.95%, CeO2), zirconium(IV) oxide (nanopowder, <100 nm of particle size, ZrO2), titanium(IV) oxide (nanopowder, 99.5%), and aluminum oxide (Al2O3) were also purchased from Sigma-Aldrich and used without further purification. The zeolite Y was synthesized by the hydrothermal method reported in the literature.17
In a glass beaker with a magnetic stirring bar, 2.5 g of support powders, such as CeO2, were dispersed into the 100 ml of 3.8 × 10−3 M HAuCl4 aqueous solution. Subsequently, 20 ml of 0.53 M lysine was added dropwise to the mixture under stirring, and after that the stirring was prolonged for 30 min. To this suspension, 10 ml of 0.35 M NaBH4 solution was added dropwise, followed by adding 10 ml of 0.3 M hydrochloric acid (to achieve a pH = 9.5). The mixture was stirred for 1 h and aged for 24 h. Then the solid was separated, washed firstly with 600 ml deionized water three times in a 250 ml centrifuge bottle to remove the chlorine ions, and the residual solution was tested by 0.1 M AgNO3 solution and no AgCl precipitate was observed. The solids were collected, dried at 60 °C for 16 h, and used directly as the photocatalyst.
Photocatalytic reduction test
The reaction was conducted in a 50 ml round-bottomed Pyrex glass flask with a sealed spigot and a magnetic stirrer. The reaction temperature was controlled by an air-conditioner within a sealed wooden box. A 500 W halogen lamp was used as the incandescent light source. The light intensity in the reaction position was set at 0.4 W cm−2 and could be adjusted by changing the distance between the reactor and the light source. The wavelength range was tuned by using various glass filters to cut off the irradiation below a certain value of wavelength.
Catalytic reduction of nitrobenzene to azoxybenzene was conducted under the argon atmosphere. Typically, 3 mmol acetophenone, 30 ml isopropanol as the solvent, and 3 ml of 0.1 M KOH solution in isopropanol were mixed in the reactor, followed by adding 100 mg of the catalyst and purging with argon gas, and then stirred during the reaction and illuminated with the incandescent light.
To hydrogenate azobenzene, generally 2 mmol azobenzene was dissolved in 20 ml isopropanol in the reactor. 100 mg of the photocatalysts and a 2 ml solution of 0.1 M KOH in isopropanol were then added. After that, air in the flask was eliminated with argon. The reaction was conducted under the illumination of an incandescent light and the reaction mixture was stirred by a magnetic stirrer.
Catalytic reduction of ketones to alcohols was conducted under the argon atmosphere. Typically, 3 mmol acetophenone was dissolved in 30 ml isopropanol, and a 3 ml solution of 0.1 M KOH in isopropanol and 100 mg of the catalyst were added into the mixture that was then stirred during the reaction and illuminated with the incandescent light.
Catalytic deoxygenation of epoxides to alkenes was conducted under the argon atmosphere. Typically, 3 mmol styrene oxide was dissolved in 30 ml isopropanol, and 3 ml of a 0.1 M KOH solution in isopropanol and 100 mg of the catalyst were added into the mixture. The mixture was stirred during the reaction and illuminated with the incandescent light.
During these reactions, 0.5 ml aliquots were collected at given irradiation time intervals and filtered through a Millipore filter (pore size 0.45 μm) to remove the catalyst particulates. Then the flask was purged with argon again for more than 3 min to remove air and then sealed. The filtrates were analyzed by an Agilent 6890 gas chromatograph with the HP-5 column. Quantities of the products and reactants were calculated from the peak areas of the compounds using calibration curves. An Agilent HP5973 mass spectrometer was used to determine and analyze the product compositions.
Characterization
TEM images were taken with a JEOL JEM-2100 transmission electron microscope employing an accelerating voltage of 200 kV. The specimens were sample powders deposited onto a copper microgrid coated with a holey carbon film. The element composition of some samples was determined by an energy-dispersive X-ray spectroscope attached on an FEI Quanta 200 scanning electron microscope. The diffuse reflectance UV-Visible spectra of the samples were recorded on a Cary 5000 spectrometer. The XPS data were recorded on an ESCALAB 250 spectrometer and Al Kα radiation was used as the X-ray source. The C1s peak at 284.5 eV was used as a reference for the calibration of the binding energy scale.
Acknowledgements
X. Ke is indebted to the Queensland Government for a Smart Futures Fellowship. The authors also gratefully acknowledge financial support from the Australia Research Council.
Notes and references
-
(a) Y. Lu, H. Yu, S. Chen, X. Quan and H. Zhao, Environ. Sci. Technol., 2012, 46, 1724 CrossRef CAS;
(b) S. Sarina, H. Y. Zhu, Z. F. Zheng, S. E. Bottle and J. Chang,
et al., Chem. Sci., 2012, 3, 2138 RSC;
(c) S. Malato, P. Fernández-Ibáňez, M. I. Maldonado, J. Blanco and W. Gernjak, Catal. Today, 2009, 147, 1 CrossRef CAS;
(d) M. Fagnoni, D. Dondi, D. Ravelli and A. Albini, Chem. Rev., 2007, 107, 2725 CrossRef CAS;
(e)
M. Montalti, A. Credi, L. Prodi and M. T. Gandolfi, Handbook of Photochemistry, CRC Press, Boca Raton, 3rd edn, 2006 Search PubMed.
-
(a) X. Chen, L. Liu, P. Y. Yu and S. S. Mao, Science, 2011, 331, 746 CrossRef CAS;
(b) M. Bowker, Green Chem., 2011, 13, 2335 RSC;
(c) G. Palmisano, E. García-López, G. Marcí, V. Loddo, S. Yurdakal, V. Augugliaro and L. Palmisano, Chem. Commun., 2010, 46, 7074 RSC;
(d) Z. G. Zou, J. H. Ye, K. Sayam and H. Arakawa, Nature, 2001, 414, 625 CrossRef CAS.
-
(a) A. Tanaka, K. Hashimoto and H. Kominami, Chem. Commun., 2011, 47, 10446 RSC;
(b) H. Kominami, A. Tanaka and K. Hashimoto, Chem. Commun., 2010, 46, 1287 RSC;
(c) S. Higashimoto, N. Kitao, N. Yoshida, T. Sakura, M. Azuma, H. Ohue and Y. Sakata, J. Catal., 2009, 266, 279 CrossRef CAS;
(d) M. Zhang, C. C. Chen, W. H. Ma and J. C. Zhao, Angew. Chem., Int. Ed., 2008, 47, 9730 CrossRef CAS.
-
(a) B. Pal, T. Torimoto, K. Okazaki and B. Ohtani, Chem. Commun., 2007, 483 RSC;
(b) A. Maldotti, L. Andreotti, A. Molinari, S. Tollari, A. Penoni and S. Cenini, J. Photochem. Photobiol., A, 2000, 133, 129 CrossRef CAS;
(c) S. O. Flores, O. Rios-Bernij, M. A. Valenzuela, I. Córdova, R. Gómez and R. Gutiėrrez, Top. Catal., 2007, 44, 507 CrossRef CAS;
(d) H. Tada, T. Ishida, A. Takao and S. Ito, Langmuir, 2004, 20, 7898 CrossRef CAS;
(e) F. Mahdavi, T. C. Bruton and Y. Li, J. Org. Chem., 1993, 58, 744 CrossRef CAS;
(f) J. L. Ferry and W. H. Glaze, Langmuir, 1998, 14, 3551 CrossRef CAS;
(g) O. Rios-Bernÿ, S. O. Flores, I. Córdova and M. A. Valenzuela, Tetrahedron Lett., 2010, 51, 2730 CrossRef.
-
(a) T. Uchiyama, H. Yoshida, Y. Kuwauchi, S. Ichikawa, S. Shimada, M. Haruta and S. Takeda, Angew. Chem., Int. Ed., 2011, 50, 10157 CrossRef CAS;
(b) H. Zhang, T. Watanabe, M. Okumura, M. Haruta and N. Toshima, Nat. Mater., 2012, 11, 49 CrossRef;
(c) Y. Zhang, X. Cui, F. Shi and Y. Deng, Chem. Rev., 2012, 112, 2467 CrossRef CAS.
-
(a) H. Y. Zhu, X. B. Ke, X. Z. Yang, S. Sarina and H. W. Liu, Angew. Chem., Int. Ed., 2010, 49, 9657 CrossRef CAS;
(b) H. Y. Zhu, X. Chen, Z. F. Zheng, X. B. Ke, E. Jaatinen, J. C. Zhao, C. Guo, T. F. Xie and D. J. Wang, Chem. Commun., 2009, 7524 RSC;
(c) X. B. Ke, S. Sarina, J. Zhao, X. G. Zhang, J. Chang and H. Y. Zhu, Chem. Commun., 2012, 48, 3509 RSC;
(d) X. G. Zhang, X. B. Ke and H. Y. Zhu, Chem.–Eur. J., 2012, 18, 8048 CrossRef CAS.
-
(a) S. Linic, P. Christopher and D. B. Ingram, Nat. Mater., 2011, 10, 911 CrossRef CAS;
(b) P. Mulvaney, Langmuir, 1996, 12, 788 CrossRef CAS;
(c) D. K. Roper, W. Ahn and M. Hoepfner, J. Phys. Chem. C, 2007, 111, 3636 CrossRef CAS;
(d) M. Jakob, H. Levanon and P. V. Kamat, Nano Lett., 2003, 3, 353 CrossRef CAS.
-
(a) T. Mitsudome, A. Noujima, Y. Mikami, T. Mizugaki, K. Jitsukawa and K. Kaneda, Angew. Chem., Int. Ed., 2010, 49, 5545 CrossRef CAS;
(b) F. Zaccheria, F. Santoro, R. Psaro and N. Ravasio, Green Chem., 2011, 13, 545 RSC;
(c) J. Ni, L. He, Y. M. Liu, Y. Cao, H. Y. He and K. N. Fan, Chem. Commun., 2011, 47, 812 RSC.
-
(a) F. Z. Su, L. He, J. Ni, Y. Cao, H. Y. He and K. N. Fan, Chem. Commun., 2008, 3531 RSC;
(b) Y. Zhu, H. Qian, B. A. Drake and R. Jin, Angew. Chem., Int. Ed., 2010, 49, 1295 CrossRef CAS.
-
(a) E. Merino, Chem. Soc. Rev., 2011, 40, 3835 RSC;
(b) H. M. Nanjundaswamy and M. A. Pasha, J. Chem. Res., 2005, 12, 772 CrossRef;
(c) A. Grirrane, A. Corma and H. Carcía, Science, 2008, 322, 1661 CrossRef CAS.
-
(a) S. Kohtani, E. Yoshioka, K. Saito, A. Kudo and H. Miyabe, Catal. Commun., 2010, 11, 1049 CrossRef CAS;
(b) K. Boujlel, P. Martigny and J. Simonet, J. Electroanal. Chem., 1983, 144, 437 CrossRef CAS.
-
(a) A. S. K. Hashmi and G. J. Hutchings, Angew. Chem., Int. Ed., 2006, 45, 7896 CrossRef;
(b) M. D. Hughes, Y. J. Xu, P. Jenkins, P. McMorn, P. Landon and D. I. Enache, Nature, 2005, 437, 1132 CrossRef CAS;
(c) M. Haruta, Catal. Today, 1997, 36, 153–166 CrossRef CAS.
-
(a) A. Primo, T. Marina, A. Corma, R. Molinari and H. García, J. Am. Chem. Soc., 2011, 133, 6930 CrossRef CAS;
(b) A. Maldotti, A. Molinari, R. Juárez and H. García, Chem. Sci., 2011, 2, 1831 RSC;
(c) H. Kominami, A. Tanaka and K. Hashimoto, Appl. Catal., A, 2011, 397, 121 CrossRef CAS.
-
(a) A. Furube, L. Du, K. Hara, R. Katoh and M. Tachiya, J. Am. Chem. Soc., 2007, 129, 14852 CrossRef CAS;
(b) M. Murdoch, G. I. N. Waterhouse, M. A. Nadeem, J. B. Metson and M. A. Keane,
et al., Nat. Chem., 2011, 3, 489 CAS.
- C. G. Silva, R. Juárez, T. Marino, R. Molinari and H. García, J. Am. Chem. Soc., 2011, 133, 595 CrossRef.
-
(a) J. P. Roth, J. C. Yoder, T. J. Won and J. M. Mayer, Science, 2001, 294, 2524 CrossRef CAS;
(b) L. Tebben and A. Studer, Angew. Chem., Int. Ed., 2011, 50, 5034 CrossRef CAS.
- B. A. Holmberg, H. T. Wang, J. M. Norbeck and Y. S. Yan, Microporous Mesoporous Mater., 2003, 59, 13 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c2gc36542a |
|
| This journal is © The Royal Society of Chemistry 2013 |
Click here to see how this site uses Cookies. View our privacy policy here.