 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceReactions of p-coumaryl alcohol model compounds with dimethyl carbonate. Towards the upgrading of lignin building blocks†
Jessica N. G. Stanleyab, Maurizio Selvaa, Anthony F. Mastersb, Thomas Maschmeyerb and Alvise Perosa*a
aDepartment of Molecular Sciences and Nanosystems, Centre for Sustainable Chemical Technologies, Università Ca’ Foscari, Venezia, Italy. E-mail: alvise@unive.it; Fax: +39 041 234 8584; Tel: +39 041 234 8958
bLaboratory of Advanced Catalysis for Sustainability, School of Chemistry F11, The University of Sydney, Sydney, NSW 2006, Australia. E-mail: thomas.maschmeyer@sydney.edu.au; Fax: +61 2 9351 3329; Tel: +61 2 9351 4504
First published on 6th September 2013
Abstract
Cinnamyl alcohol 1 and 4-(3-hydroxypropyl)phenol 2, two compounds resembling the lignin building block p-coumaryl alcohol, can be selectively transformed into different products by catalytic methodologies based on dimethyl carbonate (DMC) as a green solvent/reagent. Selectivity can be tuned as a function of the reaction temperature and of the nature of the catalyst. Basic catalysts such as K2CO3, trioctylmethylphosphonium methylcarbonate ([P8881][CH3OCOO]), and CsF/αAl2O3 promote selective transesterification of the aliphatic hydroxyl group at 90 °C. However, amphoteric solids such as alkali metal-exchanged faujasites, NaX and NaY, selectively yield the corresponding alkyl ethers at higher temperatures (165–180 °C). The phenolic hydroxyl group of 2 can be methylated similarly with the faujasites at high temperatures. This preliminary screening for selectivity illustrates reactivity trends and delineates some of what might be among the most promising synthetic pathways to upgrade lignin-derived chemical building blocks.
Introduction
As the availability and security of organic fossil resources dwindle, and environmental concerns regarding emissions from the combustion of fossil fuels continue to grow, the percentage of chemicals and fuels obtained from renewable sources, such as lignocellulosic biomass, can be expected to rise. Lignocellulose, which is more evenly distributed geographically than fossil resources,1 is comprised mainly of cellulose, hemicellulose, and lignin, with lignin generally constituting 15–30% by weight and 40% by energy content. Synthetic approaches for the conversion of lignin to chemicals are not as well developed as those for the cellulosic components, which is partly due to the recalcitrant nature of lignin that gives plants some of their strength.2 However, the unique structure of lignin as an amorphous, highly substituted, aromatic polymer makes it particularly valuable as a potential source of a variety of chemicals, especially as it is the major bio-based source of aromatics.3 Much effort is devoted to the development of new chemical technology to process lignin streams into higher value-added compounds. Aggressive depolymerisation of lignin yields BTX (benzene, toluene, xylene) chemicals, plus phenol and aliphatics that can all be used in conventional chemical processes. Selective depolymerisation could instead yield monomeric lignin aromatics, not accessible by traditional petrochemistry. Sources of this kind of monomeric lignin include pretreatment streams, such as those from the pulp and paper industries (Kraft and Sulfite pulping), as well as new feedstock streams from biorefinery schemes (e.g. organosolv, steam explosion, pyrolysis, ammonia fibre explosion, hot water, dilute acid, etc.).3,4 The products consist of lignin with a molecular weight that ranges from 50![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000–20000 for lignosulfonate from sulfite pulping, to 2000–3000 in Kraft liquors, to below 1000 for organosolv. These fractions consist of monomeric lignin molecules based on C9 phenylpropenyl units with the average number of C, H and O atoms of approximately 9, 6–9, and 2.5, respectively. The molecular complexity of these monomeric streams has prompted the use of simpler low molecular weight lignin model compounds to aid in their investigation. It has been recognised that many of the products derived from the disruption of various lignin linkages resemble p-coumaryl, coniferyl, and sinapyl alcohol (Fig. 1), and effort has been put into the conversion of these models to other target chemicals by exploiting their functionality.
000–20000 for lignosulfonate from sulfite pulping, to 2000–3000 in Kraft liquors, to below 1000 for organosolv. These fractions consist of monomeric lignin molecules based on C9 phenylpropenyl units with the average number of C, H and O atoms of approximately 9, 6–9, and 2.5, respectively. The molecular complexity of these monomeric streams has prompted the use of simpler low molecular weight lignin model compounds to aid in their investigation. It has been recognised that many of the products derived from the disruption of various lignin linkages resemble p-coumaryl, coniferyl, and sinapyl alcohol (Fig. 1), and effort has been put into the conversion of these models to other target chemicals by exploiting their functionality. | ||
| Fig. 1 p-Coumaryl alcohol, coniferyl alcohol, and sinapyl alcohol. | ||
Such an approach, however, poses the issue of the overabundance of targets that can potentially be produced. The experience of the chemical industry shows that this complexity is best handled by using broad-based technologies (selective reductions and oxidations, bond making/breaking processes, catalysis, etc.) to produce multiple outputs,5 as opposed to a ‘like-for-like’ target-based approach aimed at replacing well-established chemicals produced from fossil feedstock.6,7
A curiosity driven broad-based strategy was, therefore, in our opinion a constructive approach towards the development of new chemistry and, in the longer term, a plethora of new chemicals. Some products are already in commercial use by the chemical industry, making them interesting in the short term, while others might be structural building blocks that enable new products/applications.3
In the present study we focused our attention on two compounds resembling p-coumaryl alcohol: cinnamyl alcohol 1 and 4-(3-hydroxypropyl)phenol 2. These compounds were chosen because they not only have –OH groups that closely resemble the ones present in p-coumaryl alcohol, but they are also readily available in the pure form, and their derivatives are easily identifiable by standard analytical techniques.
Dimethylcarbonate (DMC) is an excellent reagent and solvent for upgrading these compounds with lignin chemical functionalities, especially when viewed from a ‘green’ perspective. DMC is a good alternative to traditional methylating agents such as harmful dimethylsulfate and methyl halides,8 while the only by-products obtained during methylation reactions are carbon dioxide and methanol. The latter can, in principle, be recycled to form DMC.9,10 Furthermore, DMC is one of six organic carbonates that have been identified as especially useful green solvents.11 Thus, we propose to develop chemistry with compounds that model lignin functional groups by combining their use with a green methylating agent and solvent.
A further advantage of DMC is that its reactivity can be tuned for a variety of nucleophiles.12–19 Of particular relevance to the reactivity of p-coumaryl model compounds investigated in the present case is the tuneable selectivity of DMC towards aromatic and aliphatic OH groups, depending on the catalyst and reaction conditions.13,18–21 For example, basic catalysts can be used to promote a transesterification process, that is an equilibrium reaction, to form methyl alkyl carbonates, while weak bases or alkali metal-exchanged faujasites can be used to form methyl ethers irreversibly (Scheme 1). Furthermore, the methoxycarbonylating vs. methylating selectivity of DMC can also be tuned by changing the reaction conditions,22 while basic catalysed methylation could also occur on aromatic OH groups.
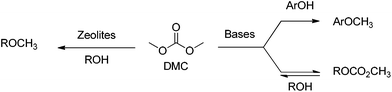 | ||
| Scheme 1 | ||
The main focus of this work was on assessing greener and easily accessible synthetic procedures for the transformation of cinnamyl alcohol 1, and 4-(3-hydroxypropyl)phenol 2, with DMC. This was done by investigating a range of catalysts, including solid K2CO3, CsF/αAl2O3, NaX, NaY and an ionic liquid (IL) [P8881][CH3OCOO], for the methylation and transesterification of the target compounds used as models of p-coumaryl alcohol. Cinnamyl alcohol, 1, was chosen as it contains a propylene side chain with a terminal OH, which is a common feature in many lignin streams.23 Likewise, 4-(3-hydroxypropyl)phenol 2 was chosen as a compound to model lignin functionalities as it has both an aliphatic OH on a propyl side chain and a phenolic OH in the para position. The focus of this study is on screening the product selectivity associated with different catalyst types and reaction conditions. Our interest was therefore focused on understanding the reactivity trends of DMC with the different OH groups, the relevant reaction equilibria, and the reaction pathways. A thorough understanding of these types of reactivities is of fundamental importance for the development of new greener processes aimed at transforming phenolic streams derived from lignin into higher value bio-based chemicals. Thus, we report that the selectivity of methylation/decarboxylation on the aliphatic and phenolic OH sites of these compounds in the presence of DMC can be tuned by changing the catalyst and/or reaction temperature. A range of products was observed of which four are highlighted, as they could be obtained selectively and isolated in good yields: cinnamyl methyl carbonate (1a, Scheme 2), cinnamyl methyl ether (1b, Scheme 2), 3-(4-hydroxyphenyl)propyl methyl carbonate (2c, Scheme 4), and 3-(4-methoxyphenyl)propyl methyl carbonate (2d, Scheme 4).
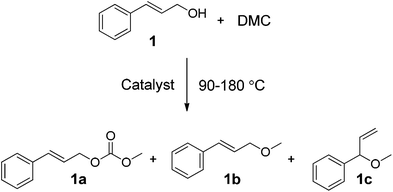 | ||
| Scheme 2 | ||
Results
Cinnamyl alcohol, 1
The reaction of DMC with cinnamyl alcohol, 1, was investigated first at 90 °C and then at 165 and 180 °C. In both cases a solution of 1 (0.3 g, 1.9 mmol) in DMC (0.36 mol, 30 mL; DMC serving both as a reagent and a solvent) was set to react in the presence of different catalysts: K2CO3, CsF/αAl2O3, NaX, NaY, or [P8881][CH3OCOO] (the weight ratio (Q) of solid catalyst:substrate was 3:1; the molar ratio of [P8881][CH3OCOO]:1 was 0.05:1). The reaction conditions for the solid catalysts in particular were chosen based on previously reported experimental conditions.18,24 The weight ratios were not optimised. The progress of the reactions was followed by GC-MS. When operating at 90 °C all reactions were carried out in glassware under atmospheric conditions. When operating at 165 and 180 °C a stainless steel autoclave was used and the mixture was flushed with N2.The study was aimed at understanding the effect of different catalysts and temperatures on the conversion of 1, in terms of its extent and the relative selectivities achievable.
Three main compounds were observed, isolated, and characterised by GC-MS and 1H NMR (1a, 1b, and 1c, Scheme 2). Unidentified by-products amounted to ≤17%. The results are listed in Tables 1 (90 °C) and 2 (165–180 °C).
| Entry | Catalyst | Qa (wt:wt) or mol:mol (IL) | Timeb/h | Conv./% | GC-MS selectivity/% | ||
|---|---|---|---|---|---|---|---|
| 1a | 1b | Othersd | |||||
| a Q is the weight (or molar) ratio between the catalyst and the substrate.b Time to reach equilibrium.c Time when the reaction stopped.d Others: unidentified by-products detected by GC-MS analyses. | |||||||
| 1 | K2CO3 | 3 | 96 | 91 | 90 | 2 | 8 |
| 2 | [P8881][CH3OCOO] | 0.05 | 5.5 | 89 | 90 | 4 | 6 |
| 3 | CsF/αAl2O3 | 3 | 50 | 94 | 81 | 3 | 17 |
| 4 | NaX | 3 | 24c | 0 | — | — | — |
| 5 | NaY | 3 | 24c | 0 | — | — | — |
| Entry | Catalyst | Qb (wt:wt) or mol:mol (IL) | Temp./°C | Conv./% | GC-MS selectivity/% | |||
|---|---|---|---|---|---|---|---|---|
| 1a | 1b | 1c | Othersc | |||||
| a All reactions lasted 3 h.b Q is the weight (or molar) ratio between the catalyst and the substrate.c Others: unidentified by-products detected by GC-MS analyses. | ||||||||
| 1 | K2CO3 | 3 | 165 | 98 | 82 | 10 | 3 | 4 |
| 2 | [P8881][CH3OCOO] | 0.05 | 165 | 71 | 86 | 5 | 1 | 8 |
| 3 | NaY | 3 | 165 | 51 | 4 | 70 | 21 | 5 |
| 4 | NaX | 3 | 165 | 85 | 29 | 45 | 21 | 6 |
| 5 | NaY | 3 | 180 | 100 | — | 91 | 8 | 2 |
| 6 | NaX | 3 | 180 | 100 | — | 56 | 39 | 5 |
For the reaction catalysed by CsF/αAl2O3 at 90 °C, a typical plot of the conversion of 1 as a function of time is shown in Fig. 2. In the case of K2CO3, [P8881][CH3OCOO], and CsF/αAl2O3 (Table 1, entries 1–3; Fig. 2), the reaction proceeded with high conversion (89–94%) and good selectivity (81–90%) towards the transesterification product, cinnamyl methyl carbonate (1a), with only traces of cinnamyl methyl ether (1b). The transesterification equilibrium was achieved in 5.5 h to 96 h, for [P8881][CH3OCOO] and K2CO3, respectively. As shown in Fig. 2 for CsF/αAl2O3, equilibrium was reached in 50 h. Isolated yields of 80–81% were reached for product 1a. In the case of the reaction with K2CO3 (entry 1, Table 1), 1a could be isolated in 95% purity (1H NMR) simply by the removal of the solid catalyst by filtration, and of excess DMC by rotary evaporation. In the case of the reaction with [P8881][CH3OCOO], 1a was isolated by flash column chromatography (fcc) on silica gel eluting with a petroleum ether–diethyl ether mixture (50:50 v/v). No attempts to optimise product isolation were made as this was beyond the scope of the work. No conversion of 1 was observed after 24 h for either of the NaX or NaY catalyst at 90 °C.
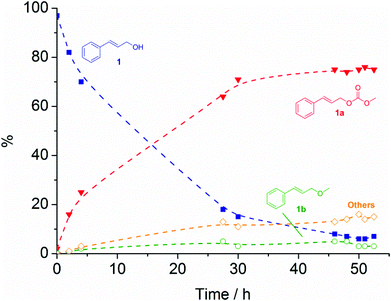 | ||
Fig. 2 Plot of the conversion of 1 ( ) and formation of 1a ( ) and formation of 1a ( ), 1b ( ), 1b ( ), and unidentified by-products ( ), and unidentified by-products ( ) at 90 °C in the presence of CsF/αAl2O3. Broken lines are shown as a visual guide only. ) at 90 °C in the presence of CsF/αAl2O3. Broken lines are shown as a visual guide only. | ||
The higher temperature regime of 165–180 °C was also investigated using K2CO3, [P8881][CH3OCOO], NaX and NaY as catalysts in an autoclave. The reaction time was 3 h. In addition to products 1a and 1b, 3-methoxy-3-phenylpropene, 1c, was also observed (Scheme 1). The results are listed in Table 2.
Just as was observed at 90 °C, 1a was the major product in the presence of K2CO3 and [P8881][CH3OCOO] at 165 °C. However, bearing in mind the different reaction times: (i) the selectivity to 1a decreased from 90% (90 °C), to 82–86% at 165 °C; (ii) in the presence of K2CO3, the conversion increased to 98% at 165 °C, compared to 91% at 90 °C; (iii) instead, at the same temperatures, in the presence of [P8881][CH3OCOO], the conversion decreased from 89% to 71%.
At 165 °C, NaY and NaX prompted conversion (51% and 85% for NaY and NaX, respectively) mainly to 1b with 70% selectivity in the presence of NaY and with 45% selectivity in the presence of NaX. A minor product, 3-methoxy-3-phenylpropene, 1c, was also observed. The transesterification product, 1a, was only obtained with 4% and 29% selectivity with NaY and NaX, respectively.
To achieve quantitative conversion in the presence of both NaY and NaX it was sufficient to increase the temperature to 180 °C. In the presence of NaY, the major product, 1b, could be obtained with 91% selectivity and 64% isolated yield (by fcc). The minor product, 1c, was also observed with 8% selectivity. In the presence of NaX, the selectivity towards the major product, 1b, decreased to 56% due to the sizable amount of the co-product, 1c (38%). The different selectivities of the two zeolites towards 1a prompted us to consider as the cause an in situ decarboxylation reaction of 1a to 1b. To corroborate such a hypothesis, cinnamyl methyl carbonate, 1a, (entry 1, Table 1) was heated in the presence of NaY under conditions identical to those reported in entry 5, Table 2, with the solvent changed from DMC to dimethoxyethane (DME). In this case, 1b was the sole observed product (Scheme 3).
 | ||
| Scheme 3 | ||
Taking into consideration the relatively large quantity of zeolites used it is desirable that the catalyst be recyclable. Thus, a series of zeolite recycling experiments was carried out. Once the experiment of entry 3 in Table 2 was completed, the solid NaY zeolite was filtered, washed with diethyl ether, and dried at 70 °C under vacuum overnight. The recovered NaY was then used to repeat the reaction, under identical conditions to those reported above (entry 3, Table 2), and the results are reported in Fig. 3 (Run 1). The recovery/reaction cycle was repeated once more. Both runs 1 and 2 showed a significant drop in the catalyst performance: after 2 runs the conversion of 1 and the selectivity to 1b decreased to 26 and 61%, respectively. However, if the used zeolite was re-calcined (400 °C, ramp rate 5 °C min−1) and used in another reaction, the catalytic activity was restored (Run 3). To probe the recyclability of the zeolite further, an additional recovery/reaction cycle was performed, followed by another re-calcination/reaction sequence. As expected, the conversion dropped in the reaction where the catalyst was simply dried under vacuum (Run 4, Fig. 3), while the activity was restored after the second re-calcination (Run 5).
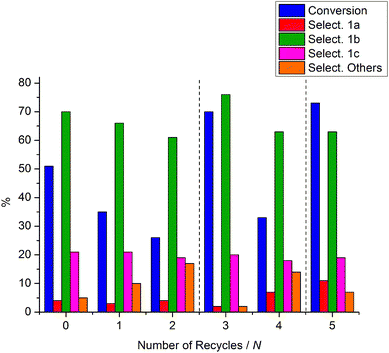 | ||
| Fig. 3 The recycling of NaY. All reactions are carried out at 165 °C for 3 h. | ||
4-(3-Hydroxypropyl)phenol, 2
The reaction of 4-(3-hydroxypropyl)phenol, 2, with DMC was tested under reaction conditions analogous to those described above, first at 90 °C at atmospheric pressure, and then at 165 and 180 °C in an autoclave under N2. A solution of 2 (0.3 g, 1.9 mmol) in DMC (0.36 mol, 30 mL) was prepared to react in the presence of different catalysts: K2CO3, CsF/αAl2O3, NaX, NaY or [P8881][CH3OCOO] (the weight ratio (Q) of solid catalyst:substrate was 3:1; the molar ratio of [P8881][CH3OCOO]:2 was 0.05:1). Here, too, the reaction conditions were chosen based on previous experience,13,19 and the weight ratios were not optimised.18,19 As with cinnamyl alcohol, 1, the study was aimed at understanding the effect on the conversion of 2 of different catalysts at different temperatures, and on controlling product selectivity. Five main products were observed, isolated, and characterised by GC-MS and 1H NMR (2a, 2b, 2c, 2d, and 2e, Scheme 4). The new product, 2d, and the product, 2b, for which there was no previously reported NMR data, were also characterised by 13C NMR. Other unidentified by-products (≤8% total yield, by GC-MS) were also detected. The results are listed in Tables 3 and 4 and in Fig. 4.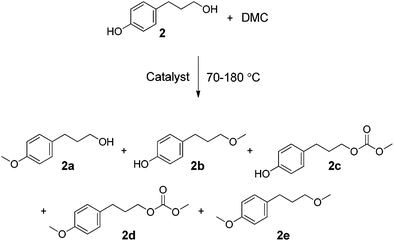 | ||
| Scheme 4 | ||
![Plot of the conversion of 2 () and formation of 2c (), 2d (), and unidentified by-products () at 90 °C in the presence of [P8881][CH3OCOO]. Broken lines are shown as a visual guide only.](/image/article/2013/GC/c3gc40334c/c3gc40334c-f4.gif) | ||
Fig. 4 Plot of the conversion of 2 ( ) and formation of 2c ( ) and formation of 2c ( ), 2d ( ), 2d ( ), and unidentified by-products ( ), and unidentified by-products ( ) at 90 °C in the presence of [P8881][CH3OCOO]. Broken lines are shown as a visual guide only. ) at 90 °C in the presence of [P8881][CH3OCOO]. Broken lines are shown as a visual guide only. | ||
| Entry | Catalyst | Qa (wt:wt) or mol:mol (IL) | Temp./°C | Timeb/h | Conv./% | GC-MS selectivity/% | |||
|---|---|---|---|---|---|---|---|---|---|
| 2a | 2c | 2d | Othersc | ||||||
| a Q is the weight (or molar) ratio between the catalyst and the substrate.b Time to reach equilibrium.c Others: unidentified by-products detected by GC-MS analyses.d Time when the reaction stopped.e Non-optimised time. Equilibrium reached between 3 and 18 h.f Non-optimised time. Equilibrium reached between 55 and 119 h. | |||||||||
| 1 | K2CO3 | 3 | 90 | 74 | 97 | — | 89 | 4 | 7 |
| 2 | [P8881][CH3OCOO] | 0.05 | 90 | 150d | 97 | — | 72 | 23 | 4 |
| 3 | CsF/αAl2O3 | 3 | 90 | 18e | 96 | — | 83 | 13 | 4 |
| 4 | NaY | 3 | 90 | 240d | 14 | 5 | 95 | 0 | 0 |
| 5 | NaX | 3 | 90 | 240d | 53 | 42 | 36 | 13 | 8 |
| 6 | CsF/αAl2O3 | 3 | 70 | 119f | 95 | — | 94 | 5 | 1 |
| Entry | Catalyst | Qb (wt:wt) or mol:mol (IL) | Temp./°C | Conv./% | GC-MS selectivity/% | |||||
|---|---|---|---|---|---|---|---|---|---|---|
| 2a | 2b | 2c | 2d | 2e | Othersc | |||||
| a All reactions lasted 3 h.b Q is the weight (or molar) ratio between the catalyst and the substrate.c Others: unidentified by-products detected by GC-MS analyses. | ||||||||||
| 1 | K2CO3 | 3 | 165 | 99 | 0 | — | 60 | 38 | — | 2 |
| 2 | [P8881][CH3OCOO] | 0.05 | 165 | 95 | 2 | — | 62 | 32 | — | 4 |
| 3 | NaY | 3 | 165 | 48 | 8 | 11 | 77 | 3 | — | 0 |
| 4 | NaX | 3 | 165 | 100 | 3 | 4 | 5 | 78 | 10 | 0 |
| 5 | NaY | 3 | 180 | 80 | 2 | 14 | 76 | 6 | — | 1 |
| 6 | NaX | 3 | 180 | 100 | 1 | 17 | 2 | 65 | 12 | 3 |
At 90 °C, in the presence of K2CO3, [P8881][CH3OCOO], and CsF/αAl2O3, the main reaction was the transesterification of the aliphatic OH of 2 with DMC, which yielded 3-(4-hydroxyphenyl)propyl methyl carbonate, 2c (entries 1–3, Table 3). With K2CO3 and CsF/αAl2O3 as catalysts conversions of 2 and selectivities to 2c were 96–97% and 83–89%, achieved after 74 h or 18 h, respectively (entries 1 and 3, Table 3). An experiment to improve selectivity using CsF/αAl2O3 was conducted at lower temperature (70 °C, entry 6, Table 3). In this case, notwithstanding a longer reaction time (119 h vs. 18 h at 90 °C), the selectivity to product 2c increased up to 94% (by GC-MS). The product 2c could be isolated with yields up to 72%.
A different behaviour was observed in the case of [P8881][CH3OCOO]. Initially, the conversion to 2c reached a maximum of 74% at 49 h and then dropped to 67% after 150 h due to the formation of 2d that increased from 16% to 26%, as shown in Fig. 4.
In the presence of NaY and NaX (entries 4 and 5, Table 3), low conversions (14 and 53% respectively) were obtained even after extended reaction times. The transesterification compound, 2c, was again the major product of the reaction catalysed by NaY. In contrast, in the presence of NaX, methylation occurred predominantly on the aromatic OH group to form 2a, while low conversion to the transesterification products 2c and 2d was observed after longer reaction times.
The experiment was then performed at 165–180 °C, conditions as above. In addition to 2a, 2c and 2d, methyl ethers 2b and 2e were also observed (Scheme 4). The results are listed in Table 4.
At 165 °C, in the presence of K2CO3 and [P8881][CH3OCOO], conversion was similar to that at 90 °C (again, bearing in mind the different reaction times); however, the product distributions were remarkably different: the selectivity towards 2c dropped because of the formation of 2d. The ratio 2c:2d was in the range of 1.5–1.8 (entries 1 and 2, Table 4). It should be noted that product 2d has not been previously reported in the literature.
With NaY and NaX as catalysts two distinct product distributions were observed. NaY yielded 2c both at 165 °C and at 180 °C, with 48% and 80% conversion and with selectivities of 77% and 76%, respectively. On the other hand, NaX achieved 100% conversion with 2d the major product both at 165 and 180 °C. The fully methylated product 2e was also present with yields of 10–12% after 3 h. To shed light on the reaction pathway of 2 with DMC in the presence of NaX at 180 °C, a preliminary kinetic profile was collected (Fig. 5), with samples taken at different reaction times between 10 min and 5 h.
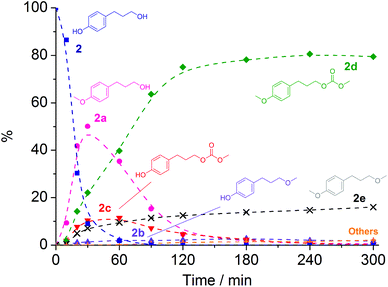 | ||
Fig. 5 Plot of the conversion of 2 ( ) and formation of 2a ( ) and formation of 2a ( ), 2b ( ), 2b ( ), 2c ( ), 2c ( ), 2d ( ), 2d ( ), 2e ( ), 2e ( ), and unidentified by-products ( ), and unidentified by-products ( ) at 180 °C in the presence of NaX. Broken lines are shown as a visual guide only. ) at 180 °C in the presence of NaX. Broken lines are shown as a visual guide only. | ||
The profile of Fig. 5 indicated that: (i) the proportion of product 2a reached a maximum of 50% in 30 min, and then dropped to less than 1% within 4 h; (ii) similarly, the proportion of product 2c went up to a maximum of 12% in 1 h, and then decreased as the reaction proceeded further; (iii) the proportion of product 2d reached 80% after 120 min, and remained steady for the next 3 hours; (iv) the proportion of product 2b remained low, reaching a maximum of 3% within 3 h and then decreasing as the reaction proceeded; and (v) the proportion of product 2e increased with time, and reached a selectivity of 16% in 5 h.
Although the concentration of 2d appeared to plateau, as shown in Fig. 5, an additional experiment was performed to determine whether 2d could react further to give 2evia an in situ decarboxylation, by analogy to the conversion of 1a to 1b. The reaction was performed at 180 °C for 3 h. Pure 2d was used as a starting material, NaX as a catalyst, and DME as a solvent in place of DMC. However, in this case 2e was not formed (Scheme 5). Instead, 2a was obtained as the major product. When an identical reaction was performed with NaX replaced with NaY, no conversion was attained at all.
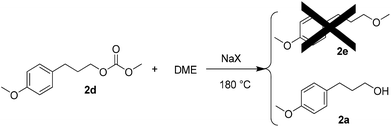 | ||
| Scheme 5 | ||
Discussion
Effect of the different catalysts at 90 °C: selective formation of carbonates 1a and 2c by transesterification
The five catalysts tested at 90 °C showed that the reactivity of 1 and 2 with DMC followed trends that could be rationalised based on the previous knowledge of base-catalysed reactions of DMC with hydroxyl-containing substrates. In particular, basic catalysts such as K2CO3, CsF/αAl2O3, and [P8881][CH3OCOO] at this lower temperature prompted a selective equilibrium reaction of 1 and 2 to yield the transesterification products at the aliphatic OH: 1a and 2c. Selectivity towards the transesterification product 2c with CsF/αAl2O3 could be improved by lowering the temperature and increasing the reaction times. This was consistent with previous investigations of reactions of dialkyl carbonates with alcohols in the presence of basic catalysts.25–27 The aromatic hydroxyl group of 2 remained relatively inert, except with [P8881][CH3OCOO]. In this case, after reaching a maximum of 74% selectivity towards 2c, the reaction progressed further in favour of the product 2d showing that this catalyst promoted the methylation of the phenolic OH. This higher reactivity of [P8881][CH3OCOO] for methylation on the phenolic OH occurred notwithstanding the lower catalyst loadings (5 mol%), compared to the other solid catalysts used in up to 3 weight excess over the substrate. This behaviour was in analogy to our recently reported results on the IL-catalysed transesterification of alcohols with dialkylcarbonates.26 No attempts were made here to recycle K2CO3, as this is an already established option.7The NaX and NaY solid zeolite catalysts are also basic, but less so than K2CO3, CsF/αAl2O3 and [P8881][CH3OCOO], and this probably accounts for the observation that at 90 °C no conversion was observed in 24 h for the reaction of DMC with 1, and that low conversion was observed for the reaction with 2 after extended reaction periods. Albeit with low conversion, in the presence of NaX, 2a was formed, followed by a separate transesterification step to form 2c. This result was opposite to that observed previously by us in the reaction of DMC with hydroxybenzyl alcohols catalysed by X and Y-faujasites.18 In that case, the benzyl OH group was more prone to methylation than the aromatic OH one. When the reaction of 2 in the presence of NaX at 90 °C was allowed to proceed further, 2d was also observed. While selectivity in this case was low, these results were in agreement with the mechanistic study undertaken at 180 °C, as described below.
In the presence of NaY at 90 °C, only 2c was observed at low conversion indicating preferential reaction at the aliphatic OH.
Effect of temperature on catalysis by K2CO3 and [P8881][CH3OCOO]
For the reaction of DMC with cinnamyl alcohol, 1, there was no benefit with increasing the temperature from 90 °C to 165 °C. While conversion increased in the presence of K2CO3 despite the shorter reaction time (entry 1, Tables 1 and 2), selectivity to the transesterification product 1a decreased with temperature in favour of the methyl derivative 1b and of compound 1c. This selectivity was in agreement with the occurrence of a decarboxylation process as reported for high temperature reactions of dialkyl carbonates over faujasite catalysts (see also the next paragraph).20 Conversion decreased in the presence of [P8881][CH3OCOO] (entry 2, Tables 1 and 2).In the reaction of DMC with 2, there was a negligible change in conversion between 90 and 165 °C (although the reactions were performed over different reaction times); however, the selectivity dropped from the sole transesterification reaction product 2c at 90 °C, to a mixture of 2c and 2d at 165 °C, the latter deriving from O-methylation competing with transesterification at the higher temperature (entries 1 and 2, Tables 3 and 4). Hardly any conversion to products 2a, 2b, and 2e was observed after 3 h.
Effect of the type of zeolite on the reaction of DMC with cinnamyl alcohol 1 at 165–180 °C: selective formation of the methyl ether 1b
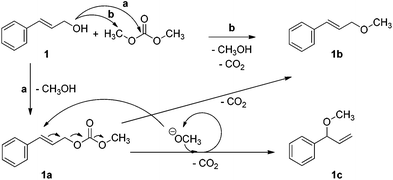
The data reported in Table 2 suggest that NaX and NaY exhibit different activities for the reaction of DMC with 1. Although both zeolites have the same framework, their Si/Al ratios are 1.2 and 2.4 for X and Y zeolites, respectively.28 This difference results in a higher abundance of active sites and a higher basicity of NaX compared to NaY, and in our case, the greater abundance of sites may be directly correlated to the higher conversion achieved with NaX.As for the selectivity, it also appears to be dependent on the nature of the site: the less basic sites in NaY favour the formation of 1b, while the stronger basic sites of NaX favour 1a. The product 1c is also formed preferentially with the more basic NaX catalyst. The result is consistent with the position of NaX in the acid–base scale proposed by Barthomeuf for faujasites,29,30 and with our previous findings on the reactivity of indolyl carboxylic acids with DMC in the presence of zeolites.17 Plausible reaction pathways that account for the formation of 1a, 1b, and 1c are shown in Scheme 6.
 | ||
| Scheme 6 | ||
Formation of 1a can occur only by a carboxymethylation pathway (a).The methyl ether 1b, instead, can be formed either by direct methylation of 1 (pathway b), or by decarboxylation of 1a.
The lack of 1a at incomplete conversion in the presence of NaY suggested that pathway (b) in Scheme 6 could be favoured over pathway (a).
The product 1c could be formed from 1a by an allyl rearrangement catalysed by methoxide, followed by decarboxylation as indicated in Scheme 6. Indirect evidence for this hypothesis was collected by heating 1a under the same experimental conditions, but in the absence of DMC as a source of methanol, and observing 1b as the sole product. The lack of 1c indicated that it was formed only when DMC was present and able to generate methoxide. As 1a is required for the formation of 1c, this explanation could also account for the lack of conversion to 1c in the presence of NaY compared to NaX. We considered it unnecessary to run a full kinetic profile of the reaction of 1 in the presence of zeolite NaX, as this pathway is less complex than the one that applies for 2 (see later), making identification of the intermediates easier.
Recycling experiments of NaY at 165 °C
Fig. 3 clearly shows that NaY can be reused for the reaction with 1 with no loss in activity after re-calcination at 400 °C in dry air, or with some loss in conversion after mild thermal treatment (70 °C, under vacuum). Recyclability is desirable as the catalyst is used in quite high amounts (although it is conceivable that the weight ratio could be dropped from Q = 3 to Q = 1.518). Nonetheless, the drop in conversion in the recycling runs 1, 2, and 4 (Fig. 3) could be explained by the absorption of water by the zeolite, which could occur during the handling of the catalyst between runs (water is not efficiently removed during the mild thermal treatment), causing hydrolysis of the cinnamyl methyl carbonate intermediate 1a, back to the starting cinnamyl alcohol. This is consistent with the restoration of the catalytic activity (Runs 3 and 5, Fig. 3) after the used zeolite was re-calcined (400 °C, ramp rate 5 °C min−1), although some fouling by organic compounds cannot be ruled out. This hydration/fouling of the zeolite could also account for the poorer selectivity after each recycle, particularly as the water could promote other reactions such as hydrolysis. The formation of coke from the initial fouling components could in principle cause loss of activity after recycle runs, and the subsequent improvement in activity after calcination is also consistent with the removal of any coke. Nonetheless, the zeolite remained white throughout the experiments and the reaction solutions were clear and colourless. Thus, although the formation of undetectable white coke on the catalyst31 cannot be ruled out, we deem it unlikely that any was formed in the present case. It is interesting to note that the selectivity towards products 1a and 1c was otherwise relatively unchanged between the recycling/regeneration runs.The reaction pathway of 2 in the presence of NaX: formation of carbonate 2d
Based on the results reported in Tables 3 and 4, and Fig. 5, the sequence for the reaction of DMC with 2 in the presence of NaX could involve three different pathways with two final products, as shown in Scheme 7. Methylation could occur first on the phenolic OH, forming 2a, followed by transesterification at the aliphatic OH to form 2d. At the same time, transesterification could occur on the aliphatic OH to form 2c, which then undergoes methylation at the phenolic OH to also form 2d. These pathways are consistent with the results obtained in the presence of NaX at 90 °C. Simultaneously, at the higher temperature, 2b could be formed from the methylation of the aliphatic OH, and then undergo methylation at the phenolic OH to form 2e as the final product.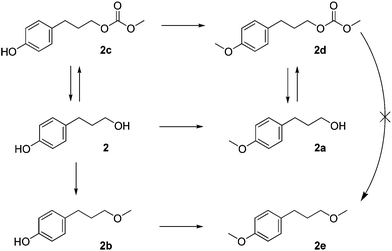 | ||
| Scheme 7 | ||
The formation of 2e
In contrast with the observed formation of 1b from the in situ decarboxylation of 1a with NaY (Scheme 3), the reaction of 2d with NaX using DME as a solvent (Scheme 5) indicates that an in situ decarboxylation of 2d (or indeed of 2c) does not occur. The data of Table 2 show that at e.g. 165 °C (compare entries 3 and 4) NaY is less active than NaX, but shows a much greater selectivity towards the decarboxylation of 1a to yield 1b. Likewise, in the reaction of 2 (Table 4), NaY is also less active than NaX. The lower basicity of NaY as compared to NaX originates from a higher Si:Al ratio. This means that fewer active sites are present and that these active sites will be able to interact more strongly with the various electron-rich species during the reaction. Both of these phenomena might lead to lower conversions. The difference in selectivity, however, is most likely due to the less basic sites as they promote decarboxylation over hydrolysis. In the case of 2b this is even more extreme due to the additional electron-donating methoxy group, which further strengthens the interactions of the substrates with the zeolite, leading to complete deactivation with NaY.Experimental
General
The following reagents were used as received: 4-(3-hydroxypropyl)phenol, 2, dimethyl carbonate (DMC), potassium carbonate (K2CO3), diethyl ether, and petroleum ether (all Aldrich). Cinnamyl alcohol, 1 (Sigma), was purified by vacuum distillation. Dimethoxyethane (DME, Aldrich) was dried by distillation over sodium and stored under nitrogen. The zeolites NaX and NaY (both Aldrich) were calcined at 400 °C (ramp rate 5 °C min−1) under dry air for 8–14 h immediately before use. [P8881][CH3OCOO] was prepared according to a method recently reported by us.19 CsF/αAl2O3 was also prepared according to the literature.27MS (EI, 70 eV) analyses were run using a GC fitted with an HP5/MS capillary column (30 m). 1H NMR spectra were recorded using a 300 MHz spectrometer. 13C NMR spectra were recorded using a 100 MHz spectrometer. CDCl3 was used as a solvent. Chemical shifts were reported in δ values downfield from TMS.
General procedure for reactions carried out in glassware
A two-necked, 50 mL round bottom glass Quickfit flask was fitted with a Sura-Seal stopper and condenser, and equipped with a magnetic stirrer. The substrate (cinnamyl alcohol, 1, or 4-(3-hydroxypropyl)phenol, 2, 1.9 mmol) and dimethyl carbonate (0.36 mol, 30 mL) were added to the flask. The mixture was allowed to stir at RT for ∼5 min and a sample was taken (∼0.1 mL). The catalyst (K2CO3, CsF/αAl2O3, NaX, NaY or [P8881][CH3OCOO]) [the weight ratio (Q) of solid catalyst:substrate was 3:1; the molar ratio of [P8881][CH3OCOO]:substrate was 0.05:1]) was then added, and the reaction was run at 90 °C with stirring at 800 rpm. Samples (∼0.1 mL) were taken at regular intervals using a syringe through one arm of the reactor. All samples taken from the reactor were diluted with diethyl ether, and either centrifuged to remove the solid catalyst (K2CO3, CsF/αAl2O3, NaX, NaY) or passed over silica gel F60 to remove the ionic liquid catalyst ([P8881][CH3OCOO]), and the progress of the reaction was followed by GC-MS. The reaction was generally run until equilibrium was established (5.5–119 h), as determined by GC-MS.General procedure for reactions carried out in an autoclave
A stainless-steel autoclave (120 mL internal volume) was charged with the substrate (cinnamyl alcohol, 1, or 4-(3-hydroxypropyl)phenol, 2, 1.9 mmol), dimethyl carbonate (0.36 mol, 30 mL) and the catalyst (K2CO3, [P8881][CH3OCOO], NaX and NaY) [the weight ratio (Q) of solid catalyst:substrate was 3:1; the molar ratio of [P8881][CH3OCOO]:substrate was 0.05:1]). Before the reaction, the autoclave was purged at RT with N2 to remove air. The autoclave was then electrically heated to the desired temperature (165–180 °C), with the reaction mixture kept under magnetic stirring throughout the reaction. After 3 h, the autoclave was cooled to RT, purged to remove CO2, and opened. A sample of the reaction mixture (∼0.1 mL) was taken, diluted with diethyl ether, either centrifuged to remove the solid catalyst (K2CO3, NaX, NaY) or passed over silica gel F60 to remove the ionic liquid catalyst ([P8881][CH3OCOO]), and the progress of the reaction was followed by GC-MS.Reaction carried out in an autoclave with sampling
A stainless-steel autoclave (240 mL internal volume), fitted with a Swagelok tap for sampling, was charged with the substrate (4-(3-hydroxypropyl)phenol, 2, 3.8 mmol), dimethyl carbonate (0.72 mol, 60 mL) and NaX [the weight ratio (Q) of zeolite:substrate was 3:1]. Before the reaction, the autoclave was purged at RT with N2 to remove air. The autoclave was then electrically heated to 180 °C, with the reaction mixture kept under magnetic stirring throughout the reaction. Samples (∼0.3 mL) were taken by opening the Swagelok tap at 10, 20, 30, 60, 90, 120, 180, 240, and 300 min. All samples taken from the reactor were diluted with diethyl ether, the solid catalyst removed by centrifugation, and the progress of the reaction was analysed by GC-MS. After 5 h, the autoclave was cooled to RT, purged to remove CO2, and opened.Recycling of NaY
A series of recycling experiments was carried out for NaY for the reaction of DMC with cinnamyl alcohol 1. Once the experiment of entry 3 in Table 2 was completed, the solid NaY zeolite was removed by filtration, washed with diethyl ether, and dried at 70 °C under vacuum overnight. The recovered NaY was then used in a subsequent reaction under identical conditions. The recovery/reaction cycle was repeated once more. After the second recycle, the solid NaY was re-calcined fluxing with air at 400 °C (ramp rate 5 °C min−1) for 12 h, and used for a subsequent reaction. The recovery/reaction was then repeated one more time, followed by another re-calcination and final re-use of the catalyst.The isolation and characterisation of products obtained from cinnamyl alcohol, 1, using solid catalysts
Crude cinnamyl methyl carbonate, 1a, cinnamyl methyl ether, 1b, and 3-methoxy-3-phenylpropene, 1c, were isolated by simple filtration of the solid and removal of DMC by rotary evaporation. Cinnamyl methyl ether, 1b, and 3-methoxy-3-phenylpropene, 1c, were further purified by flash column chromatography on silica gel F60 (eluent: petroleum ether–diethyl ether in 9:1 or 95:5 v/v). The products were characterised by GC-MS and 1H NMR.The isolation and characterisation of products obtained from cinnamyl alcohol, 1, using [P8881][CH3OCOO]
Cinnamyl methyl carbonate, 1a, was isolated and purified by flash column chromatography on silica gel F60 (eluent: petroleum ether–diethyl ether in 50:50 v/v), and characterised by GC-MS and 1H NMR.The isolation and characterisation of products obtained from 4-(3-hydroxypropyl)phenol, 2
3-(4-Hydroxyphenyl)propyl methyl carbonate 2c was isolated and purified by flash column chromatography on silica gel F60 (eluent: petroleum ether–diethyl ether in 50:50 v/v). 4-(3-Methoxypropyl)phenol, 2b, was isolated and purified by flash column chromatography on silica gel F60 (eluent: petroleum ether–diethyl ether in 30:70 v/v). 3-(4-Methoxyphenyl)-1-propanol, 2a, 3-(4-methoxyphenyl)propyl methyl carbonate, 2d, and 1-methoxy-4-(3-methoxypropyl)benzene, 2e, were isolated and purified by flash column chromatography on silica gel F60 (gradient dilution, eluent: petroleum ether–diethyl ether in 30:70–50:50 v/v). All products were characterised by GC-MS and 1H NMR. The new product, 3-(4-methoxyphenyl)propyl methyl carbonate, 2d, and the product for which there were no previously reported NMR data in the literature, 4-(3-methoxypropyl)phenol, 2b, were also characterised by 13C NMR.Conclusions
In order to establish synthetic procedures to upgrade phenolic streams derived from lignin processing, the easily accessible catalysts K2CO3, CsF/αAl2O3, NaX, NaY and an ionic liquid (IL) [P8881][CH3OCOO] were studied for the methylation and transesterification of cinnamyl alcohol 1, and 4-(3-hydroxypropyl)phenol 2, selected as functional model compounds of p-coumaryl alcohol. The results indicated that a straightforward, efficient and selective catalytic upgrading was possible with DMC as a solvent/reagent, and that derivatives with largely different chemical properties compared to the starting molecules (e.g. polarity, hydrogen-bonding ability, etc.) can be obtained, making them potentially interesting in their own right.The basic catalysts K2CO3, CsF/αAl2O3, and [P8881][CH3OCOO] yielded the aliphatic carbonates 1a and 2c at 90 °C, by transesterification, with high conversions (>90%) and selectivity (>80%).
The corresponding methyl ethers were instead accessible at higher temperature in the presence of faujasites NaY or NaX as catalysts. For example, cinnamyl methyl ether 1b was formed with 100% conversion and 91% selectivity from 1 with NaY at 180 °C.
The zeolites displayed different reactivity for 1 and 2. With cinnamyl alcohol 1, the more basic NaX favoured reaction at the carbonyl carbon of DMC forming the methyl carbonate intermediate 1a, followed by in situ decarboxylation to the methyl ether 1b. Instead, NaY promoted direct reaction towards 1b, presumably because DMC reacts more favourably at the methyl carbon without passing through the methyl carbonate intermediate.
With 4-(3-hydroxypropyl)phenol 2, NaX was also more active than NaY as a catalyst. Two separate reaction sequences occurred: the fully methylated product 2e was formed through the direct methylation of both the aliphatic and aromatic hydroxyls, while transesterification of the aliphatic hydroxyl and aromatic OH methylation with DMC yielded 2d.
Several green aspects can be recognised: (i) DMC is non-toxic and is used both as a reagent and a solvent; (ii) NaY and NaX are safe catalysts that can be recovered, reactivated, and re-used without any loss of conversion or selectivity; (iii) methanol and CO2 are the only by-products and can be recycled to form DMC; and (iv) the model compounds themselves are an inherently renewable, environmentally friendly feedstock.
Acknowledgements
We are grateful to the Italian Ministry of Education, Universities and Research (MIUR) for funding through the MIUR Cooperlink program. JNGS acknowledges the receipt of an Australian Postgraduate Award and a Dr Joan R. Clark Research Scholarship. TM acknowledges the ARC for a Future Fellowship.Notes and references
- A. Demirbas, Prog. Energ. Combust., 2007, 33, 1–18 CrossRef CAS.
- J. Zakzeski, A. L. Jongerius, P. C. Bruijnincx and B. M. Weckhuysen, ChemSusChem, 2012, 5, 1602–1609 CrossRef CAS PubMed.
- J. Zakzeski, P. C. A. Bruijnincx, A. L. Jongerius and B. M. Weckhuysen, Chem. Rev., 2010, 110, 3552–3599 CrossRef CAS PubMed.
- J. J. Bozell, J. E. Holladay, D. Johnson and J. F. White, Top Value Added Candidates from Biomass, Volume II: Results of Screening for Potential Candidates from Biorefinery Lignin, Pacific Northwest Laboratory, Richland, WA, 2007 Search PubMed.
- J. J. Bozell and G. R. Petersen, Green Chem., 2010, 12, 539–554 RSC.
- P. N. R. Vennestrom, C. M. Osmundsen, C. H. Christensen and E. Taarning, Angew. Chem., Int. Ed., 2011, 50, 10502–10509 CrossRef CAS PubMed.
- P. Y. Dapsens, C. Mondelli and J. Perez-Ramirez, ACS Catal., 2012, 2, 1487–1499 CrossRef CAS.
- M. Selva and A. Perosa, Green Chem., 2008, 10, 457–464 RSC.
- D. Delledonne, F. Rivetti and U. Romano, J. Organomet. Chem., 1995, 488, C15–C19 CrossRef CAS.
- N. Keller, G. Rebmann and V. Keller, J. Mol. Catal. A: Chem., 2010, 317, 1–18 CrossRef CAS.
- B. Schaffner, F. Schaffner, S. P. Verevkin and A. Borner, Chem. Rev., 2010, 110, 4554–4581 CrossRef CAS PubMed.
- M. Selva, C. A. Marques and P. Tundo, J. Chem. Soc., Perkin Trans. 1, 1994, 1323–1328 RSC.
- A. Bomben, M. Selva, P. Tundo and L. Valli, Ind. Eng. Chem. Res., 1999, 38, 2075–2079 CrossRef CAS.
- M. Selva, Synthesis, 2003, 2872–2876 CrossRef CAS.
- M. Selva, P. Tundo and T. Foccardi, J. Org. Chem., 2005, 70, 2476–2485 CrossRef CAS PubMed.
- M. Selva and P. Tundo, J. Org. Chem., 2006, 71, 1464–1470 CrossRef CAS PubMed.
- M. Selva, P. Tundo, D. Brunelli and A. Perosa, Green Chem., 2007, 9, 463–468 RSC.
- M. Selva, E. Militello and M. Fabris, Green Chem., 2008, 10, 73–79 RSC.
- M. Fabris, V. Lucchini, M. Noe, A. Perosa and M. Selva, Chem.–Eur. J., 2009, 15, 12273–12282 CrossRef CAS PubMed.
- M. Selva, M. Fabris and A. Perosa, Green Chem., 2011, 13, 863–872 RSC.
- M. Selva, V. Benedet and M. Fabris, Green Chem., 2012, 14, 188–200 RSC.
- P. Tundo and M. Selva, Acc. Chem. Res., 2002, 35, 706–716 CrossRef CAS PubMed.
- J. Zakzeski, A. L. Jongerius and B. M. Weckhuysen, Green Chem., 2010, 12, 1225–1236 RSC.
- M. Selva, M. Noè, A. Perosa and M. Gottardo, Org. Biomol. Chem., 2012, 10, 6569–6578 CAS.
- P. Tundo, F. Trotta, G. Moraglio and F. Ligorati, Ind. Eng. Chem. Res., 1988, 27, 1565–1571 CrossRef CAS.
- M. Selva, M. Noè, A. Perosa and M. Gottardo, Org. Biomol. Chem., 2012, 10, 6569–6578 CAS.
- J.-M. Clacens, D. Genuit, B. Veldurthy, G. Bergeret, L. Delmotte, A. Garcia-Ruiz and F. Figueras, Appl. Catal., B, 2004, 53, 95–100 CrossRef CAS.
- For morphological details and other properties of NaY and NaX faujasites, see: (a) F. Schwochow and L. Puppe, Angew. Chem., Int. Ed. Engl., 1975, 14, 620 CrossRef; (b) G. C. Bond, in Heterogeneous Catalysis Principles and Applications, Oxford University Press, New York, USA, 2nd edn, 1987, pp. 104–110 Search PubMed; and ref. 29.
- D. Barthomeuf, J. Phys. Chem., 1984, 88, 42–45 CrossRef CAS.
- B. Su and D. Barthomeuf, Stud. Surf. Sci. Catal., 1995, 94, 598–605 CrossRef CAS.
- R. Schlogl, in Preparation of Solid Catalysts, ed. G. Ertl, H. Knozinger and J. Weitkamp, Wiley-VCH, Weinheim, 1999 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: 1H NMR and MS spectra of all reported compounds and 13C NMR spectra of new compounds. See DOI: 10.1039/c3gc40334c |
| This journal is © The Royal Society of Chemistry 2013 |