Accurate determination of S in organic matrices using isotope dilution ICP-MS/MS
Lieve
Balcaen
a,
Glenn
Woods
b,
Martín
Resano
c and
Frank
Vanhaecke
*a
aGhent University, Department of Analytical Chemistry, Krijgslaan 281-S12, B-9000, Ghent, Belgium. E-mail: Lieve.Balcaen@UGent.be; Frank.Vanhaecke@UGent.be
bAgilent Technologies UK Ltd., 5500 Lakeside, Cheadle Royal Business Park, SK8 3GR Stockport, Cheshire, UK. E-mail: glenn_woods@agilent.com
cUniversity of Zaragoza, Faculty of Sciences, Department of Analytical Chemistry, Pedro Cerbuna 12, E-50009 Zaragoza, Spain. E-mail: mresano@unizar.es
First published on 12th November 2012
Abstract
Accurate determination of low levels of S in organic matrices by means of isotope dilution ICP-single quadrupole MS (e.g., for quantification of S components in reverse phase HPLC-ICP-MS experiments) is often not feasible. This work describes an accurate, sensitive and fast analytical method for the determination of S in organic matrices by means of a recently developed ‘triple quadrupole’ ICP-MS instrument, operated in MS/MS mode. The added value of the MS/MS approach for this application has been clearly visualised by varying the width of the bandpass of the first quadrupole analyzer from “fully open” (standard mode) down to single mass width (MS/MS mode). As a proof-of-concept, a biodiesel reference material has been analysed for its S content with the proposed method, using isotope dilution for calibration, and the results obtained were in excellent agreement with the certified value (within experimental uncertainty).
1 Introduction
The development of accurate, sensitive and fast analytical methods for the determination of low levels of S in organic matrices is of great importance in several application fields, e.g., S determination in fuels, biological materials and S-containing drugs. Many authors have already reported on the need for the development of such methods, to be able to (i) meet the increasing demands of governmental agencies (such as ASTM and ISO) concerning the maximum sulfur content in fuels1–4 and (ii) quantify S-containing components after a chromatographic separation.5–9ICP-MS is known as a very sensitive detection technique, which is capable of measuring a large variety of elements with a high sample throughput and which gives access to isotopic information.10 However, S determination in organic matrices by means of ICP-MS is not that straightforward, due to the following reasons: (i) occurrence of spectral overlap for all isotopes of S (Table 1); (ii) the high ionization potential of S and (iii) lower plasma robustness when introducing organics into the ICP.
| Analyte | Abundance (%) | Ions causing spectral interference |
|---|---|---|
| 32S+ | 95.04 | 16O16O+, 14N18O+, 15N16O1H+ |
| 33S+ | 0.75 | 32S1H+, 16O16O1H+, 16O17O+, 15N18O+, 14N18O1H+ |
| 34S+ | 4.20 | 33S1H+, 16O18O+ |
| 32S16O+ | 95.04 | 48Ti+, 48Ca+, 36Ar12C+ |
| 33S16O+ | 0.75 | 49Ti+, 32S17O+ |
| 34S16O+ | 4.20 | 50Ti+, 50Cr+, 50V+, 38Ar12C+, 36Ar14N+, 32S18O+, 33S17O+ |
In the literature, different approaches have been described to deal with these issues.11 The amount of organic material introduced into the ICP can be reduced by using a low-flow nebulizer, a smaller i.d. injector tube, a cooled spray chamber (or combinations thereof), or an alternative sample introduction system such as an electrothermal vaporizer.12 To minimize carbon deposition on instrument components (torch and sampling cone), oxygen gas can be admixed into the spray chamber or the auxiliary gas. To overcome the problem of spectral overlap, a large variety of methods have been proposed. The application of a double-focusing sector field (SF) ICP-MS, operated at medium mass resolution (R = 4000), can be seen as the most universal approach,6,13 but this type of instrument comes at a higher cost than quadrupole-based instruments, and is therefore less prevalent. Another disadvantage accompanying this elegant approach is a 10-fold reduction in ion transmission efficiency and thus, signal intensity. That is why several authors have suggested alternative strategies for tackling the problem of spectral interferences by means of quadrupole-based ICP-MS. Recently, Donati et al. and Amais et al.4,14 have reported on the use of the interference standard method (IFS). The IFS method requires neither instrument modification, nor addition of reaction gases, but is based on the concept that the use of argon-containing ions as internal standard allows better correction methods for spectral interferences caused by polyatomic ions, thus improving the accuracy of determination using ICP-QMS.
For some sample types, S can also be measured practically interference-free with a quadrupole-based ICP-MS instrument equipped with a collision/reaction cell.15–17 On the one hand, one can aim at a reduction of the spectral background at the sulphur isotope masses, e.g., by using Xe as a cell gas.17 As also the transmission of S+ ions is simultaneously reduced, this method was reported to be suitable for the determination of S concentrations in the range of 10 to 50 mg L−1 only. Another approach is to use oxygen as a cell gas to convert S+ ions into SO+ ions, which can be detected at m/z 48, 49 and 50 (for 32S, 33S and 34S, respectively).15 Detection limits as low as 0.2 μg L−1 can be obtained with this method (via32S16O+). However, also this approach is not sufficient for organic matrices or samples containing Ti or high levels of Ca due to the occurrence of spectral interferences, certainly when accurate results are required for more than one S isotope (e.g., for isotope ratio measurements) (Table 1). For example, in an earlier study,6 we aimed at revealing reactive metabolites of a Cl-containing candidate drug via reverse phase HPLC hyphenated to ICP-mass spectrometry. As reactive metabolites were expected to bind to glutathione (a natural S-containing nucleophile), co-elution of Cl and S is indicative of the formation of such reactive species. In earlier work involving quantification of the metabolites of a Br-containing candidate drug via reverse phase HPLC-ICP-MS, it was demonstrated that on-line species-unspecific isotope dilution counteracts the effect of gradient elution on the sensitivity and provides accurate results.18,19 However, as described before, in the case of S, the situation is more complex as spectral interferences may jeopardize the utility of this approach. One of the possibilities suggested to tackle this problem was the use of oxygen as a cell gas in an ICP-DRC-QMS instrument. However, in the organic matrix under investigation, this approach still does not allow the use of isotope dilution as the signal of 34S16O+ at a mass-to-charge ratio of 50 suffers from overlap with the signals from 38Ar12C+, 36Ar14N+, 33S17O+ and 32S18O+.6 Therefore, the use of SF-ICP-MS was compulsory to obtain accurate results for this application.
Very recently however, a new type of quadrupole-based ICP-MS instrument has been introduced onto the market. This instrument is referred to as a triple quadrupole or an ICP-QQQ set-up by the manufacturer, although the terminology “triple quadrupole” is not entirely correct, as in-between the two quadrupole analyzers, an octopole-based collision/reaction cell is located. For the first time ever, an ICP-MS instrument can be operated in MS/MS mode.9 The first quadrupole prevents all off-mass ions from entering the cell, allowing more controlled and efficient interference removal in reaction mode, regardless of sample type.
The work reported in this paper aimed at evaluating whether it is possible to develop a fast, sensitive and accurate IDMS method for the determination of S in organic matrices by means of ICP-MS/MS. As a proof-of-concept, the technique was successfully applied to a S-containing reference material, NIST SRM 2773 (biodiesel).
2 Experimental
2.1 Instrumentation
All measurements were carried out with an Agilent 8800 ICP-MS instrument (Agilent Technologies, Japan). In this instrument, an octopole-based collision/reaction cell is located in-between two quadrupole analyzers (Fig. 1). The width of the bandpass of the first quadrupole analyzer can be varied from ‘fully open’ down to single mass width. The octopole collision/reaction cell can be used in vented mode or can be pressurized with either a collision gas (to selectively slow down polyatomic ions or to induce collision-induced dissociation), a reactive gas (to selectively react with either the interfering or the target ion to attain interference-free conditions) or a mixture of both. The bandpass width of the second quadrupole analyzer is traditionally set at one mass unit. The major advantage of such an ICP-QQQ instrument is enhanced control over the chemical resolution of spectral overlap owing to the double mass selection in MS/MS mode. In the experiments reported in this paper, this capability was relied upon to provide interference-free conditions for monitoring the signals of both 32S and 34S (for isotope dilution purposes) in a very demanding matrix. An overview of the most important instrumental parameters is presented in Table 2.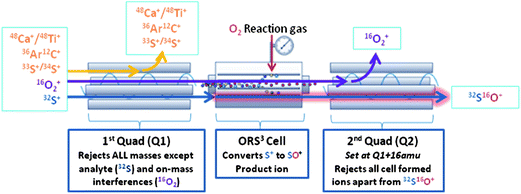 | ||
| Fig. 1 Schematic representation of the operating principle of the ICP-QQQ system, functioning in MS/MS mode, leading to an interference-free determination of 32S (as 32S16O+). | ||
| Parameter | Value |
|---|---|
| RF power | 1450 W |
| Carrier gas flow rate | 0.98 L min−1 |
| O2 option gas flow rate | 75 mL min−1 |
| Spray chamber temperature | −5 °C |
| O2 reaction gas flow rate | 0.4 mL min−1 |
| Q1 bias | −2 V |
| Octopole bias | −9 V |
| Q2 bias | −18 V |
2.2 Samples and reagents
The reference material NIST SRM 2773 (biodiesel produced from animal fat), obtained from the National Institute of Standards and Technology (USA), was used to check the accuracy of the method.The 98.8% 34S spike (sodium sulfate) used for isotope dilution was obtained from Isoflex (USA) and the single element 10 g kg−1 inorganic S standard solution (used for preparing calibration standards and for reverse ICP-IDMS) from SPEX CertiPrep (USA).
Only high-purity reagents were used for sample preparation. Water was purified by means of a Direct Q-3 Milli-Q system (Millipore, USA), while HNO3 (pro analysis, ChemLab, Belgium) was further purified by subboiling distillation. All samples and standards were diluted in absolute ethanol (USP grade for analysis, Fisher Scientific, UK), containing 0.14 M HNO3.
2.3 Sample preparation
To check the linearity of the calibration curves, sulphur standards with concentrations ranging between 0 and 850 μg kg−1 were prepared, by diluting a 10 g kg−1 S single element standard solution with ethanol.For the isotope dilution experiment, a stock solution A (approximately 2.25 mg g−1 S) of the spike material was prepared by dissolving 0.1 g of the powder in 10 mL of 0.14 M HNO3. Subsequently, 250 μL of spike solution A was mixed with 65 μL of a S standard solution of natural isotopic composition (1 g kg−1) and further diluted to a volume of 25 mL with 0.14 M HNO3. In this way, the 34S enrichment of the spike was reduced to ∼90%, which allows a more accurate and precise characterisation of the spike. The concentration of this ‘diluted’ 34S-spike solution (solution B) was determined to be 0.711 μmol g−1 34S by reverse ICP-IDMS using a S standard solution of natural isotopic composition.
Approximately 1 g of the sample (NIST SRM 2773) was directly weighed into a 15 mL polypropylene tube and an accurately weighed amount of spike solution B (∼0.2 g) was added and the solution was further diluted to 25 mL with ethanol. Three different mixtures of sample and spike were prepared to allow evaluation of the reproducibility of the method. Blanks were obtained using exactly the same procedure, but without addition of the biodiesel sample.
3 Results and discussion
3.1 Optimization of ICP-QQQ for S detection in an organic matrix
The ICP-QQQ instrument can be operated in three different modes, more specifically (i) the single-quad mode where Q1 only functions as an ion guide (further described as the ‘standard’ mode), (ii) the single-quad mode where Q1 acts as a bandpass filter, with the bandpass sufficiently wide to allow both S+ and SO+ ions to pass through the system (further described as the ‘bandpass’ mode), and (iii) the MS/MS mass-shift mode where Q1 is set to the precursor ion mass and Q2 is set to the mass of a target reaction product ion (further described as ‘MS/MS’ mode).The fact that the instrument can be used in these 3 different modes makes it very well suited for a large variety of applications, as well as for clearly demonstrating the strength of using the MS/MS mode.
As a first test, the linearity of the calibration curves obtained for a set of S-standards (with concentrations ranging between 0 and 850 μg kg−1), diluted with pure ethanol, was evaluated for the three different modes of operation.
In the standard mode, S-intensities were measured at m/z ratios of 32, 33 and 34 (Q2), while in the bandpass and MS/MS modes, oxygen was added as a reactive gas into the octopole cell to convert the S+ ions into SO+ ions, which were measured at the corresponding m/z ratios of 48, 49 and 50 (Q2). In the MS/MS mode, the corresponding values for Q1 were 32, 33 and 34, respectively (precursor ions).
To obtain stable plasma conditions during the analysis of organic solvents, some instrument modifications were done (compared to the standard instrument set-up). To reduce the amount of organic vapour entering the plasma, the Peltier-cooled spray chamber was cooled down to −5 °C and the standard injector tube (2.5 mm internal diameter) was replaced by a 1 mm i.d. injector. To prevent carbon-buildup in the instrument, O2 was added (as a 20% mixture in Ar) to the spray chamber. For maximum sensitivity, the ion lenses were optimised for the different plasma conditions when running organics.
The O2 reaction gas flow rate was optimized by introducing (1) an ethanol blank solution and (2) a S-standard in ethanol and evaluating both the signal intensity and the signal-to-background ratio as a function of the cell gas flow rate. As can be seen in Fig. 2, the cell gas flow rate has little influence on the signal-to-background ratio, while the signal intensity itself clearly reaches a maximum at an O2 gas flow rate of ∼0.4 mL min−1. On the basis of Poisson counting statistics, it can be expected that higher signal intensities lead to a better isotope ratio precision. This was demonstrated by calculating the average RSD value for 10 replicate measurements of the 32S16O+/34S16O+ isotope ratio in biodiesel samples, at different O2 cell gas flow rates. RSD values of 0.60%, 0.43% and 0.60% were obtained at an O2 gas flow rate of 0.15, 0.4 and 0.7 mL min−1, respectively. This trend is in good agreement with the corresponding theoretical RSD values that can be calculated on the basis of counting statistics (0.43%, 0.31% and 0.42% RSD, respectively). Therefore, a cell gas flow rate of 0.4 mL min−1 was selected for all further S measurements.
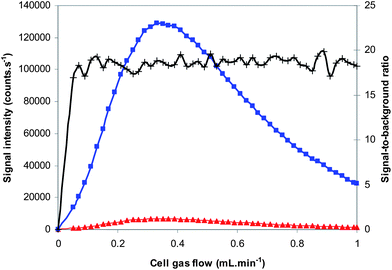 | ||
| Fig. 2 Optimization of the cell gas (O2) flow rate. Signal intensities are shown as a function of the cell gas flow rate for an ethanol blank (red triangles) and a 175 μg kg−1 S standard in ethanol (blue squares). The signal-to-background ratio is given in black (crosses) and the corresponding values can be read on the right Y-axis. | ||
From the calibration curves presented in Fig. 3, it is clear that an accurate determination of S in standard mode is almost impossible, as a consequence of the (oxygen-based) spectral overlap which is always present (see Table 1). The situation clearly improves when the instrument is operated in “reaction mode” and O2 is added into the cell. While the linearity of the calibration curves for both the bandpass and the MS/MS mode is very good, an offset between the calibration curves is clearly visible (mainly for m/z 49 and 50). This shows that there is still a considerable spectral overlap at these mass-to-charge ratios when the instrument is operated in the bandpass mode, a finding that corresponds to the data obtained by De Wolf et al.6 However, in MS/MS mode, the blank values are drastically reduced and the signal intensities obtained at m/z ratios of 48, 49 and 50 follow the natural isotopic pattern of S. This indicates that – by using the MS/MS mode – S can be determined (nearly) interference-free in organic matrices via, at least, its two main isotopes. The limits of detection obtained for this method were calculated based on the standard deviation of the response (s) and the slope of the calibration curve (m), according to the formula: LOD = 3 (s m−1). The standard deviation of the response was determined based on the standard deviation of the y-intercept of the regression lines. In this way, LOD values of 5, 4 and 7 μg kg−1 (or 4, 3 and 6 μg L−1) were obtained via32S16O+, 33S16O+ and 34S16O+, respectively. The closeness in terms of LOD for S isotopes showing large variation in their natural isotopic abundance illustrates that the S concentration in the ethanol used (contamination) is the limiting factor. When LOD calculations are only based on 3 times the internal standard deviation (i.e. the standard deviation for repeated measurements of one calibration blank), much lower values are obtained via32S16O+ (0.5 μg kg−1 or 0.4 μg L−1) and 34S16O+ (2 μg kg−1 or 1.6 μg L−1), but not via33S16O+ (6 μg kg−1 or 5 μg L−1). The instrumental LODs calculated in this way reflect the differences in natural isotopic abundance among the S isotopes. Although the amount of LOD-values, obtained for S in organic matrices, that is reported in the literature is rather limited, some values are given for comparison. Smith et al.,20 Sulyok et al.21 and De Wolf et al.6 published the values of 16 μg L−1, 100 μg L−1, and 10 μg L−1, respectively, via32S16O+. However, a much higher value was obtained via34S16O+ (300 μg L−1).6 The use of sector field ICP-MS allowed us to reduce these LOD values to 1 μg L−1 and 2 μg L−1via32S+ and 34S+, respectively.
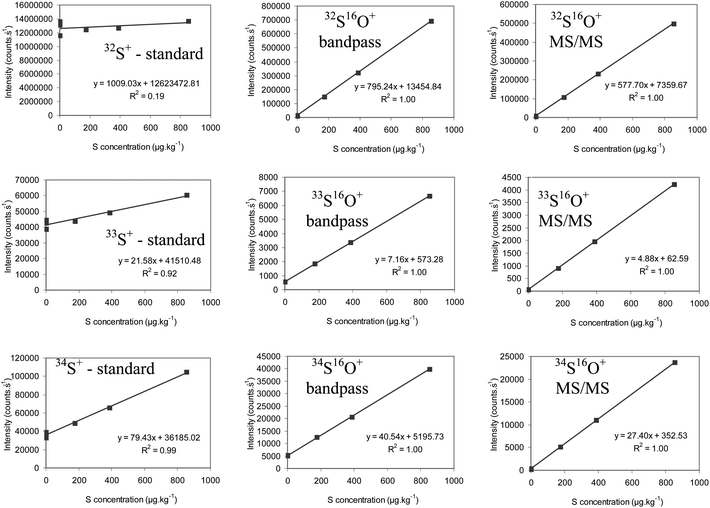 | ||
| Fig. 3 Calibration curves obtained for 32S+, 33S+ and 34S+ (standard mode) and for 32S16O+, 33S16O+ and 34S16O+ (bandpass mode and MS/MS mode) for a series of standards with concentrations ranging between 0 and 850 μg kg−1. | ||
The strength of the technique becomes even more apparent when S has to be determined in a matrix, containing a considerable amount of Ca or Ti (Table 1). When the instrument is operated in the bandpass mode, the Ca and Ti ions will pass through Q1, enter the cell and will also be transmitted by Q2, such that they are detected at the same nominal mass-to-charge ratio as the SO+-ions. However, in MS/MS mode, where the first quadrupole is set to the S precursor ion mass (with unit mass resolution), the Ca and Ti ions are rejected from the ion beam and are therefore not entering the cell and Q2 (Fig. 1). This is demonstrated in Fig. 4, where spectral peaks are shown for (i) an ethanol blank and (ii) an ethanol blank containing 50 μg L−1 Ca and Ti, in the mass range of 47.55 to 50.55 amu. For each solution, spectral peaks are shown for both the bandpass mode (A) and the MS/MS mode (B). It can be seen that in the bandpass mode, the signal intensities obtained at m/z ratios of 48, 49 and 50 clearly increase when Ca and Ti are added to the sample, while in MS/MS mode no significant difference can be noticed between both solutions. Moreover, when comparing the signal intensities obtained for the ethanol blank in bandpass and MS/MS modes (C), those in MS/MS mode are lower and only in this mode the spectrum displays the natural isotopic pattern of S. This again proves that the blank values obtained in bandpass mode can not only be attributed to the S present in the blanks, but also partially to interfering species.
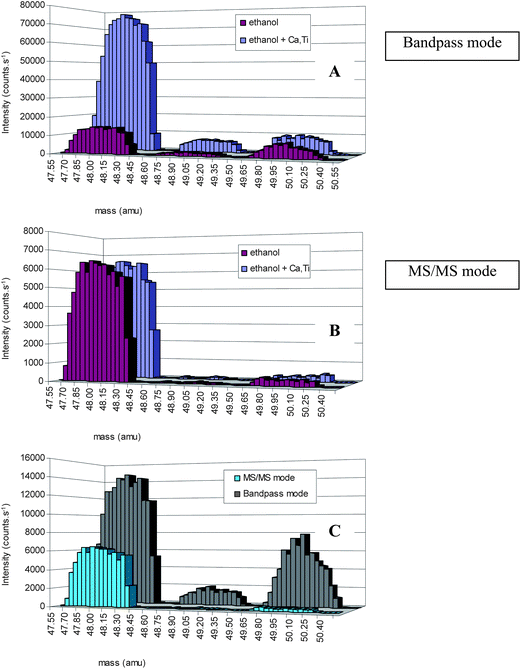 | ||
| Fig. 4 Comparison of the spectra obtained for the mass range from 47.55 to 50.55 amu for (i) an ethanol blank and (ii) an ethanol blank, containing Ca and Ti when operating the instrument in the bandpass mode (A) and the MS/MS mode (B). (C) shows the comparison for the ethanol blank solution in both bandpass and MS/MS modes. | ||
3.2 Determination of S in NIST SRM 2773 (biodiesel) by means of isotope dilution ICP-MS/MS
In order to meet the aim of this work and as a proof-of-concept for the MS/MS approach described above, an isotope dilution ICP-MS/MS method was developed for the determination of S in the reference material NIST SRM 2773. This material is a commercial 100% biodiesel produced from animal feedstocks and comes with a certified mass fraction value for S of 7.39 ± 0.39 μg kg−1. According to the procedure described by Chaves et al.3 the only sample pretreatment step that is required for biodiesel samples is simple dilution in ethanol.As the spike material was enriched in 34S, the 32S/34S ratio was used for the ID approach (with 32S and 34S measured at m/z 48 (32S16O+) and m/z 50 (34S16O+), respectively). All raw signal intensities were blank-corrected with those obtained for an ethanol blank. No mass bias correction was performed because all isotope ratios were determined experimentally and mass discrimination can be assumed to be constant. Three different blends of SRM sample + spike and two blends of ethanol blank + spike have been analysed by means of ID-ICP-MS/MS. The average blank value was calculated to be 0.011 μg g−1 and was subtracted from the results obtained. The results from the S determination by ID-ICP-MS/MS are summarized in Table 3. The results obtained for each of the three SRM samples agree well with the certified value within the experimental uncertainty. Moreover, an evaluation of the 95% confidence interval shows that the method not only results in accurate, but also in reproducible results.
| Sample | Concentration | Certified value |
|---|---|---|
| SRM 2773 – 1 | 7.234 μg g−1 | |
| SRM 2773 – 2 | 7.227 μg g−1 | |
| SRM 2773 – 3 | 7.231 μg g−1 | |
| Average | 7.231 μg g−1 | 7.39 ± 0.39 μg g−1 |
| Standard deviation | 0.003 μg g−1 | |
| 95% Confidence interval | 7.231 μg g−1 ± 0.015 μg g−1 |
4 Conclusions and outlook
A novel method has been described for the determination of S in organic matrices by means of ICP-MS/MS. Several authors have reported before on the determination of S after converting the S+ ions into SO+ ions by means of oxygen as a reaction gas. However, in organic matrices and/or matrices containing Ca, Ti or Cr, this approach did not provide accurate results, at least not when isotope dilution or isotope ratio analysis was aimed at. In this communication, it has been shown that ICP-MS/MS is a very promising technique to deal with this problem. As a proof-of-concept, the technique was successfully applied to the S determination in a biodiesel reference material.Although only biodiesel has been analysed in this initial study, it may be assumed that a very large variety of samples can be analyzed for their S content in the same way. As has been described in the introduction, one of the application fields of S isotope ratio determination in organic matrices via ICP-MS/MS will be the use of on-line species-unspecific isotope dilution for quantification of S-containing materials via reverse phase HPLC-ICP-MS.
Acknowledgements
The work performed by Lieve Balcaen has been financially supported by the Fund for Scientific Research Flanders (FWO-Vlaanderen). Martín Resano acknowledges the Spanish Ministry of Science and Innovation (Project CTQ2009-08606).References
- J. Heilmann, S. F. Boulyga and K. G. Heumann, Accurate determination of sulfur in gasoline and related fuel samples using isotope dilution ICP-MS with direct sample injection and microwave-assisted digestion, Anal. Bioanal. Chem., 2004, 380, 190–197 CrossRef CAS.
- J. Heilmann and K. Heumann, Sulfur trace determination in petroleum products by isotope dilution ICP-MS using direct injection by thermal vaporization (TV-ICP-IDMS), Anal. Bioanal. Chem., 2009, 393, 393–397 CrossRef CAS.
- E. S. Chaves, M. T. C. de Loos-Vollebregt, A. J. Curtius and F. Vanhaecke, Determination of trace elements in biodiesel and vegetable oil by inductively coupled plasma optical emission spectrometry following alcohol dilution, Spectrochim. Acta, Part B, 2011, 66, 733–739 CrossRef CAS.
- R. S. Amais, G. L. Donati and J. A. Nobrega, Interference standard applied to sulfur determination in biodiesel microemulsions by ICP-QMS, J. Braz. Chem. Soc., 2012, 23, 797–803 CrossRef CAS.
- C. Rappel and D. Schaumloffel, The role of sulfur and sulfur isotope dilution analysis in quantitative protein analysis, Anal. Bioanal. Chem., 2008, 390, 605–615 CrossRef CAS.
- K. De Wolf, L. Balcaen, E. Van De Walle, F. Cuyckens and F. Vanhaecke, A comparison between HPLC-dynamic reaction cell-ICP-MS and HPLC-sector field-ICP-MS for the detection of glutathione-trapped reactive drug metabolites using clozapine as a model compound, J. Anal. At. Spectrom., 2010, 25, 419–425 RSC.
- J. G. Martinez-Sierra, F. M. Sanz, P. H. Espilez, R. Santamaria-Fernandez, J. M. M. Gayon and J. I. G. Alonso, Evaluation of different analytical strategies for the quantification of sulfur-containing biomolecules by HPLC-ICP-MS: application to the characterisation of S-34-labelled yeast, J. Anal. At. Spectrom., 2010, 25, 989–997 RSC.
- C. Losada, J. J. Alberti, J. Saurina and S. Sentellas, Determination of S-containing drug metabolites from in vitro and in vivo metabolism studies by using LC-ICP/MS, Anal. Bioanal. Chem., 2012, 404, 539–551 CrossRef CAS.
- S. D. Fernández, N. Sugishama, J. R. Encinar and A. Sanz-Medel, Triple Quad ICPMS (ICPQQQ) as a new tool for absolute quantitative proteomics and phosphoproteomics, Anal. Chem., 2012, 84, 5851–5857 CrossRef.
- F. Vanhaecke, L. Balcaen and D. Malinovsky, Use of single-collector and multi-collector ICP-mass spectrometry for isotopic analysis, J. Anal. At. Spectrom., 2009, 24, 863–886 RSC.
- L. Balcaen, B. De Samber, K. De Wolf, F. Cuyckens and F. Vanhaecke, Hyphenation of reverse-phase HPLC and ICP-MS for metabolite profiling – application to a novel antituberculosis compound as a case study, Anal. Bioanal. Chem., 2007, 389, 777–786 CrossRef CAS.
- M. Resano, M. Verstraete, F. Vanhaecke, L. Moens and J. Claessens, Direct determination of sulfur in Bisphenol A at ultratrace levels by means of solid sampling-electrothermal vaporization-ICP-MS, J. Anal. At. Spectrom., 2001, 16, 793–800 RSC.
- N. Jakubowski, T. Prohaska, L. Rottmann and F. Vanhaecke, Inductively coupled plasma- and glow discharge plasma-sector field mass spectrometry – Part I: tutorial: fundamentals and instrumentation, J. Anal. At. Spectrom., 2011, 26, 693–726 RSC.
- G. L. Donati, R. S. Amais and J. A. Nobrega, Interference standard: a new approach to minimizing spectral interferences in inductively coupled plasma mass spectrometry, J. Anal. At. Spectrom., 2011, 26, 1827–1832 RSC.
- D. R. Bandura, V. I. Baranov and S. D. Tanner, Detection of ultratrace phosphorus and sulfur by quadrupole ICPMS with dynamic reaction cell, Anal. Chem., 2002, 74, 1497–1502 CrossRef CAS.
- C. H. Yang and S. J. Jiang, Determination of B, Si, P and S in steels by inductively coupled plasma quadrupole mass spectrometry with dynamic reaction cell, Spectrochim. Acta, Part B, 2004, 59, 1389–1394 CrossRef.
- P. R. D. Mason, K. Kaspers and M. J. van Bergen, Determination of sulfur isotope ratios and concentrations in water samples using ICP-MS incorporating hexapole ion optics, J. Anal. At. Spectrom., 1999, 14, 1067–1074 RSC.
- L. I. L. Balcaen, B. De Samber, K. De Wolf, F. Cuyckens and F. Vanhaecke, Hyphenation of reverse-phase HPLC and ICP-MS for metabolite profiling-application to a novel antituberculosis compound as a case study, Anal. Bioanal. Chem., 2007, 389, 777–786 CrossRef CAS.
- B. Meermann, M. Bockx, A. Laenen, C. Van Looveren, F. Cuyckens and F. Vanhaecke, Speciation analysis of bromine-containing drug metabolites in feces samples from a human in vivo study by means of HPLC/ICP-MS combined with on-line isotope dilution, Anal. Bioanal. Chem., 2012, 402, 439–448 CrossRef CAS.
- C. J. Smith, I. D. Wilson, L. Weidolf, F. bou-Shakra and M. Thomsen, Enhanced detection of sulphur and phosphorous containing compounds in HPLC-inductively coupled plasma mass spectrometry using chemical resolution via hexapole-based reaction with oxygen, Chromatographia, 2004, 59, S165–S170 CAS.
- M. Sulyok, S. Hann, C. G. Hartinger, B. K. Keppler, G. Stingeder and G. Koellensperger, Two dimensional separation schemes for investigation of the interaction of an anticancer ruthenium(III) compound with plasma proteins, J. Anal. At. Spectrom., 2005, 20, 856–863 RSC.
- M. Berglund and M. E. Wieser, Isotopic compositions of the elements 2009 (IUPAC Technical Report), Pure Appl. Chem., 2011, 83, 397–410 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2013 |