 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Regeneration-on-a-chip? The perspectives on use of microfluidics in regenerative medicine
Björn
Harink†
a,
Séverine
Le Gac†
b,
Roman
Truckenmüller
a,
Clemens
van Blitterswijk
a and
Pamela
Habibovic
*a
aDepartment of Tissue Regeneration, MIRA Institute for Biomedical Engineering and Technical Medicine, PO Box 217, 7500AE Enschede, The Netherlands. E-mail: bjornharink@gmail.com; p.habibovic@utwente.nl; Fax: +31 53 489 2150; Tel: +31 53 489 3400
bBIOS, The Lab-on-a-Chip Group, MESA+ Institute for Nanotechnology, University of Twente, Enschede, The Netherlands. E-mail: s.legac@utwente.nl
First published on 31st May 2013
Abstract
The aim of regenerative medicine is to restore or establish normal function of damaged tissues or organs. Tremendous efforts are placed into development of novel regenerative strategies, involving (stem) cells, soluble factors, biomaterials or combinations thereof, as a result of the growing need caused by continuous population aging. To satisfy this need, fast and reliable assessment of (biological) performance is sought, not only to select the potentially interesting candidates, but also to rule out poor ones at an early stage of development. Microfluidics may provide a new avenue to accelerate research and development in the field of regenerative medicine as it has proven its maturity for the realization of high-throughput screening platforms. In addition, microfluidic systems offer other advantages such as the possibility to create in vivo-like microenvironments. Besides the complexity of organs or tissues that need to be regenerated, regenerative medicine brings additional challenges of complex regeneration processes and strategies. The question therefore arises whether so much complexity can be integrated into microfluidic systems without compromising reliability and throughput of assays. With this review, we aim to investigate whether microfluidics can become widely applied in regenerative medicine research and/or strategies.
 Björn Harink | Björn Harink received his BSc in Biomedical Engineering in 2006 and his MSc in 2008 in Materials, Cellular and Tissue Engineering, with focus on Micro- and Nanotechnology, at the University of Twente, Enschede, the Netherlands. During his studies, he worked on chip-based isoelectric focusing for biomarker discovery in auto-immune disease at the MESA+ Institute for Nanotechnology. Currently, he is concluding his doctoral training at the department of Tissue Regeneration of the MIRA Institute for Biomedical Engineering and Technical Medicine, University of Twente, where he is working on applying microfluidics for novel high-throughput approaches in combinatorial factor screening in biomaterials research. |
 Séverine Le Gac | Séverine Le Gac was trained as a chemist at ESPCI in Paris in 1996–2000, and she received her PhD degree from the University of Sciences and Technologies of Lille (France) with honors (2004). She was a post-doctoral fellow in the BIOS, Lab on a Chip group (Twente University, The Netherlands). Currently, she is assistant professor at the same university and director for the Nanomedicine program of the MESA+ Institute for Nanotechnology. Her research focuses on the development of microfluidic devices as enabling tools for medical applications. |
 Roman Truckenmüller | Roman Truckenmüller holds an engineering diploma and a doctorate in engineering science from the Universities of Stuttgart and Karlsruhe, Germany, respectively. After working for Siemens, Erlangen, and at the Institute of Microstructure Technology of the Karlsruhe Institute of Technology, he currently works as an assistant professor at the Department of Tissue Regeneration of the University of Twente, The Netherlands. His research focuses on micro- and nanoscale three-dimensional polymer film forming and functionalization technologies and their biomedical applications, with a particular focus on engineering of complex artificial cellular microenvironments and niches using the aforementioned technologies. |
 Clemens van Blitterswijk | Clemens A. van Blitterswijk is a Professor of Tissue Regeneration at the University of Twente, Enschede, the Netherlands. He chairs one of Europe’s leading groups in the field of tissue engineering and regenerative medicine. He graduated as a cell biologist from Leiden University in 1982 and received a PhD in 1985 from the same university. He has authored or co-authored over 350 scientific publications and acts as inventor or co-inventor on over 100 patent applications. Today most of his research deals with tissue engineering and regenerative medicine forming a unique basis of multidisciplinary research between the materials and life sciences. |
 Pamela Habibovic | Pamela Habibovic is an associate professor with tenure at the Department of Tissue Regeneration at MIRA-Institute for Biomedical Technology and Technical Medicine of the University of Twente in Enschede, the Netherlands. She obtained a BEng (2000) in Chemical Engineering and Industrial Management from The Hague University for Applied Sciences, her PhD (2005) in Biomedical Engineering from the University of Twente and carried out postdoctoral studies in biomaterials for regenerative medicine at Children’s Hospital Boston, USA, and at McGill University, Montreal, Canada. Her research interests include synthetic bone graft substitutes, bioinorganics and high-throughput approaches in biomaterials research. |
Introduction
While the world population is continuously aging, demand for novel strategies to repair and regenerate damaged and diseased tissues and organs has tremendously grown. Transplantation of patient's own tissue is still considered the best option in most cases; however, limited availability becomes an increasingly problematic issue, especially for elderly patients. As a consequence, the field of regenerative medicine (RM) has emerged to address these currently growing demands.RM is a highly multidisciplinary field, with a number of aims and distinguishing features. This is obvious from the definition proposed by Daar and Greenwood, in an attempt to facilitate understanding among various stakeholders. The authors defined RM as “an interdisciplinary field of research and clinical applications focused on the repair, replacement or regeneration of cells, tissues or organs to restore impaired function resulting from any cause, including congenital defects, disease, trauma and aging. It uses a combination of several converging technological approaches, both existing and newly emerging, that moves it beyond traditional transplantation and replacement therapies. The approaches often stimulate and support the body's self-healing capacity. These approaches may include, but are not limited to, the use of soluble molecules, gene therapy, stem and progenitor cell therapy, tissue engineering and the reprogramming of cell and tissue types”.1
This definition clearly demonstrates different facets researchers working in the RM field need to deal with. While the final aim of RM research is improvement of the existing strategies and clinical applications, understanding of fundamental mechanisms of developmental, (stem) cell and molecular biology is imperative to reach this goal. A variety of research models is required in the field, differing not only in the type of tissue or organ to be regenerated, but also in the cause of dysfunction or damage. And finally, strategies toward regeneration are multiple, including cells, soluble factors, natural and synthetic biomaterials, and combinations thereof. For all these different aspects in RM research, (i) cell/tissue/organ type, (ii) cause of damage and (iii) regenerative strategy, reliable models are needed that resemble the in vivo situation as closely as possible, which comes with a high level of complexity.
Next to this quest for improving the quality of the existing research models, a recurring issue encountered in the field of RM is a need to considerably increase the rate at which new approaches are developed and implemented, while decreasing the cost thereof. This implies notably fast and reliable assessment of (biological) performance to select potentially interesting candidates, but also to rule out poor ones at an early stage of development, something that is not possible when using classical research approaches.
Microfluidics, defined by Whitesides as “the science and technology of systems that process or manipulate small (10−9 to 10−18 liters) amounts of fluids, using channels with dimensions of tens to hundreds of micrometers”,2 offers an extensive toolbox that may be useful for developing novel, more representative in vitro models for RM research. Microfluidic devices have already been used as platforms for cell-based screens, in particular for studying fundamental biological processes and for drug testing, owing to advantages they offer over classical cell culture systems: temporal and spatial control over fluids and physical parameters, and integration of sensors to obtain direct and in situ read-out. Moreover, microfluidics may provide a new avenue to accelerate research in the field of RM, as it has proven its maturity for the realization of high-throughput screening (HTS) platforms,3 through development of multiplexed platforms, parallelization of the assays as well as automation.
While microfluidics as technology is obviously attractive for many reasons, it is important to investigate whether and, if so, how it can be routinely utilized in RM research. One of the questions that needs to be answered is whether the biological complexity of real tissues, including heterogeneous cell population, extracellular matrix (ECM), chemical and physical cues in 3D, and systemic effects, can be implemented in microfluidic devices. Recent work on engineered cellular microenvironments and in particular organs-on-chips suggest that this certainly is possible.4,5 Equally important is the question whether the cause and the nature of the injury can be mimicked in a reliable way. Also timing is important: can such culture systems run long enough to study clinically relevant regeneration? And lastly, is it possible to investigate different regenerative strategies in microfluidic devices? While these systems are probably suitable for drug-based therapies and, to a certain extent, cell therapies, introduction of bioactive, natural or synthetic 3D biomaterials into the system may cause issues such as the loss of transparency of the device, flow regime retention, and if applicable, limit control over gradients and their stability. The aspect of biomaterials should not be ignored in this context as the need for synthetic alternatives to natural tissue and biological approaches, which suffer from issues of immunogenicity, lot-to-lot variability, high cost and, most importantly, limited availability, is tremendously growing. Fig. 1 illustrates why novel research tools are needed in RM and where microfluidic tools can make a valuable contribution to the field.
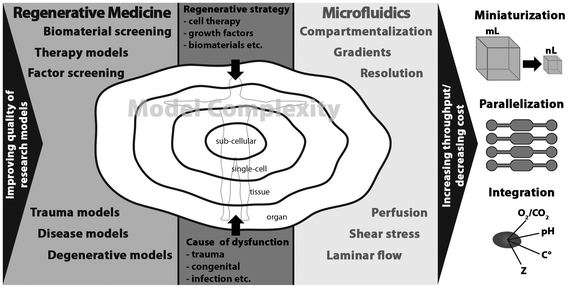 | ||
| Fig. 1 The needs of regenerative medicine research and the tools microfluidics offers to meet these needs. | ||
With this review, we aim at providing the state-of-the-art of the application of microfluidics in RM research. After an overview on the potential of microfabricated and microfluidic tools to advance research in RM, we will present examples of established microfluidic models for neuronal, vascular, musculoskeletal and hepatic regeneration. Possibilities and limitations of these techniques will be discussed in view of requirements from the RM field. Finally, we will give our view on the future perspectives of microfluidics for RM, and highlight the remaining challenges that have to be overcome before microfluidics can become a commonly applied tool for RM research.
Properties of microfluidic systems and their applicability to RM research
Lab-on-a-chip technology and microengineering approaches, both derived from the microelectronics field, provide a unique and unprecedented toolbox to be used in cell biology and related fields, including RM. As mentioned before, in order to improve quality of the RM research models, it is important to both mimic the cell-biological microenvironment which presents a high level of confinement and to incorporate soluble or surface-bound gradients and natural or synthetic materials to reach a high level of tissue/organ complexity. To increase the rate at which research is performed, the development of HTS systems is of great value, and the integration of read-out sensors into such systems enables direct feedback on the cell state and microenvironment. In the following paragraphs, these different features offered by microfluidic systems are presented.Physical cell microenvironment
Microfluidic devices present a high level of confinement, which resembles the environment cells experience in vivo. Compared to classical open microwells, these confined, closed and convection-free vessels enable local accumulation of substances secreted by cells, and have proven consequently to be more efficient to study autocrine–paracrine signaling.6 Furthermore, the micrometer-sized structures are characterized by a larger surface-to-volume ratio, which offers a higher level of control over various physical parameters, such as temperature or gas concentrations in solution (e.g., oxygen tension).7 This capability has notably been exploited to create hypoxic conditions,8 which are particularly important to recapitulate ischemia as found in injured tissues. A simple approach to deliver well-defined oxygen amounts in the cell culture medium has been reported, which relies on the use of a membrane-based oxygenator:9 this device consists of a three-layer structure with a thin gas-permeable PDMS membrane placed between a fluidic channel and a gas channel. The oxygen tension is precisely controlled in the gas channel, and thanks to the gas-permeability of PDMS, the same oxygen conditions are found in the fluidic channel.Similarly, the predictability and control of flows, due to their laminar character, offer new experimentation schemes. First, cells can be cultured under dynamic conditions with continuous perfusion of fresh medium, and they can be subjected to biologically relevant shear stresses.10 This experimentation scheme is not conceivable in classical culture platforms, where cells are grown in a static environment, without any active super- or perfusion. At the same time, microfluidic platforms can be designed so that nutrient delivery and gas exchange are solely governed by diffusion,11 circumventing thereby shear stress and associated issues10 and reproducing conditions found in vivo. Next, cells and microtissues can be exposed simultaneously to different flow compositions in a microfluidic platform, by exploiting the laminar character of the flow.12 This approach has been applied to create a temperature step in a microchannel, which has provided new understanding on the development of Drosophila melanogaster embryos and on the importance of the temperature on the process of embryogenesis.13,14 More recently, an embryonic body has been cultured at the interface between a differentiating solution and standard culture medium to induce neural cellular differentiation in half of the tissue while leaving the other half undifferentiated.15
Finally, the utilization of microfluidic culture conditions is highly attractive to rapidly exchange the fluid in the cell surrounding, which is precluded in standard microwells, and to control the cell microenvironment in a temporal manner.16
Soluble gradients
In vivo, chemical signals are mainly found in the form of gradients, which elicit highly different cell responses than simple bulk addition. For instance, during embryogenesis, organ and tissue development and, similarly, during regeneration and wound healing, gradients of morphogens or their repetitive periodic patterns are responsible for cell recruitment and ECM production and organization.17,18 Conventional gradient generators, such as Zigmond19 and Dunn20 systems consisting of side-by-side or concentric chambers connected by a narrow bridge are poorly controlled, irreproducible, and unquantifiable. In contrast, the laminar character of the flows at the microscale facilitates the generation of continuous, stable and precise gradients. Two approaches are mostly used to generate gradients of soluble compounds in microfluidic systems:21 (i) serial dilutions22 between a solution containing a soluble factor of interest and a “buffer” or (ii) via diffusion from a source structure, e.g., a chamber or a channel, to a sink,23 this often occurring through a barrier with a high fluidic resistance such as an array of channels with a low square-micron cross-section24 or a hydrogel material.25 As is illustrated by examples in the section on various tissues, this capability to generate gradients has enabled so far the study of chemotaxis,26 outgrowth of axons in neuronal cells22 or filopodia in endothelial cells,27 as well as the determination of optimal culture conditions by varying the concentration of specific factors in growth medium.28Material-related considerations
In a similar way as they respond to (bio)chemical factors and gradients thereof, cells are highly sensitive to the mechanical properties of the substrate they are grown on, and possible variations in its stiffness/softness. Initially, microfluidic systems have been fabricated from rigid materials such as silicon and glass, for which mature microfabrication processes derived from the microelectronic field were available, for both the realization of structures and substrate assembly. Slightly later, polymer materials entered the field, those being photo- or heat-curable such as SU-8 epoxy, polyimide photoresist and the polydimethylsiloxane (PDMS) elastomer,29–31 respectively, and thermoplastics such as polymethylmethacrylate, polycarbonate, polystyrene (PS),32 cyclic-olefin-copolymers, or Teflon®.33 Interestingly, PDMS has rapidly become the most popular substrate to realize cell culture platforms since it is biocompatible, gas-permeable, transparent, cheap, its processing does not require any dedicated cleanroom environment, and it lends itself well to the realization of integrated valves.34 However, PDMS suffers from a number of limitations, as recently acknowledged: it is highly hydrophobic; its porous structure works as a “sponge” for small and hydrophobic compounds, resulting in osmolality and concentration shifts in the cell environment; small oligomers can be released from the bulk PDMS into the solution in the devices; it is highly gas-permeable, which impedes the creation of hypoxic conditions;32 and its deformability makes it challenging to reliably realize either low micrometer-sized structures or shallow and wide channels or chambers, or even to align and bond a PDMS layer with another structured substrate.35–37 In that context, PS, of which commercially available culture dishes are made and which is fully characterized for cell culture experiments, is gaining interest35 even though it is gas-impermeable. Alternatively, biopolymers such as silk fibroin have been utilized to build microfluidic devices intended for biological experiments.38 More details on these material-related aspects of microfluidic systems can be found in elegant reviews by Berthier et al.35 and Bettinger and Borenstein.39In addition to these materials, soft polymer substrates which are frequently encountered in classical biological experiments, such as gelatin,40 hydrogels,41,25 and silk fibroin,42,43 have also been employed to fabricate relatively simple microfluidic structures, as discussed in the next sections. The main interest in these materials lies in their tunable mechanical properties which are obtained by tailoring their composition and polymerization conditions. This capability is notably exploited to introduce mechanical gradients into microfluidic systems and to study influence of stiffness on cell fate, as demonstrated by Lutolf and co-workers.44 Furthermore, soluble active factors can be encapsulated in the polymer matrix and progressively released in a controlled manner during experiments.45 Alternatively, the material can be pre-loaded with cells. Finally, biological response to these polymers can be tuned through embedding of functional groups into the backbone of the material.46 Such soft materials are usually processed either by using a combination of molding and polymerization techniques comparable to soft-lithography47 with the polymerization being initiated using, e.g., light, heat, pH or salt concentrations, or by soft embossing.48
Surface-bound chemical signals
The chemical nature of the substrate plays an important role, and interactions with the ECM environment are essential for the proper functioning of the cells. Surface coating is routinely applied in standard culture dishes as a uniform layer covering the whole dish. While this approach enables the control of cell adhesion, it is not suitable to screen various ECM components, especially in a combinatorial manner, to understand the influence of the cell–ECM interaction on the cell fate and to find optimal ECM conditions due to the number of independent dishes required and the price of ECM proteins. Using microfabrication and microprinting techniques, any kind of molecules can be patterned on a substrate along well-defined geometries. As a result, in a single culture dish, a large amount of ECM conditions can be tested, and their influence on the cell fate assessed. This micropatterning approach has brought valuable knowledge on the influence of the shape, surface area and chemical nature of the patterns on the cell behavior. It has for example been shown that surface area directly correlates with cell viability and growth rate,49 as well as with cell differentiation into various tissue lineages.50 Furthermore, Bhatia and co-workers have systematically screened mixtures of various ECM components, such as laminin and collagen, for their ability to promote human mesenchymal stromal cells (hMSC) to differentiate into hepatic lineages.51From single cell to sophisticated organ models
Microfluidic devices have already been applied to a variety of cell-based in vitro systems including individual cells,52 cell monolayers, complex and sophisticated multicellular tissues53 or even organ-like models.5,4Single-cell-level experiments are of particular interest in the field of RM to extend our knowledge on stem cells, in the quest to create artificial niches to preserve cell stemness,54–56 and to ultimately be able to control cell fate. Microengineering approaches and lab-on-a-chip technology have enabled generation of single-cell platforms, as discussed in several reviews,52,57–59 in opposition to standard laboratory approaches which typically deal with large cell populations. In a microfluidic format, individual cells can be isolated from a large population, trapped in dedicated structures, which enables to both follow their fate60,61 and analyze their content, possibly in a parallel manner.60,62,63 Lutolf and co-workers, for example, systematically studied the influence of various parameters such as substrate stiffness,57,64,65 cell–cell interactions and ECM proteins on hMSC differentiation and mouse neural stem cell self-renewal.44 Similarly, Chen. et al. have recently trapped an MCF-7 cancer cell in a microfluidic chamber and allowed it to expand in situ into a tissue/spheroid filling the microchamber.66 While developed as a cancer model, this approach could also be used to grow microtissues starting from a handful primary cells isolated from a patient, to study the process of injury and effect of regenerative strategies.
At the opposite side of the spectrum, studies on 3D cellular aggregates or microtissues, which are currently gaining significant interest,67 also greatly benefit from the utilization of microfabrication and lab-on-a-chip technology. Where standard hanging-drop68 and rotary bioreactor-based69,70 microspheroid formation techniques fail for the large-scale preparation of microtissues with homogeneous size and shape, microfabricated well arrays,71 microfluidic channels72,73 or droplet platforms74 have proven their suitability for the spontaneous, rapid and massive generation of microtissues with a highly controlled size and shape.67 These microengineering approaches have notably been applied to the field of cancer research for drug screening assays,75 as well as for creating elementary building blocks which upon successive self-assembly can give rise to more complex tissues with a clinically relevant size,71 or even to get new insights into biological processes such as angiogenesis. When generated in a microfluidic format, such microtissues have been successfully employed to study cell–cell interactions and the ability of specific cell lines to form co-culture multicellular aggregates.76 In another approach, microtissues have been prepared by directly including cells into a hydrogel.77 A main advantage of this hydrogel-based strategy is its suitability to generate tissue structures with a great variety of shapes78 when using photolithography-like polymerization, including high-aspect ratio structures like long fibers,79 which are not possible to create using conventional microfabrication techniques. Furthermore, such hydrogel microtissues also lend themselves well to self-assembly processes to generate larger pieces of tissue.80 Finally, by combining cellular systems with microfabricated structures, different groups have been able to create sophisticated models that emulate the organ-physiological architecture.81–84 Owing to their complexity, these organs-on-chip models have found applications for drug screening and nanotoxicology assays, as biologically relevant alternatives to animal experimentation, or for understanding of particular diseases, but their use in RM research is still limited.
Large-scale integration platforms for HTS
While the features of microfluidic systems discussed so far are mainly useful to improve quality of the in vitro models for RM, the most important advantage of microfluidic devices to accelerate progress of RM research is their high level of integration. This can be seen two-fold: on one hand, for the parallelization of the assays via the multiplexing of the device, in the same way as 96-well plates include 96 individual vessels for independent assays, and, on the other hand, for the implementation of a series of successive steps on one single platform. For both types of integration, it is essential to compartmentalize the fluids and cells. This is realized by either adding valves85,34 or using droplet-based microfluidics.86,87 Interestingly, this on-chip compartmentalization strategy is considered as a promising alternative to robotic fluidic handling.The realization of robust valves in microfluidic platforms has long been a major challenge from a fabrication point of view. However, nowadays, a few standard strategies are routinely used: (i) the Quake's valves made using multilayer soft-lithography,34 which exist in the push-up and push-down “flavors”; (ii) “normally closed” valves85 consisting of a thin polymer membrane sandwiched between two substrates; and (iii) the pin Braille valves.88 Interestingly, these valves, originally aimed at isolating small fluidic chambers, can serve a few other purposes such as pumping89 using a peristaltic approach, mixing88,90 (while pumping), or fluid metering.91 Adding valves in a microfluidic platform enables to drastically reduce the number of fluidic connections for a given multiplexed device, as well as between devices while increasing the multiplexing and integration capability. Furthermore, as discussed in recent reviews,92,93 combining series of valves in a smart way enables to decrease the number of actuation/control lines for microfluidic large-scale integration (mLSI).92 This mLSI strategy has proven to be very promising for a wide spectrum of applications. In the field of RM, it enables screening of a great variety of soluble factors and their concentrations, and assessment of their influence on cell growth and fate.
Concerning HTS in microfluidic systems, it is worth mentioning that, while these systems are highly suitable for parallelization and assay integration, issues exist with compatibility with classical high-throughput equipment, including robotic fluid handling for 96-, 384-, or 1536-well plates, and with conventional assay and biological read-out equipment. Thus, despite the expected decrease in cost due to miniaturization and reduced reagent and biological material use, to compete with the existing industry, screening in microfluidic systems must first be adopted more routinely by different fields to justify the required initial investment in suitable automated systems. Alternatively, microfluidic systems should be designed in such a way that they are compatible with standard laboratory equipment.
Integrated sensors for cell culture monitoring
Integration in microfluidic platforms also includes the implementation of smart capabilities or sensors realized using microelectromechanical system (MEMS) technology to precisely monitor in situ, in real-time and in a non-invasive way the cell microenvironment and the cell activity, a feature which is also of great importance when increasing throughput of screening. Of particular interest in this context for the field of RM94 are (i) oxygen sensors to regulate the oxygen tension in the cell surrounding or to follow the cell respiratory activity;95 (ii) or general sensors to control the physical parameters (e.g., carbon dioxide, temperature, pH) in the cell culture medium; (iii) sensors to measure the cellular stress level and the production of reactive oxygen species (ROS); (iv) sensors to determine the cell metabolism; (v) electrochemical sensors to track the chemical activity of neurons and neural tissues;96 and (vi) sensors to determine in a non-invasive way the differentiation status of stem cells by detecting specific markers for differentiation such as alkaline phosphatase97 or to assess proper functioning of the tissues by quantifying proteins secreted by the tissue such as albumin in the case of liver. Sensing generally relies on either electrochemical/electrical principles or on optical read-out, and sometimes includes enzymatic degradation processes for metabolic measurements of specific substrates (e.g., glucose, lactate or pyruvate). Jeong and co-workers reported an electrochemical sensor for long-term monitoring of alginate-based 3D lung cellular models for viability assays, which could be applied for the detection of any electro-active species.98 In another approach, pancreatic islets were encapsulated in an alginate-based shell including oxygen-sensitive fluorescent moieties, using a microfluidic droplet-based platform.99 This HTS approach, which can bring valuable information on the pancreatic islet activity, can easily be applied for the detection of other analytes than oxygen alone. Finally, Krommenhoek et al. reported an integrated sensor device with a footprint of <1 cm2 to monitor in parallel different parameters in a bioreactor such as the temperature, the pH, the oxygen concentration, and the biomass.100 Although this device has originally been developed for yeast culture, it could easily be employed in an integrated bioreactor for closely monitoring the growth environment of microtissues.With this brief overview of properties of microfluidic devices, we have attempted to indicate how they can contribute to advancement of RM research. Table 1 summarizes some important differences between microfluidic and classical cell culture models. In the next section, we will discuss some examples of microfluidic systems which have been developed for RM research, specifically to study neuronal, vascular, musculoskeletal and hepatic regeneration.
| Conventional | Microfluidics |
|---|---|
| Cell microenvironment | |
| No confinement (open wells). | Confinement (closed systems). |
| Limited level of spatial control (e.g. only single-well or trans-well systems). | High level of spatial control (e.g. compartmentalization for co-culture, 3-dimensionality and sub-cellular resolution). |
| No fluid control (only static or chaotic). | High level of control over fluids (e.g. laminar flow, perfusion, and temporal control over fluid exchange). |
| Limited possibilities for creating physical stimuli. | Various physical stimuli possible (e.g. stiffness, shear, compression). |
| Low temporal and spatial control over chemical stimuli (only bulk addition). | Possibility to create highly defined spatial and temporal chemical stimuli (e.g. soluble or surface gradients). |
| Established and characterized culture substrate materials. | Limited characterization of applied substrate materials and limited use of biologically characterized materials. |
| Biological read-out | |
| Compatible with conventional standardized biological assays. | Compatibility issues with conventional standardized biological assays. |
| Compatible with established read-out equipment. | Compatibility issues with established read-out equipment. |
| Comparable to large amount of data from historical experiments. | Low number of available historical experiments limits comparison. |
| Limited possibilities for in situread-out of biological processes. | Possibility to integrate sensors and assays for in situread-out of biological processes. |
| High-throughput screening (HTS) | |
| High reagent and biological (cell) material use in HTS setting. | Reduced reagent and biological (cell) material use in HTS setting. |
| Limited possibilities to parallelize and integrate assays. | Highly applicable to parallelization and integration of assays. |
| Compatible to conventional high-throughput (robotics) equipment. | Not compatible with conventional high-throughput (robotics) equipment. |
Neuronal regeneration
Microfluidic systems have a long history in the field of neuronal regeneration. Therefore, this application illustrates particularly well advancements in microfluidic technology to better meet biological demands.When studying neuronal degeneration and regeneration, the geometry of the microenvironment is of utmost importance. Enclosed and separated compartments are usually employed to study axon outgrowth and their response to stimuli. Compartmentalized devices such as Campenot chambers (Fig. 2) have for instance been employed for that purpose.101,102 In these devices, a fluoropolymer divider is attached to a standard culture dish using vacuum grease. Neuronal bodies are plated in the central compartment, while axons can grow out to surrounding compartments, through either the vacuum grease sealing or “rough” scratches in the surface of the culture dish previously generated with a knife. Although these systems have provided new insights into neuronal development and degeneration,102 they suffer from a number of limitations including limited resolution at the scale of the size of neurons, cumbersome assembly with high risk for leakage, and easy axon growth disruption upon the slightest mechanical strain.
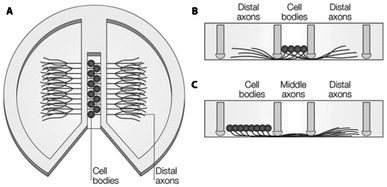 | ||
| Fig. 2 Schematic diagram of a Campenot chamber. (A) Top-view with cell-bodies in the center and axons spreading to the outer chambers by scratches in the surface or through vacuum grease. (B) Side-view of situation in A. (C) Alternative seeding possibility from the left chamber, so the middle part of the axons can be exposed to treatment, separately. Reproduced with permission, copyright 2005 Macmillan Publishers Ltd.102 | ||
The emergence of microtechnology has brought up alternative microfluidics-based systems, with dimensions that could be precisely controlled. For instance, compartmentalized systems can easily be realized using soft lithography rapid prototyping in PDMS with micron-sized structures such as grooves through which axon growth is guided, while neuronal bodies are retained.103–106 Interestingly, the same design has enabled cell co-culture, each compartment being used for a different cell type, as well as cell–cell interaction studies. Such systems have been notably applied to study the creation of synapses between neurons (Fig. 3),107,108 chronic excitotoxin-dependent axon degeneration, excessive stimulation by neuro-transmitters,106 and degeneration induced by paclitaxel, a mitotic inhibitor.109
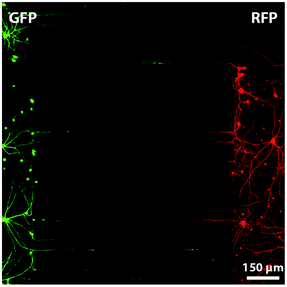 | ||
| Fig. 3 Fluorescent microscopy image of a compartmentalized microfluidic device in which two chambers are connected with micro-grooves of 7.5 μm × 3 μm × 900 μm. Neurons, from rat hippocampus, on the left produce Green Fluorescent Protein (GFP) and neurons on the right Red Fluorescent Protein (RFP). Such a system allows the investigation and manipulation of synapses between neurons. Reproduced with permission, copyright 2010 Elsevier.103 | ||
After a third compartment has been added between the two initial chambers, the axonal part of the neurons has been locally exposed to a flow of detergent, creating thereby precise “injuries”, with the rationale of mimicking trauma-induced degeneration.104
In an even more complex device, neurons were co-cultured with glial cells, like astrocytes or Schwann cells, introduced in another compartment, while a fourth chamber was employed to flow acrylamide to induce axotomy. Thereafter, neuron regeneration was studied, and, interestingly, axons showed a higher tolerance to acrylamide than neuronal bodies, especially compared to reported toxicity values for standard culture. This observation could be explained by the fact that microfluidics enabled local delivery of toxins to either the axon or the neuronal body.110 Alternatively, axotomy was achieved using a femto-second laser for localized heat-induced ablation.111
A different strategy to guide axons and study their outgrowth is known as the Bonhoeffer strip assay, which relies on specific chemical patterns to promote or inhibit axon growth.112 While this assay enables to identify inhibiting factors for axon outgrowth, this technique, where neurons are simply plated, exhibits low reproducibility, and, furthermore, neurons are randomly oriented. By combining microfluidics with chemical patterning of polylysine and aggrecan to promote and guide axon growth, or to alter it, better cell alignment was achieved.113 Furthermore, nutrients are supplied in such a system using flow regimes that resemble the in vivo situation, and compounds can precisely and specifically be delivered to different cell subpopulations.
As biological and chemical cues predominantly occur in the form of gradients in native tissue, an extensive amount of research has focused on the creation of gradients of factors that promote neuron growth,22,114 guide it,115 stimulate cellular differentiation,22 establish synapses,116,117 or induce diseases.118 Gradients were generated through various approaches, using a resistance mixer network,22 hydrogel-based barriers,116,117,119 or arrays of high-resistance microchannels,115,118 some of which being fully compatible with standard compartmentalized devices.118 The influence of mechanical gradients on neurite growth was similarly studied in an H-shaped channel configuration.120 The device was filled with a collagen gel, and a gradient of cross-linking agent was created across the connecting channel, resulting thereby in a gradient of gel stiffness. Seeding neural cells in the middle of the cross-channel allowed for studying the influence of the collagen gel stiffness on axon outgrowth. Whereas an updated version was employed to study gradients of adhesive ligands on the collagen gel.121 In another approach to assess the sensitivity of neural cells to mechanical forces, cells were grown on a stretchable PDMS membrane; this notably enabled the investigation of stretch-related growth of integrated axons,122 dynamic stretch injury of axons,123 mechanical breaking of microtubules124 and localized mechanotransduction on sensory nerves.125
As an advantage over the so far discussed 2D culture approaches, 3D neuronal tissues or neurospheres are supposed to more closely resemble the in vivo environment of neurons, making the study of their function more relevant. A microfluidic device with compartment chambers separated by micropillars was employed to trap spheroids derived from aggregates of adipose tissue-derived stem cells (ATSC). The tissues were subsequently stimulated to differentiate into neurospheres, with neurons sprouting through the pillar network.28,126 A similar approach was utilized for the co-culture of Schwann cells (SC) derived from human embryonic stem cells (hESC) with hESC-derived neurospheres to obtain spheroid formation.127
A number of attempts to develop HTS systems have been reported by combining microfluidics with microarray technology. For example, Shi and co-workers demonstrated a microarray platform with microfluidic channel connections in a 96-well plate format for screening the effect of small molecules on synaptogenesis.128 This system is a particularly good example where microfluidics is made compatible with conventional laboratory equipment for well-plate culture dishes, while offering HTS solutions to search for potentially interesting factors for neural regeneration. In another example, a microfluidic concentration gradient generator network with multiple downstream culture chambers was used to screen for optimal combinations of soluble factors to induce differentiation of rat MSCs into Schwann cells.129
Although all the microfluidic systems discussed so far do have the potential to become valuable RM models, they have predominantly been used for studying fundamentals of degeneration and drug screening for prevention of degeneration. Interestingly, one of the early reported devices combined microfluidics and microengineering and aimed at creating a retinal–neural interface in an attempt to create a true RM model.130 In this work, the authors developed an artificial synapsis system by micropatterning substrates to guide neurite growth, with localized neurotransmitter delivery while using soft materials. It is envisioned that more of such systems will appear in the future specifically for the purpose of studying regenerative strategies.
Vascular regeneration and wound healing
Vascularization is of great importance in regenerative medicine for proper oxygen and nutrient supply, and most cells in the human body are not much further than 100–200 μm from a capillary.131 Without proper vascularization, tissue constructs of larger than 200–400 μm are not viable because of oxygen and nutrient depletion. Therefore, new blood vessel formation is a relevant part of every regenerative strategy. Since the dimensions in microfluidic conduits are comparable to those of natural microvessels, which typically range from a few micrometers to tenths of millimeters, microdevices are particularly attractive to realize capillary vessels or to study the processes of vasculogenesis and angiogenesis. For instance, an in vitro microvessel network has been successfully created from collagen type-1 gel using soft lithography techniques (Fig. 4).132 After device fabrication, human umbilical vein endothelial cells (HUVEC) were seeded in the 100 μm × 100 μm microchannels, and left to attach and proliferate on their walls to yield an endothelialized lumen, and the channels were thereafter perfused with culture medium or whole blood. To study interactions between HUVECs and perivascular cells, the latter were added to the collagen gel before device fabrication. Upon treatment with growth factors such as vascular endothelial growth factor (VEGF), which is a signal protein produced by cells at low-oxygen or hypoxic conditions as well as a well-known angiogenic factor, sprouting angiogenic structures were observed in the gel matrix. Similarly, the same microdevice has successfully been applied to study cell–cell interactions between pericytes and smooth muscle cells, and as a model for thrombosis upon chemical induction of blood clogging in the created microvessels. This artificial capillary network is of utmost interest not only to test materials and compounds for RM strategies but also as a potential implant candidate. Alternatively, microfluidic devices have been produced by combining two different types of gels, each containing one cell type (fibroblasts or endothelial cells). The aim was to provide additional versatility in creating ECM microenvironments in the bulk and the channel network in one device to investigate implantable candidates and to study cell–cell interactions.133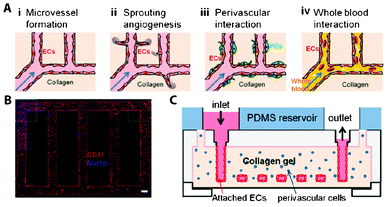 | ||
| Fig. 4 Gel-based 3D microvascular network made of collagen type-I gel. (A) Schematic representation of research possibilities on this platform. (B) Fluorescent microscopy image of human umbilical vein endothelial cells (HUVEC) on the walls of the gel-based networks, stained for the nuclei (blue) and CD31 (red), an angiogenic marker. (C) Schematic side-view representation of the microvascular networks. Reproduced with permission, copyright 2012 National Academy of Sciences, USA.132 | ||
To recreate the perivascular stem cell niche134 as a model for vasculogenesis, a microfluidic approach was applied where HUVECs and stromal cells were co-cultured in parallel gel lanes. Specifically, the device system included five independent lanes, the three middle lanes consisting of fibrin gel while the external lanes were kept for medium perfusion. When cells were separately added to lanes two and four, the formation of vessel-like structures into the middle gel lane was observed. Interestingly, this study was one of the first to demonstrate vascular generation with lumen formation and an actual hollow capillary network inside a microfluidic system by simply co-culturing cells in gel without any patterning of the cells in a microchannel network.
As mentioned before, VEGF gradients are known to elicit angiogenic sprouting. To study this phenomenon, Shamloo et al. designed a simple three-channel gradient device, with cells being cultured in the middle channel as a monolayer and exposed to gradients of VEGF created upon diffusion from the side channels via an array of low-micrometer channels.10 VEGF gradients have alternatively been created across a 3D gel structure in a 3-lane microfluidic device:27,135–138 collagen type-1 gel phase in the middle lane was exposed to a medium flow on one side, and VEGF-supplemented medium on the other side, to yield a VEGF gradient in the gel. Cells cultured on the wall of the gel on the lower end of the gradient migrated in the gel structure to 3D blood vessels. This platform has additionally been employed for a variety of other studies to quantitatively analyze vascular growth on an endothelial monolayer using VEGF alone27 or in combination with either the angiogenic regulator sphingosine-1-phosphate (S1P)136 or ANG-1, a co-factor known to be involved in stabilizing vessels.137 In the same platform, co-culture of endothelial cells in collagen gel with fibroblasts in alginate beads yielded capillary bed-like structures.139
Sprouting of vessels relies on the migration of endothelial cells; this process has been studied separately in devices focusing on wound healing models. In a classical wound healing assay, which was one of the earliest methods to study directional cell migration in vitro after “injury”,140 a scratch is made with a sharp object in a cell monolayer to remove cells along a sub-millimeter-to-millimeter-sized line. Thereafter, the rate and efficiency of cell migration to close this artificial wound is monitored. This assay has proven to be interesting for testing the effects of drugs as well as co-culture settings on cell proliferation and migration; however, it suffers from a poor reproducibility due to the uncontrolled way “damages” are realized in the cell monolayers. In that context, laminar flows, as found at the micrometer scale, are particularly attractive to create wounds in a highly controlled manner.141,142 For instance, van der Meer et al. and Felder et al. employed a 3-phase flow configuration, the center solution containing trypsin, to promote cell detachment from the surface in a well-defined way only in the middle of the channel while leaving cells on the side unaffected.141,142
For vascular regeneration, microfluidics can provide improved in vitro models mainly for fundamental research on diseases such as thrombosis and for testing RM strategies such as soluble compounds and combinations of hydrogel materials. Furthermore, hydrogel-based microdevices could be employed as implantable constructs for organ repair, and they provide a strategy for connecting artificial organs to the vascular network.
Musculoskeletal regeneration
Developing reliable models to study regeneration of musculoskeletal tissues, including bone, cartilage and skeletal muscle, presents additional challenges of complex 3D architecture and strong dependence on mechanical stimuli such as compression and stretching, since these tissues are part of the human locomotion system, giving rigidity and mobility to the human body.Relatively simple culture devices have been proposed to investigate the effect of microfluidic confinement and continuous perfusion on osteogenesis. For example, devices were developed containing a single microchannel in which osteoblasts, bone-forming cells, were cultured and continuously perfused with osteogenesis-inducing factors, such as dexamethasone, bone morphogenetic protein-2 (BMP-2) or a combination of both factors, to study their effect on osteogenic differentiation, as compared to static cell culture systems.143–145 Similarly, Leclerc et al. studied the effect of perfusion with different shear stress intensities on the behavior of murine osteoblasts146 in a 3D microfabricated capillary network. Interestingly, elevated levels of alkaline phosphatase, a marker for osteogenic differentiation, were found in the microfluidic format, either upon exposure to flow or simply under static conditions, as compared to static culture in 2D flasks. These examples demonstrated that confinement already has an influence on cell differentiation, which was further promoted in the presence of a shear flow. In a recent review, Riehl and Lim have discussed in detail these differences between macro- and microfluidic in vitro systems for skeletal RM research.147
In another approach to evaluate the effect of mechanical stimuli on the process of osteogenesis in a high-throughput manner, Moraes et al. built a microfluidic-based compression array, specifically designed to expose cells encapsulated in a hydrogel to mechanical strain (Fig. 5).148,149 Separate polyethylene glycol (PEG) hydrogel plugs loaded with murine MSCs were formed in a microfluidic chamber using photopolymerization. Application of a pressure on a PDMS membrane led to compression of the hydrogel plugs, and subsequently of the cells and nuclei. This and similar high-throughput platforms are likely to provide valuable information on the effect of compression on cellular differentiation by using various hydrogels.
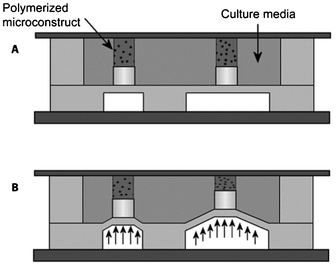 | ||
| Fig. 5 High-throughput screening platform for compression analysis of cells in hydrogel materials. (A) Schematic representation of the compression array at rest and (B) in compressed state. Reproduced with permission, copyright 2010 Elsevier.148 | ||
Not only mechanical signals are important to steer osteogenesis or chondrogenesis, but chemical signals can also contribute to differentiation processes. To investigate this, a 3D microtissue was generated from primary human bone marrow-derived MSCs between two rows of pillars in a microfluidic channel to study the process of osteogenesis.150,126 After one week of exposure to osteoinductive chemical stimuli in the platform, calcium deposition was observed, which indicates bone formation.
The influence of insulin growth factor 1 (ILGF-1) on chondrocyte proliferation was studied in separate chambers made from collagen gel, in a concentration-dependent manner.151 For that purpose, an ILGF-1 gradient was generated upstream to the culture chamber using a microfluidic resistor mixer network.
In an attempt to introduce a high-throughput strategy for screening relevant biomaterials and their effect on 3D culture of osteoblasts, various biomaterials were deposited using inkjet printing in independent microfluidic chambers. Thereafter, MC3T3-E1 osteoblastic cells were seeded in the chamber, and tested for their ability to form mineral nodules (Fig. 6).152 In a further study, the same system was employed to investigate the effect of bacteria and antibiotics on the process of osteogenesis, as well as biofilm-related infections, which are frequently the reason for failure of, for example, orthopedic implants.153,154
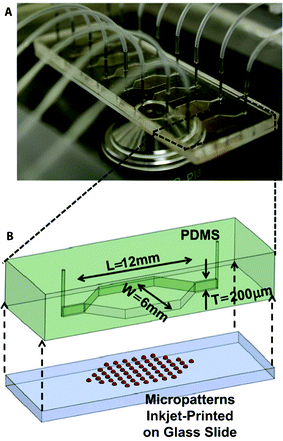 | ||
| Fig. 6 High-throughput screening platform for cell–biomaterial interactions, using parallel microfluidic chambers with different inkjet printed materials. (A) Photograph of the microfluidic platform, depicting multiple chambers. (B) Schematic representation of a single chamber with printed micropatterns. Reproduced with permission, copyright 2012 Elsevier.152 | ||
During embryonic development of muscular tissue, or myogenesis, myoblasts fuse together to form myotubes, which are early skeletal muscle fibers. This myoblast-to-myotube fusion was emulated in microfluidic format by Folch and co-workers using a long-term culture strategy.155 In a first step, cells were seeded on a patterned surface combining fibronectin linear structures with a PEG cell-repellent coating, to guide the attachment of murine myoblast cells (C2C12) along specific lines in a microfluidic chamber. After 7 days of culture under diffusion-based perfusion, myotubes were formed in the chamber along the fibronectin lines. This device was particularly useful to study the mechanisms behind synaptogenesis, after local delivery of agrin and neureglin-1, both known to be involved in neuro-muscular junctions during development, mimicking the path-finding dynamics between muscle cells and neurons.156–158 In contrast to conventional culture approaches, the microfluidic format enabled single myotube interaction study in a highly reproducible way. Finally, this platform also proved to be amenable to HTS assays for the simultaneous study of multiple factors.157
To study myogenesis itself over prolonged periods, microbioreactors proved to be particularly attractive. Figallo et al. proposed a PDMS-based device having the footprint of a standard microscope slide and containing 12 independent wells. These wells acted as independent bioreactors159 in which C2C12 cells were kept in culture for up to 10 days. Compartmentalization into individual bioreactors while limiting fluidic connections was reported using another strategy relying on a pin Braille display, serving the purposes of creating valves and of pumping fluids. This system was notably applied for highly automated and multiplexed myogenesis study,88 with cell seeding and reagents mixed using the pin Braille display, and dynamic culture conditions achieved at various shear rates.
In the musculoskeletal system, damage often occurs in more than one tissue, making regeneration of defects an extra complex process. For example, osteochondral defects require regeneration of both bone and cartilage tissue, and when replacing a ligament, integration of ligament tissue into surrounding bone is as important for the success of the procedure as the quality of ligament itself. Therefore, combinations of individual musculoskeletal tissues into one system is expected to be highly valuable for RM research purposes.
Hepatic regeneration
In vitro liver models have received much attention owing to the important role of this organ in processes of metabolism and detoxification, with the motivation to develop relevant and functional alternatives to animal experiments for HTS of drugs, chemicals, nanoparticles, etc.160 In that context, liver tissue models are also combined with models of other target tissues for inter-organ interaction studies.161,162 However, from a RM point of view, engineering liver tissue is only driven by the fact that in cases of liver failure, transplantation is the only available option, since no maintenance therapy exists. Since a few reviews were published in the last years on microfluidic liver in vitro models, only selected examples are presented in this section and the reader is referred to these reviews for complementary information.161,163,164One of the earlier attempts to use micromachining to create liver tissue was reported by Kaihara and colleagues.165 They applied microfabricated vascular networks in silicon and glass substrates coated with Matrigel™ or Vitrogen™ as templates to grow endothelial cells and hepatocytes monolayers. These monolayers, which were shown to maintain their albumin production, were lifted from the platform after 4–5 days of culture, and folded as 3D vascularized tissues prior to implantation into rats.
Since this seminal work aiming at regeneration, a variety of microbioreactors has been described for 2D and 3D culture of hepatocytes under perfusion conditions, for long-term culture,166–173 and sometimes subsequent coupling to a gradient generator for concentration-dependent toxicity studies.174,175 In general, the use of a microfluidic format is accompanied by an enhancement in liver function compared to conventional culture, as measured by albumin/urea production163 and relevant gene expression.176 However, direct exposure of the cells to the perfusion proved to lead to cell damage; therefore, most of the reported reactors contain a porous membrane between the medium flow and the cell culture for diffusion-based and shear-free delivery of fresh nutrients to the tissues. For instance, Ostrovidov et al. employed a PDMS membrane functioning as a scaffold for the growth of hepatocytes, while providing maximum surface area for perfusion on the opposite side.171 With this approach, the authors demonstrated formation of hepatic cellular aggregates which were viable for more than two weeks. Alternatively, etched silicon172 or polymer membranes177 have been reported for the same purpose. Using the same perfusion-based culture approach through a porous membrane, Griffith and co-workers developed a multiplexed platform compatible with standard well-plate equipment; the device included 12 independent microreactors in which primary hepatocytes could be kept in 3D culture for several weeks, while maintaining important liver specific functions.172,178
In another approach, 3D hepatocyte tissues were combined with microfabricated PDMS structures recapitulating the liver sinusoidal space that is naturally made from endothelial cells.82 Medium was perfused in a microchannel separated from the cell culture chamber by the microfabricated liver sinusoid. Functional liver tissue was obtained, after seeding of hepatocytes, and culture was demonstrated for over 7 days. In a more refined device, rat primary hepatocytes or human Hep G2/C3A cells were cultured in connection to a rat vasculature via a membrane.179 The model, which was tested for short-term survival and function maintenance, was seen as a promising ex vivo model for clinical settings.
Hepatospheres or hepatocyte-base spheroids were also reported as an in vitro approach to culture liver cells, while keeping their functions.180 As for other tissues, hepatospheres were formed in microfabricated well arrays180 or microfluidic devices.115 For instance, culture of hepatospheres in microchannels equipped with microwells enabled to keep their geometry and function, in a parallelized fashion for HTS, while assessing the effect of flow, and testing co-culture.181–184
The different liver models presented are excellent candidates for drug screening at first, but for the future it is envisioned that assembling and implanting such microtissues may support or overtake certain liver functions as an RM strategy.
Besides liver, systems for kidney and lung/airways are well known examples of tissues built by employing microfluidics and other microengineering technologies, predominantly to test a specific function of the organ or for drug screening, rather than as a model to test regenerative strategies. For example, microfluidic systems were used to study renal cell behavior under influence of shear stress and chemical gradients.185–187 Huh and co-workers demonstrated a microfluidic device to investigate lung injury by fluid mechanical stresses,81 as well as mechanical stretching, and used the system as a model to test for toxic aerosols.83 Kniazeva et al. demonstrated a microfluidic approach for a respiratory assist device, using high surface-to-volume ratio of microfluidic channel networks in a gas-permeable silicone material.188
Future perspectives
Examples of platforms used as a model to study regenerative processes in neuronal, vascular, musculoskeletal and hepatic applications which we have discussed so far are illustrative of the advantages of microfluidics over classical, static cell cultures in a Petri dish. The power to predict and control flows has been utilized for purposes of creating artificial tissue ‘defects’, biologically relevant shear stresses, gradients of compounds of interest, and manipulation of cell orientation and movement, all with high precision. Besides, examples of parallelization demonstrated exciting opportunities to increase screening throughput by a multitude of what is achieved in conventional settings.While fluid regimes applied are often very smart and create well-defined gradients, the features of most platforms are relatively simple in terms of geometry and cell population. Cells are often cultured as a monolayer, on the bottom of a channel/chamber or on a membrane. Experiments are predominantly performed on one cell-type, and when two cell-types are involved, they are either separated in the device or mixed in a random manner. Experimental results from such studies are undoubtedly useful to obtain some fundamental information on cell–cell interactions or response of cells to (bio)chemicals, but the question remains if they are sophisticated enough to test and develop regenerative strategies. This question is highly relevant considering that even in the case of a comparatively simple injury like skin wound, damage involves much more than a monolayer and one cell type. Other tissues like, for example, bone are even more complex owing to their well-defined 3D structure but also because steps leading to complete regeneration of bone tissue, including, for example callus formation and mineralization, are multiple.
For these reasons, models that combine the 3D geometrical complexity including ECM and cell heterogeneity with the already discussed advantages of microfluidics seem like the way to go in order for microfluidics to become a standard tool in the RM research. But is this feasible?
Organs-on-chips, developed as advanced in vitro models with the aim to mimic the potential key-aspects of human physiology with respect to a certain tissue or organ, and combining realistic biological read-out with simplicity, low cost, high throughput and reproducibility, may potentially make a large impact on RM research. For this, in contrast to conventional cell culture, microfluidic chips provide features such as organ-level organization of cells or tissues, physiological gradients of growth factors or cytokines, shear stress from pulsatile fluid flow or cyclic stretch from elastic membranes. While we have briefly described some of such models in the previous section, a more detailed review of various examples of successful organs-on-chips has recently been published by Baker.4
As mentioned earlier, the existing organs-on-chips are excellent models to study fundamental physiological processes and for drug/toxicity screens, but do they meet the needs of RM research? In a conventional approach for organs-on-chips, the major cells or tissues contributing to the overall function of a certain organ are cultured in separate microfluidics compartments, and through connections between the compartments, fundamental physiological processes are studied upon exposure to stimuli. As is the case for ‘regular’ on-chip systems, the cells or tissues in the compartments are mainly cultured in comparatively poorly defined environments. These have simple 2½D geometries as derived from anisotropic micro-structuring processes and are made from materials which are biocompatible or inert. To increase the potential relevance of such systems for studying regenerative processes, it would be useful for example to engineer more complex artificial cellular microenvironments. These engineered environments should have hierarchical multi-scale 3D or curved geometries such as the unique structure of the hepatic cord of the liver,189 supporting a corresponding spatial organization and consequently communication of the cells as it is similarly found in the vast majority of the mammalian tissues. Within each compartment, heterogeneous populations of cells could be created by co-culture of cells in the form of simultaneous cell culture in the same environment,190 physically separated,110 or in a unique configuration,191 to provide tissue organization and function, or to recreate an artificial cell niche. But also compartments themselves could possibly be positioned in such a way that they more closely resemble the three-dimensionality of native tissue. By doing so, an environment would be created in which cells can be cultured for longer, clinically relevant time periods to allow for studying all processes leading to successful regeneration. In such, more complex systems, it is also envisioned that some of the effects of the immune system during regeneration could be mimicked. These effects are of great importance for the natural process of regeneration of any tissue, and, yet, they are lacking in all available in vitro models. Increasing structural complexity of model tissues or organs may bring along issues of inadequate oxygen and nutrient supply and additional active perfusion or engineering of artificial vessels may be required.
Most importantly, such 3D models should be suitable to test any type of regenerative strategy of interest. While testing of growth factor-based therapies will probably be most easily applied, therapies including bioactive materials, either alone or as tissue engineered constructs may pose great challenges. Such biomaterials can be of any of the three material types, metals, ceramics or polymers, depending on the tissue to be regenerated, which means a much larger variation compared to the materials frequently used in microfluidic systems. Needless to say, these materials do not meet requirements of transparency, gas-permeability and processability, making their introduction into microfluidic systems not trivial. Furthermore, these bioactive materials dynamically interact with the biological systems, through protein adsorption, degradation, etc., which makes it imperative to study the level of miniaturization required to have them match the on-chip microenvironment, but also to integrate them into the device in a relevant way. Concerning the latter, coating technologies offer a relatively simple solution, although the aspect of 3D is partially lost. But also microfluidic systems themselves can be applied to develop gradients or arrays of relevant biomaterials to be studied, for example as demonstrated by Burdick et al.192 and Zaari et al.193
Surely, 3D microenvironments with heterogeneous cell populations, room for ECM production over a longer period of time, possibility to create relevant tissue injuries and study regeneration by any type of regenerative strategy, without compromising the advantages of microfluidic systems in general, and possibility to increase throughput of screening in particular would be a dream come true to anyone working in the field of RM.
This increase in complexity will undoubtedly also introduce challenges regarding applicability and reliability of assays, which may not be suitable for that level of complexity in 3D. Van der Meer and van den Berg recognized this issue and suggested that enhancement of complexity should be accompanied by further technological advancements in terms of integration of microelectrical, micromechanical and microfluidic components.5 While the authors identified biologists, toxicologists and the pharmaceutical industry as end-users of such advanced organs-on-chips systems, we believe that they may be of great interest to scientists developing regenerative strategies as well.
Acknowledgements
PH acknowledges financial support by Innovative Research Incentives Scheme Veni (#10236) of the Netherlands Organization for Scientific Research (NWO).References
- A. Daar and H. Greenwood, J. Tissue Eng. Regener. Med., 2007, 1, 179–184 CrossRef CAS.
- G. M. Whitesides, Nature, 2006, 442, 368–373 CrossRef CAS.
- S. J. Maerkl, Integr. Biol., 2009, 1, 19–29 RSC.
- M. Baker, Nature, 2011, 471, 661–665 CrossRef CAS.
- A. D. van der Meer and A. van den Berg, Integr. Biol., 2012, 4, 461–470 RSC.
- H. Yu, C. M. Alexander and D. J. Beebe, Lab Chip, 2007, 7, 726–730 RSC.
- P. C. Thomas, S. R. Raghavan and S. P. Forry, Anal. Chem., 2011, 83, 8821–8824 CrossRef CAS.
- M. Csete, Ann. N. Y. Acad. Sci., 2005, 1049, 1–8 CrossRef CAS.
- A. P. Vollmer, R. F. Probstein, R. Gilbert and T. Thorsen, Lab Chip, 2005, 5, 1059–1066 RSC.
- A. Shamloo, D. Ph, H. Xu and S. Heilshorn, Tissue Eng. A, 2012, 18, 320–330 CrossRef CAS.
- P. Lee and P. Hung, Biotechnol. Bioeng., 2006, 94, 5–14 CrossRef CAS.
- S. Takayama, E. Ostuni, P. LeDuc, K. Naruse, D. E. Ingber and G. M. Whitesides, Nature, 2001, 411, 1016 CrossRef CAS.
- E. M. Lucchetta, M. S. Munson and R. F. Ismagilov, Lab Chip, 2006, 6, 185–190 RSC.
- E. Lucchetta, J. Lee, L. Fu, N. Patel and R. Ismagilov, Nature, 2005, 434, 1134–1138 CrossRef CAS.
- W.-T. Fung, A. Beyzavi, P. Abgrall, N.-T. Nguyen and H.-Y. Li, Lab Chip, 2009, 9, 2591–2595 RSC.
- J. Warrick, I. Meyvantsson, J. Ju and D. J. Beebe, Lab Chip, 2007, 7, 316–321 RSC.
- T. Tabata and Y. Takei, Development, 2004, 131, 703–712 CrossRef CAS.
- H. L. Ashe and J. Briscoe, Development, 2006, 133, 385–394 CrossRef CAS.
- S. Zigmond, J. Cell Biol., 1977, 75, 606–616 CrossRef CAS.
- D. Zicha, G. A. Dunn and A. F. Brown, J. Cell Sci., 1991, 99, 769–775 Search PubMed.
- T. M. Keenan and A. Folch, Lab Chip, 2008, 8, 34–57 RSC.
- B. G. Chung, L. A. Flanagan, S. W. Rhee, P. H. Schwartz, A. P. Lee, E. S. Monuki and N. L. Jeon, Lab Chip, 2005, 5, 401–406 RSC.
- C. W. Frevert, G. Boggy, T. M. Keenan and A. Folch, Lab Chip, 2006, 6, 849–856 RSC.
- T. M. Keenan, C.-H. Hsu and A. Folch, Appl. Phys. Lett., 2006, 89, 114103 CrossRef.
- S.-Y. Cheng, S. Heilman, M. Wasserman, S. Archer, M. L. Shuler and M. Wu, Lab Chip, 2007, 7, 763–769 RSC.
- N. L. Jeon, H. Baskaran, S. K. W. Dertinger, G. M. Whitesides, L. Van de Water and M. Toner, Nature Biotechnol., 2002, 20, 826–830 CAS.
- G. S. Jeong, S. Han, Y. Shin, G. H. Kwon, R. D. Kamm, S.-H. Lee and S. Chung, Anal. Chem., 2011, 83, 8454–8459 CrossRef CAS.
- J. Choi, S. Kim, J. Jung, Y. Lim, K. Kang, S. Park and S. Kang, Biomaterials, 2011, 32, 7013–7022 CrossRef CAS.
- C. S. Effenhauser, G. J. Bruin, A. Paulus and M. Ehrat, Anal. Chem., 1997, 69, 3451–3457 CrossRef CAS.
- D. C. Duffy, J. C. McDonald, O. J. Schueller and G. M. Whitesides, Anal. Chem., 1998, 70, 4974–4984 CrossRef CAS.
- J. C. Mcdonald, D. C. Duffy, J. R. Anderson and D. T. Chiu, Electrophoresis, 2000, 21, 27–40 CrossRef CAS.
- G. Mehta, J. Lee, W. Cha, Y.-C. Tung, J. J. Linderman and S. Takayama, Anal. Chem., 2009, 81, 3714–3722 CrossRef CAS.
- K. Ren, W. Dai, J. Zhou, J. Su and H. Wu, Proc. Natl. Acad. Sci. U. S. A., 2011, 108, 8162–8166 CrossRef CAS.
- M. A. Unger, C. Hou-Pu, T. Thorsen, A. Scherer and S. R. Quake, Science, 2000, 288, 113–116 CrossRef CAS.
- E. Berthier, E. W. K. Young and D. Beebe, Lab Chip, 2012, 12, 1224–1237 RSC.
- K. J. Regehr, M. Domenech, J. T. Koepsel, K. C. Carver, S. J. Ellison-Zelski, W. L. Murphy, L. A. Schuler, E. T. Alarid and D. J. Beebe, Lab Chip, 2009, 9, 2132–2139 RSC.
- M. W. Toepke and D. J. Beebe, Lab Chip, 2006, 6, 1484–1486 RSC.
- C. J. Bettinger, K. M. Cyr, A. Matsumoto, R. Langer, J. T. Borenstein and D. L. Kaplan, Adv. Mater., 2007, 19, 2847–2850 CrossRef CAS.
- C. J. Bettinger and J. T. Borenstein, Soft Matter, 2010, 6, 4999 RSC.
- A. Paguirigan and D. J. Beebe, Lab Chip, 2006, 6, 407–413 RSC.
- N. W. Choi, M. Cabodi, B. Held, J. P. Gleghorn, L. J. Bonassar and A. D. Stroock, Nat. Mater., 2007, 6, 908–915 CrossRef CAS.
- C. J. Bettinger, K. M. Cyr, A. Matsumoto, R. Langer, J. T. Borenstein and D. L. Kaplan, Adv. Mater., 2007, 19, 2847–2850 CrossRef CAS.
- B. Kundu, R. Rajkhowa, S. C. Kundu and X. Wang, Adv. Drug Deliv. Rev., 2013, 65, 457–470 CrossRef CAS.
- S. Gobaa, S. Hoehnel, M. Roccio, A. Negro, S. Kobel and M. Lutolf, Nat. Methods, 2011, 8, 949–957 CrossRef CAS.
- P. S. Lienemann, M. P. Lutolf and M. Ehrbar, Adv. Drug Delivery Rev., 2012, 64, 1078–1089 CrossRef CAS.
- S. Allazetta, S. Cosson and M. P. Lutolf, Chem. Commun., 2011, 47, 191–193 RSC.
- Y. Xia, E. Kim, X. Zhao, J. Rogers, M. Prentiss and G. Whitesides, Science, 1996, 273, 347–349 CAS.
- S. Kobel, M. Limacher, S. Gobaa, T. Laroche and M. P. Lutolf, Langmuir, 2009, 25, 8774–8779 CrossRef CAS.
- C. S. Chen, M. Mrksich, S. Huang, G. M. Whitesides and D. E. Ingber, Biotechnol. Prog., 1998, 14, 356–363 CrossRef CAS.
- R. McBeath, D. Pirone and C. Nelson, Dev. Cell, 2004, 6, 483–495 CrossRef CAS.
- C. Flaim, S. Chien and S. Bhatia, Nat. Methods, 2005, 2, 119–125 CrossRef CAS.
- S. Le Gac and A. van den Berg, Trends Biotechnol., 2010, 28, 55–62 CrossRef CAS.
- J. Rouwkema, N. C. Rivron and C. A. van Blitterswijk, Trends Biotechnol., 2008, 26, 434–441 CrossRef CAS.
- S. Kobel and M. P. Lutolf, Biotechniques, 2010, 48, ix–xxi CrossRef.
- S. Kobel and M. P. Lutolf, Curr. Opin. Biotechnol., 2011, 22, 690–697 CrossRef CAS.
- A. Ranga and M. P. Lutolf, Curr. Opin. Cell Biol., 2012, 24, 236–244 CrossRef CAS.
- C. E. Sims and N. L. Allbritton, Lab Chip, 2007, 7, 423–440 RSC.
- R. Trouillon, M. K. Passarelli, J. Wang, M. E. Kurczy and A. G. Ewing, Anal. Chem., 2013, 85, 522–542 CrossRef CAS.
- H. Yin and D. Marshall, Curr. Opin. Biotechnol., 2012, 23, 110–119 CrossRef CAS.
- D. Di Carlo, L. Y. Wu and L. P. Lee, Lab Chip, 2006, 6, 1445–1449 RSC.
- S. A. Kobel, O. Burri, A. Griffa, M. Girotra, A. Seitz and M. P. Lutolf, Lab Chip, 2012, 12, 2843–2849 RSC.
- F. T. G. van den Brink, E. Gool, J.-P. Frimat, J. Bomer, A. van den Berg and S. Le Gac, Electrophoresis, 2011, 32, 3094–3100 CrossRef CAS.
- K. S. Phillips, H. H. Lai, E. Johnson, C. E. Sims and N. L. Allbritton, Lab Chip, 2011, 11, 1333–1341 RSC.
- J. S. Marcus, W. F. Anderson and S. R. Quake, Anal. Chem., 2006, 78, 3084–3089 CrossRef CAS.
- P. M. Gilbert, K. L. Havenstrite, K. E. G. Magnusson, A. Sacco, N. A. Leonardi, P. Kraft, N. K. Nguyen, S. Thrun, M. P. Lutolf and H. M. Blau, Science, 2010, 329, 1078–1081 CrossRef CAS.
- Y.-C. Chen, P. Ingram, X. Lou and E. Yoon, 16th International MicroTAS Proceedings, 2012, 106–108 Search PubMed.
- G. Mehta, A. Y. Hsiao, M. Ingram, G. D. Luker and S. Takayama, J. Controlled Release, 2012, 164, 192–204 CrossRef CAS.
- J. M. Kelm and M. Fussenegger, Trends Biotechnol., 2004, 22, 195–202 CrossRef CAS.
- M. Ingram, G. Techy and R. Saroufeem, In Vitro Cell. Dev. Biol., 1997, 33, 459–466 CAS.
- S. L. Nyberg, J. Hardin, B. Amiot, U. A. Argikar, R. P. Remmel and P. Rinaldo, Liver Transplant., 2005, 11, 901–910 CrossRef.
- N. C. Rivron, E. J. Vrij, J. Rouwkema, S. Le Gac, A. van den Berg, R. K. Truckenmüller and C. A. van Blitterswijk, Proc. Natl. Acad. Sci. U. S. A., 2012, 109, 6886–6891 CrossRef CAS.
- Y. Torisawa, B. Chueh, D. Huh, P. Ramamurthy, T. M. Roth, K. F. Barald and S. Takayama, Lab Chip, 2007, 7, 770–776 RSC.
- H.-J. Jin, Y.-H. Cho, J.-M. Gu, J. Kim and Y.-S. Oh, Lab Chip, 2011, 11, 115–119 RSC.
- L. Yu, M. C. W. Chen and K. C. Cheung, Lab Chip, 2010, 10, 2424–2432 RSC.
- Y.-C. Tung, A. Y. Hsiao, S. G. Allen, Y. Torisawa, M. Ho and S. Takayama, Analyst, 2011, 136, 473–478 RSC.
- Y. Torisawa, B. Mosadegh, G. D. Luker, M. Morell, K. S. O'Shea and S. Takayama, Integr. Biol., 2009, 1, 649–654 RSC.
- B. G. Chung, K.-H. Lee, A. Khademhosseini and S.-H. Lee, Lab Chip, 2012, 12, 45–59 RSC.
- A. Khademhosseini and R. Langer, Biomaterials, 2007, 28, 5087–5092 CrossRef CAS.
- T. Takei, N. Kishihara, S. Sakai and K. Kawakami, Biochem. Eng. J., 2010, 49, 143–147 CrossRef CAS.
- U. A. Gurkan, S. Tasoglu, D. Kavaz, M. C. Demirel and U. Demirci, Adv. Healthcare Mater., 2012, 1, 149–158 CrossRef CAS.
- D. Huh, H. Fujioka, Y.-C. Tung, N. Futai, R. Paine, J. B. Grotberg and S. Takayama, Proc. Natl. Acad. Sci. U. S. A., 2007, 104, 18886–18891 CrossRef CAS.
- P. Lee, P. Hung and L. Lee, Biotechnol. Bioeng., 2007, 97, 1340–1346 CrossRef CAS.
- D. Huh, B. D. Matthews, A. Mammoto, M. Montoya-Zavala, H. Y. Hsin and D. E. Ingber, Science, 2010, 328, 1662–1668 CrossRef CAS.
- C. Moraes, G. Mehta, S. C. Lesher-Perez and S. Takayama, Ann. Biomed. Eng., 2012, 40, 1211–1227 CrossRef.
- W. H. Grover, M. G. von Muhlen and S. R. Manalis, Lab Chip, 2008, 8, 913–918 RSC.
- M. T. Guo, A. Rotem, J. A. Heyman and D. A. Weitz, Lab Chip, 2012, 12, 2146–2155 RSC.
- J. Baret, V. Taly, M. Ryckelynck, C. A. Merten and A. D. Griffiths, Med. Sci., 2009, 25, 627–632 Search PubMed.
- W. Gu, X. Zhu, N. Futai, B. S. Cho and S. Takayama, Proc. Natl. Acad. Sci. U. S. A., 2004, 101, 15861–15866 CrossRef CAS.
- C. L. Hansen, M. O. A. Sommer and S. R. Quake, Proc. Natl. Acad. Sci. U. S. A., 2004, 101, 14431–14436 CrossRef CAS.
- N. Bontoux, L. Dauphinot, T. Vitalis, V. Studer, Y. Chen, J. Rossier and M.-C. Potier, Lab Chip, 2008, 8, 443–450 RSC.
- C. L. Hansen, E. Skordalakes, J. M. Berger and S. R. Quake, Proc. Natl. Acad. Sci. U. S. A., 2002, 99, 16531–16536 CrossRef CAS.
- J. Melin and S. R. Quake, Annu. Rev. Biophys. Biomol. Struct., 2007, 36, 213–231 CrossRef CAS.
- B. Mosadegh, T. Bersano-Begey, J. Y. Park, M. A. Burns and S. Takayama, Lab Chip, 2011, 11, 2813–2818 RSC.
- B. Starly and A. Choubey, Ann. Biomed. Eng., 2008, 36, 30–40 CrossRef.
- S. M. Grist, L. Chrostowski and K. C. Cheung, Sensors, 2010, 10, 9286–9316 CrossRef CAS.
- C. Amatore, S. Arbault, Y. Bouret, B. Cauli, M. Guille, A. Rancillac and J. Rossier, ChemPhysChem, 2006, 7, 181–187 CrossRef CAS.
- M. Takeda, H. Shiku, K. Ino and T. Matsue, Analyst, 2011, 136, 4991–4996 RSC.
- S. H. Jeong, D. W. Lee, S. Kim, J. Kim and B. Ku, Biosens. Bioelectron., 2012, 35, 128–133 CrossRef CAS.
- W. Chen, M. Lisowski, G. Khalil, I. R. Sweet and A. Q. Shen, PLoS One, 2012, 7, e33070 CAS.
- E. E. Krommenhoek, J. G. E. Gardeniers, J. G. Bomer, X. Li, M. Ottens, G. W. K. van Dedem, M. Van Leeuwen, W. M. van Gulik, L. A. M. van der Wielen, J. J. Heijnen and A. van den Berg, Anal. Chem., 2007, 79, 5567–5573 CrossRef CAS.
- R. B. Campenot, Proc. Natl. Acad. Sci. U. S. A., 1977, 74, 4516–4519 CrossRef CAS.
- L. S. Zweifel, R. Kuruvilla and D. D. Ginty, Nat. Rev. Neurosci., 2005, 6, 615–625 CrossRef CAS.
- A. M. Taylor, D. C. Dieterich, H. T. Ito, S. A. Kim and E. M. Schuman, Neuron, 2010, 66, 57–68 CrossRef CAS.
- D. Kilinc, J.-M. Peyrin, V. Soubeyre, S. Magnifico, L. Saias, J.-L. Viovy and B. Brugg, Neurotoxic. Res., 2011, 19, 149–161 CrossRef.
- W. W. Liu, J. Goodhouse, N. L. Jeon and L. W. Enquist, PLoS One, 2008, 3, e2382 Search PubMed.
- K. A. Hosie, A. E. King, C. A. Blizzard, J. C. Vickers and T. C. Dickson, ASN Neuro, 2012, 4, 47–57 CrossRef CAS.
- J. W. Park, B. Vahidi, A. M. Taylor, S. W. Rhee and N. L. Jeon, Nat. Protoc., 2006, 1, 2128–2136 CrossRef CAS.
- A. M. Taylor and N. L. Jeon, Crit. Rev. Biomed. Eng., 2011, 39, 185–200 CrossRef.
- I. H. Yang, R. Siddique, S. Hosmane, N. Thakor and A. Höke, Exp. Neurol., 2009, 218, 124–128 CrossRef CAS.
- L. Li, L. Ren, W. Liu, J.-C. Wang, Y. Wang, Q. Tu, J. Xu, R. Liu, Y. Zhang, M.-S. Yuan, T. Li and J. Wang, Anal. Chem., 2012, 84, 6444–6453 CrossRef CAS.
- Y. Kim, K. Karthikeyan, S. Chirvi and D. P. Davé, Lab Chip, 2009, 9, 2576–2581 RSC.
- J. Walter, S. Henke-Fahle and F. Bonhoeffer, Development, 1987, 101, 909–913 CAS.
- B. Vahidi, J. W. Park, H. J. Kim and N. L. Jeon, J. Neurosci. Methods, 2008, 170, 188–196 CrossRef CAS.
- C. J. Wang, X. Li, B. Lin, S. Shim, G.-L. Ming and A. Levchenko, Lab Chip, 2008, 8, 227–237 RSC.
- N. Bhattacharjee, N. Li, T. M. Keenan and A. Folch, Integr. Biol., 2010, 2, 669–679 RSC.
- A. Kunze, M. Giugliano, A. Valero and P. Renaud, Biomaterials, 2011, 32, 2088–2098 CrossRef CAS.
- A. Kunze, A. Valero, D. Zosso and P. Renaud, PLoS One, 2011, 6, e26187 CAS.
- A. Kunze, R. Meissner, S. Brando and P. Renaud, Biotechnol. Bioeng., 2011, 108, 2241–2245 CrossRef CAS.
- C. R. Kothapalli, E. van Veen, S. de Valence, S. Chung, I. K. Zervantonakis, F. B. Gertler and R. D. Kamm, Lab Chip, 2011, 11, 497–507 RSC.
- H. G. Sundararaghavan, G. A. Monteiro, B. L. Firestein and D. I. Shreiber, Biotechnol. Bioeng., 2009, 102, 632–643 CrossRef CAS.
- H. G. Sundararaghavan, S. N. Masand and D. I. Shreiber, J. Neurotrauma, 2011, 28, 2377–2387 CrossRef.
- B. J. Pfister, A. Iwata, D. F. Meaney and D. H. Smith, J. Neurosci., 2004, 24, 7978–7983 CrossRef CAS.
- D. H. Smith, J. A. Wolf, T. A. Lusardi, V. M. Lee and D. F. Meaney, J. Neurosci., 1999, 19, 4263–4269 CAS.
- M. D. Tang-Schomer, A. R. Patel, P. W. Baas and D. H. Smith, FASEB J., 2010, 24, 1401–1410 CrossRef CAS.
- Y.-W. Lin, C.-M. Cheng, P. R. Leduc and C.-C. Chen, PLoS One, 2009, 4, e4293 Search PubMed.
- S.-M. Ong, C. Zhang, Y.-C. Toh, S. H. Kim, H. L. Foo, C. H. Tan, D. van Noort, S. Park and H. Yu, Biomaterials, 2008, 29, 3237–3244 CrossRef CAS.
- L. Ziegler, S. Grigoryan, I. H. Yang, N. V. Thakor and R. S. Goldstein, Stem Cell Rev., 2011, 7, 394–403 CrossRef.
- P. Shi, M. A. Scott, B. Ghosh, D. Wan, Z. Wissner-Gross, R. Mazitschek, S. J. Haggarty and M. F. Yanik, Nat. Commun., 2011, 2, 510 CrossRef.
- X. Tian, S. Wang, Z. Zhang and D. Lv, PLoS One, 2012, 7, e42804 CAS.
- M. C. Peterman, N. Z. Mehenti, K. V. Bilbao, C. J. Lee, T. Leng, J. Noolandi, S. F. Bent, M. S. Blumenkranz and H. A. Fishman, Artif. Organs, 2003, 27, 975–985 CrossRef CAS.
- R. K. Jain, P. Au, J. Tam, D. G. Duda and D. Fukumura, Nat. Biotechnol., 2005, 23, 821–823 CrossRef CAS.
- Y. Zheng, J. Chen, M. Craven, N. W. Choi, S. Totorica, A. Diaz-Santana, P. Kermani, B. Hempstead, C. Fischbach-Teschl, J. A. López and A. D. Stroock, Proc. Natl. Acad. Sci. U. S. A., 2012, 109, 9342–9347 CrossRef CAS.
- B. M. Gillette, J. A. Jensen, B. Tang, G. J. Yang, A. Bazargan-Lari, M. Zhong and S. K. Sia, Nat. Mater., 2008, 7, 636–640 CrossRef CAS.
- B. Carrion, C. P. Huang, C. M. Ghajar, S. Kachgal, E. Kniazeva, N. L. Jeon and A. J. Putnam, Biotechnol. Bioeng., 2010, 107, 1020–1028 CrossRef CAS.
- S. Chung, R. Sudo, I. K. Zervantonakis, T. Rimchala and R. D. Kamm, Adv. Mater., 2009, 21, 4863–4867 CrossRef CAS.
- W. A. Farahat, L. B. Wood, I. K. Zervantonakis, A. Schor, S. Ong, D. Neal, R. D. Kamm and H. H. Asada, PLoS One, 2012, 7, e37333 CAS.
- Y. Shin, J. S. Jeon, S. Han, G.-S. Jung, S. Shin, S.-H. Lee, R. Sudo, R. D. Kamm and S. Chung, Lab Chip, 2011, 11, 2175–2181 RSC.
- R. Sudo, S. Chung, I. K. Zervantonakis, V. Vickerman, Y. Toshimitsu, L. G. Griffith and R. D. Kamm, FASEB J., 2009, 23, 2155–2164 CrossRef CAS.
- J. M. Chan, I. K. Zervantonakis, T. Rimchala, W. J. Polacheck, J. Whisler and R. D. Kamm, PLoS One, 2012, 7, e50582 CAS.
- C.-C. Liang, A. Y. Park and J.-L. Guan, Nat. Protoc., 2007, 2, 329–333 CrossRef CAS.
- M. Felder, P. Sallin, L. Barbe, B. Haenni, A. Gazdhar, T. Geiser and O. Guenat, Lab Chip, 2012, 12, 640–646 RSC.
- A. D. van der Meer, K. Vermeul, A. A. Poot, J. Feijen and I. Vermes, Am. J. Physiol.: Heart Circ. Physiol., 2010, 298, H719–725 CrossRef CAS.
- K. Jang, K. Sato, K. Igawa, U. Chung and T. Kitamori, Anal. Bioanal. Chem., 2008, 390, 825–832 CrossRef CAS.
- Y.-T. Xiao, L.-X. Xiang and J.-Z. Shao, Biochem. Biophys. Res. Commun., 2007, 362, 550–553 CrossRef CAS.
- D. Rickard, T. Sullivan and B. Shenker, Dev. Biol., 1994, 161, 218–228 CrossRef.
- E. Leclerc, B. David, L. Griscom, B. Lepioufle, T. Fujii, P. Layrolle and C. Legallaisa, Biomaterials, 2006, 27, 586–595 CrossRef CAS.
- B. Riehl and J. Lim, Cells, 2012, 1, 1225–1245 CrossRef CAS.
- C. Moraes, G. Wang, Y. Sun and C. A. Simmons, Biomaterials, 2010, 31, 577–584 CrossRef CAS.
- C. Moraes, J.-H. Chen, Y. Sun and C. A. Simmons, Lab Chip, 2010, 10, 227–234 RSC.
- Y.-C. Toh, C. Zhang, J. Zhang, Y. M. Khong, S. Chang, V. D. Samper, D. van Noort, D. W. Hutmacher and H. Yu, Lab Chip, 2007, 7, 302–309 RSC.
- Y. Li, J. Qin, B. Lin and W. Zhang, Tissue Eng., Part C, 2010, 16, 1267–1275 CrossRef CAS.
- J.-H. Lee, Y. Gu, H. Wang and W. Y. Lee, Biomaterials, 2012, 33, 999–1006 CrossRef CAS.
- J.-H. Lee, H. Wang, J. B. Kaplan and W. Y. Lee, Acta Biomater., 2010, 6, 4422–4429 CrossRef CAS.
- J. Lee and H. Wang, Tissue Eng., Part C, 2010, 17, 39–48 CrossRef.
- A. Tourovskaia, X. Figueroa-Masot and A. Folch, Lab Chip, 2005, 5, 14–19 RSC.
- T. F. Kosar, A. Tourovskaia, X. Figueroa-Masot, M. E. Adams and A. Folch, Lab Chip, 2006, 6, 632–638 RSC.
- A. Tourovskaia, X. Figueroa-Masot and A. Folch, Nat. Protoc., 2006, 1, 1092–1104 CrossRef CAS.
- A. Tourovskaia, N. Li and A. Folch, Biophys. J., 2008, 95, 3009–3016 CrossRef CAS.
- E. Figallo, C. Cannizzaro, S. Gerecht, J. A. Burdick, R. Langer, N. Elvassore and G. Vunjak-Novakovic, Lab Chip, 2007, 7, 710–719 RSC.
- L. Shintu, R. Baudoin, V. Navratil, J.-M. Prot, C. Pontoizeau, M. Defernez, B. J. Blaise, C. Domange, A. R. Péry, P. Toulhoat, C. Legallais, C. Brochot, E. Leclerc and M.-E. Dumas, Anal. Chem., 2012, 84, 1840–1848 CrossRef CAS.
- P. M. van Midwoud, E. Verpoorte and G. M. M. Groothuis, Integr. Biol., 2011, 3, 509–521 RSC.
- Y. Imura, E. Yoshimura and K. Sato, Anal. Sci., 2012, 28, 197–199 CrossRef CAS.
- R. Baudoin, A. Corlu, L. Griscom, C. Legallais and E. Leclerc, Toxicol. in Vitro, 2007, 21, 535–544 CrossRef CAS.
- V. N. Goral and P. K. Yuen, Ann. Biomed. Eng., 2012, 40, 1244–1254 CrossRef.
- S. Kaihara, J. Borenstein, R. Koka, S. Lalan, E. R. Ochoa, M. Ravens, H. Pien, B. Cunningham and J. P. Vacanti, Tissue Eng., 2000, 6, 105–117 CrossRef CAS.
- X. Ju, D. Li, N. Gao, Q. Shi and H. Hou, Biotechnol. J., 2008, 3, 383–391 CrossRef CAS.
- J. H. Sung, J. Choi, D. Kim and M. L. Shuler, Biotechnol. Bioeng., 2009, 104, 516–525 CrossRef CAS.
- A. Carraro, W.-M. Hsu, K. M. Kulig, W. S. Cheung, M. L. Miller, E. J. Weinberg, E. F. Swart, M. Kaazempur-Mofrad, J. T. Borenstein, J. P. Vacanti and C. Neville, Biomed. Microdevices, 2008, 10, 795–805 CrossRef.
- E. Leclerc, Y. Sakai and T. Fujii, Biomed. Microdevices, 2003, 5, 109–114 CrossRef CAS.
- E. Leclerc, Y. Sakai and T. Fujii, Biochem. Eng. J., 2004, 20, 143–148 CrossRef CAS.
- S. Ostrovidov, J. Jiang and Y. Sakai, Biomed. Microdevices, 2004, 6, 279–287 CrossRef CAS.
- M. J. Powers, K. Domansky, M. R. Kaazempur-mofrad, A. Kalezi, A. Capitano, A. Upadhyaya, P. Kurzawski, K. E. Wack, D. B. Stolz, R. Kamm and L. G. Griffith, Biotechnol. Bioeng., 2002, 78, 257–269 CrossRef CAS.
- R. Baudoin, G. Alberto, P. Paullier, C. Legallais and E. Leclerc, Sens. Actuators, B, 2012, 173, 919–926 CrossRef CAS.
- N. Ye, J. Qin, X. Liu, W. Shi and B. Lin, Electrophoresis, 2007, 28, 1146–1153 CrossRef CAS.
- N. Ye, J. Qin, W. Shi, X. Liu and B. Lin, Lab Chip, 2007, 7, 1696–1704 RSC.
- J. M. Prot, C. Aninat, L. Griscom, F. Razan, C. Brochot, C. G. Guillouzo, C. Legallais, A. Corlu and E. Leclerc, Biotechnol. Bioeng., 2011, 108, 1704–1715 CrossRef CAS.
- B. Altmann, S. Giselbrecht, K.-F. Weibezahn, A. Welle and E. Gottwald, Biomed. Mater., 2008, 3, 034120 CrossRef CAS.
- K. Domansky, W. Inman, J. Serdy, A. Dash, M. H. M. Lim and L. G. Griffith, Lab Chip, 2010, 10, 51–58 RSC.
- W.-M. Hsu, A. Carraro, K. M. Kulig, M. L. Miller, M. Kaazempur-Mofrad, E. Weinberg, F. Entabi, H. Albadawi, M. T. Watkins, J. T. Borenstein, J. P. Vacanti and C. Neville, Ann. Surg., 2010, 252, 351–357 CrossRef.
- S. F. Wong, D. Y. No, Y. Y. Choi, D. S. Kim, B. G. Chung and S.-H. Lee, Biomaterials, 2011, 32, 8087–8096 CrossRef CAS.
- K. Nakazawa, Y. Izumi, J. Fukuda and T. Yasuda, J. Biomater. Sci., Polym. Ed., 2006, 17, 859–873 CrossRef CAS.
- J. Fukuda and K. Nakazawa, Tissue Eng., 2005, 11, 1254–1262 CrossRef CAS.
- S.-A. Lee, D. Y. No, E. Kang, J. Ju, D.-S. Kim and S.-H. Lee, Lab Chip, 2013 10.1039/c3lc50197c.
- Y.-C. Toh, T. C. Lim, D. Tai, G. Xiao, D. van Noort and H. Yu, Lab Chip, 2009, 9, 2026–2035 RSC.
- R. Baudoin, L. Griscom, M. Monge, C. Legallais and E. Leclerc, Biotechnol. Prog., 2007, 23, 1245–1253 CAS.
- K.-J. Jang, H. S. Cho, D. H. Kang, W. G. Bae, T.-H. Kwon and K.-Y. Suh, Integr. Biol., 2011, 3, 134–141 RSC.
- K.-J. Jang and K.-Y. Suh, Lab Chip, 2010, 10, 36–42 RSC.
- T. Kniazeva, J. C. Hsiao, J. L. Charest and J. T. Borenstein, Biomed. Microdevices, 2011, 13, 315–323 CrossRef CAS.
- N. Ferrell, K. B. Ricci, J. Groszek, J. T. Marmerstein and W. H. Fissell, Biotechnol. Bioeng., 2012, 109, 797–803 CrossRef CAS.
- J. Park, H. Koito, J. Li and A. Han, Biomed. Microdevices, 2009, 11, 1145–1153 CrossRef CAS.
- C.-T. Ho, R.-Z. Lin, W.-Y. Chang, H.-Y. Chang and C.-H. Liu, Lab Chip, 2006, 6, 724–734 RSC.
- J. Burdick, A. Khademhosseini and R. Langer, Langmuir, 2004, 20, 8–11 CrossRef.
- N. Zaari, P. Rajagopalan, S. K. Kim, A. J. Engler and J. Y. Wong, Adv. Mater., 2004, 16, 2133–2137 CrossRef CAS.
Footnote |
| † Authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2013 |