Flavoenzymes: Versatile catalysts in biosynthetic pathways
Christopher T.
Walsh
* and
Timothy A.
Wencewicz
Department of Biological Chemistry and Molecular Pharmacology, Harvard Medical School, 240 Longwood Ave., Boston, MA 02115, USA. E-mail: christopher_walsh@hms.harvard.edu; Fax: +617-432-0438; Tel: +617-432-1715
First published on 10th October 2012
Abstract
Covering: up to the end of May 2012
Riboflavin-based coenzymes, tightly bound to enzymes catalyzing substrate oxidations and reductions, enable an enormous range of chemical transformations in biosynthetic pathways. Flavoenzymes catalyze substrate oxidations involving amine and alcohol oxidations and desaturations to olefins, the latter setting up Diels–Alder cyclizations in lovastatin and solanapyrone biosyntheses. Both C4a and N5 of the flavin coenzymes are sites for covalent adduct formation. For example, the reactivity of dihydroflavins with molecular oxygen leads to flavin-4a-OOH adducts which then carry out a diverse range of oxygen transfers, including Baeyer–Villiger type ring expansions, olefin epoxidations, halogenations via transient HOCl generation, and an oxidative Favorskii rerrangement during enterocin assembly.
 Christopher T. Walsh | Christopher T. Walsh, born in 1944, majored in biology at Harvard and completed his PhD in biochemistry in the group of Fritz Lipmann at The Rockefeller University. After spending two years in the laboratory of Robert H. Abeles at Brandeis University as a Helen Hay Whitney post-doctoral fellow he joined the MIT Faculty (1972–1987) and since then has been at Harvard Medical School. He served as Chair of the Department of Chemistry at MIT (1978–1982), Chair of the Department of Biological Chemistry & Molecular Pharmacology at Harvard Medical School (1987–1995), and President of Dana-Farber Cancer Institute (1992–1995). His research interests lie in deciphering the molecular logic and enzymatic machinery of natural product biosynthesis. |
 Timothy A. Wencewicz | Timothy A. Wencewicz, born in 1983, graduated from Southeast Missouri State University in 2006 with a B.S. in chemistry and applied mathematics. He obtained his PhD in 2011 under the supervision of Marvin J. Miller at the University of Notre Dame as a NIH-sponsored Chemistry-Biochemistry-Biology Interface Program fellow where he focused on developing pathogen-targeted antibacterial drug delivery technologies based on bacterial siderophores. He is currently a postdoctoral fellow at Harvard Medical School in the laboratory of Christopher T. Walsh where his research has focused on studying the biosynthesis of unusual, nonproteinogenic amino acids. |
1 Introduction
The biosynthesis of many classes of small molecule natural products, including polyketides, peptide scaffolds, and isoprenoids involve directed condensation of monomeric building blocks in a series of chain elongation reactions leading to release of nascent product frameworks.1,2 Further processing of the nascent scaffolds can occur during one or more elongation cycles or can occur in a set of post-assembly enzymatic tailoring steps.3,4 Among the common tailoring paradigms are acylations, glycosylations, methylations, and redox reactions.The redox transformations are typically oxidations of scaffolds (e.g. hydroxylation, epoxidation, oxygen insertions), but less often can be reductive, for example, in the release of peptidyl thioesters from nonribosomal peptide synthetase assembly lines as aldehydes or alcohols.5,6 While nicotinamide coenzymes (NAD(P)H/NAD(P)) are the most common coenzymes involved in redox transformations in primary metabolic pathways, flavin-dependent enzymes are heavily utilized in secondary pathways and they are the focus of this review, as they enable a broad swath of chemistry. In turn flavins and iron, both in heme and nonheme contexts, overlap in their abilities to reductively activate and insert oxygen into natural product scaffolds; we will note the cases where flavin coenzymes provide sufficient activation.
2 Canonical features of flavoenzymes
In contrast to nicotinamide coenzymes, which typically dissociate from partner enzymes after each catalytic turnover,7 flavin coenzymes are bound so tightly (mostly noncovalently but some covalent linkages to the apoproteins are known) that, except in rare purposeful exceptions, they do not dissociate from their protein partners.8 This tight binding has likely evolved due to the lability of dihydroflavins (FMN and FAD) to undergo rapid, uncontrolled autoxidation outside the controlled microenvironments of enzyme active sites.9 In turn, it means that flavoenzymes operate in each catalytic cycle by way of two half reactions.10 In the first a substrate is oxidized while the bound oxidized flavin (most of the time FAD rather than FMN) becomes reduced; this is sometimes referred to as the reductive half reaction (Fig. 1a). In a second reoxidative half reaction, the reoxidation of the reduced flavin back to the oxidized form occurs as some cosubstrate is reduced (Fig. 1b). Thus the flavins act as true coenzymes not cosubstrates (unlike stoichiometric NADH oxidation to NAD) and do not show up in reaction stoichiometries.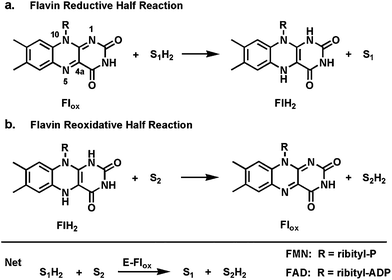 | ||
| Fig. 1 Two redox half reactions in flavoenzyme catalysis: (a) flavin reductive half reaction; (b) dihydroflavin reoxidative half reaction. | ||
The tricyclic isoalloxazine ring system of FMN and FAD offers great redox versatility, explaining why flavoproteins sit at so many cellular redox crossroads.10 Both one-electron and two-electron redox manifolds are kinetically and thermodynamically accessible (Fig. 2), so the one-electron reduced semiquinone (FlH˙) and the two-electron reduced dihydroflavin (FlH2) are both biologically relevant. Flavoproteins can thus serve as step down transformers between two-electron only manifolds (e.g. the hydride-donating (NAD(P)H), and obligate one-electron redox partners (e.g. FeIII/FeII).
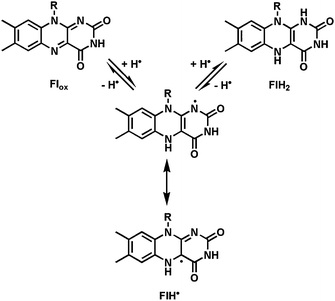 | ||
| Fig. 2 Three kinetically and thermodynamically accessible redox states: oxidized flavin (Flox); the one-electron reduced semiquinone (FlH˙); the two-electron reduced dihydroflavin (FlH2). | ||
From the point of view of mechanism, depending on the chemical features of the cosubstrates, flavin-containing enzymes can generate and react with substrate carbanion equivalents, radical equivalents, and hydride equivalents. The one-electron reactivity of both the FlH2 and FlH˙ oxidation states is the key determinant in allowing these enzyme-coenzyme systems to react with O2, in contrast to air-stable NAD(P)H coenzymes.7,11
Covalent adducts form in some flavoenzyme-mediated transformations between sites on the isoalloxazine ring of the coenzyme and a reacting center of the substrate. In particular N5 is a site of hydride addition for Flox and a site of hydride ejection from FlH2 in the reverse direction.12 N5 is also the site of covalent addition by substrate carbanions.13 In parallel, the bridgehead C4a position is the site of covalent connection with thiols14 and also with radical oxygen species, such as superoxide equivalents to yield 4a-peroxides (Fig. 3).9 We will note the double-headed reactivity of the Fl-4a-OOH intermediates in specific cases below. A small set of flavoenzymes have the isoalloxazine ring covalently tethered to an amino acid side chain (e.g. His or Cys) through the 8a-methyl or C6, indicating that those atoms can be electrophilic.15
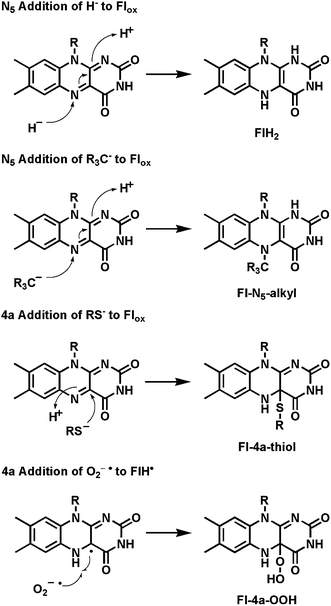 | ||
| Fig. 3 N5 and bridgehead C4a as sites of electron entry and exit from the tricyclic isoalloazine ring of flavin coenzymes: covalent 4a-thiol and 4a-hydroperoxide adducts. | ||
3 Common types of redox transformations catalyzed by flavoenzymes
3.1 Reductive half reactions
In the reductive half reactions, four types of cosubstrates are typically oxidized by two-electrons, as noted in Fig. 4a. Substrates with acid groupings (free acids or, more easily, thioesters) are desaturated to the conjugated carbonyl products (e.g. succinate to fumarate). These typically start with removal of one of the prochiral methylene hydrogens from Cα as a proton. The Cβ-H could be ejected as a hydride equivalent or it could move as a hydrogen atom, as suggested for acyl CoA dehydrogenases,16 reflecting net oxidation of the Cα carbanion/thioester enolate in two one-electron steps. A second large category of oxidations occur at H-C-XH centers, where X=O (alcohol), N (amine) or S (thiol). There are also flavin-dependent aldehyde (RCHO) oxidases, generating carboxylic acid products. Depending on the nature of the heteroatom, reaction manifolds can be viewed as involving (a) H+ and substrate carbanion, (b) H˙ and C˙ radicals, or (c) H− and a carbonium ion like transition state, within this large substrate class, with hydride transfer as the most common mechanism. The third group encompasses oxidation of dithiols to disulfides, typically involving Fl-4a-thiol adducts as schematized in Fig. 3. The last major group involves a large family of flavoenzymes oxidizing NADPH to NADP, by hydride transfer from the reduced nicotinamide coenzymes to N5 of the bound FAD.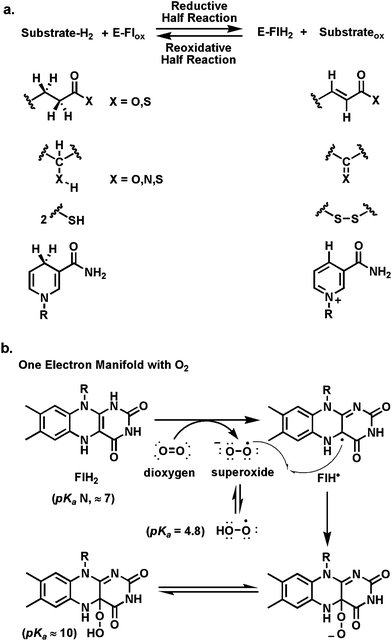 | ||
| Fig. 4 Scope of flavoenzyme redox transformations: (a) E-Flox ⇔ E-FlH2; (b) reoxidative half reaction of FlH2 with O2via one-electron transfer and recombination of FLH˙ and O2−˙. | ||
3.2 Reoxidative half reactions
For most flavoenzymes the second half reaction involves directed reoxidation of bound FADH2/FMNH2, generated by the suite of reductive half reactions noted above. These could involve formal reversal of the first half reactions. One such fate, in the second half reaction, is a net transfer of reducing equivalents from NADPH or NADH. This involves hydride transfer back out from N5 of FlH2 and represents the two-electron at a time manifold. Fumarate reductases in anaerobic bacteria create succinate as the end product as they dump electrons from their respiratory chains. Glutathione reductases spend NADPH to generate bound FlH2, which in turn drives the reduction of the oxidized intermolecular disulfide form of glutathione back to the thiol-containing tripeptide (GSH) to high levels of that thiol in cells. The one-electron manifold is seen in such enzymes as succinate dehydrogenases, which during reoxidation of FlH2 transfer one electron at a time to iron-sulfur clusters, in a separate domain (three in succinate dehydrogenase before electron transfer to a membrane soluble quinone).Of particular interest for flavoenzyme transformations in biosynthetic pathways is the one-electron reoxidation manifold for reaction with O2. This is formulated as one-electron transfer from FlH2 to yield superoxide anion (one-electron reduced dioxygen, pKa = 4.8)) and the flavin semiquinone, FlH˙ (Fig. 4b), followed by radical recombination at the bridgehead C4a to yield the 4a-flavin hydroperoxide.9,17 We will note four distinct fates for this peroxide in specific examples below.
The simplest outcome is release of H2O2 by elimination and proton transfer, a net flavoenzyme oxidase reaction, as Flox is regenerated for the next catalytic cycle. Alternatively cleavage of the O–O bond in the flavin-4a-OOH can occur with transfer of an electrophilic oxygen atom to an electron rich cosubstrate (e.g. in phenol hydroxylations to catechols). This is a monooxygenase outcome. We will note two additional variant fates for the Fl-4a-OOH: conversion to an HOCl equivalent in halogenase active sites for substrate halogenation; and behavior of the peroxyflavin as a nucleophilic equivalent in Baeyer–Villigerase transformations. Many of the interesting biosynthetic tailoring reactions that result in oxygen transfer to nascent product scaffolds involve directed reactivity of the tightly bound Fl-4a-OOH equivalent along these four outcomes in the reoxidative half reactions.
4 Biosynthetic niches for flavoprotein oxidoreductases: The reductive half reaction
4.1 Flavin-mediated desaturations to conjugated olefins
The flavoenzyme-mediated desaturation of adjacent sp3 carbon centers αβ to a carbonyl group in substrates is a key energy yielding step in the citric acid cycle (succinate to fumarate). This process is also central to the harvesting of energy in fatty acyl CoA β-oxidations. The reverse, saturation of the αβ-enoyl-S-carrier proteins by FAD-containing enoyl-ACP reductases, is the physiologic mechanism for adding hydrogens and electrons to be stored in the long methylene chains of saturated fatty acyl-S-ACP during fatty acid biosynthesis. These enoyl reductases are important catalysts in both fatty acid and polyketide scaffold constructions. Acyl carrier proteins (ACPs) are 8–10 kDa proteins where a specific serine side chain has been posttranslationally modified with the phosphopantetheine moiety from coenzyme A to provide a reactive thiol arm on which to assemble and grow the acyl chains.18,19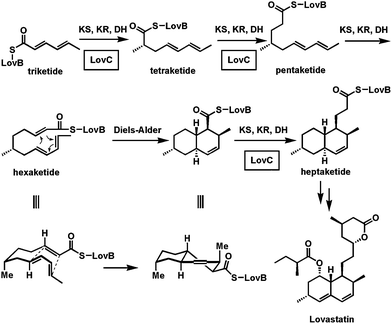 | ||
| Fig. 5 Selective action of the in trans enoyl reductase LovC during elongation of the triketide to heptaketide intermediate in lovastatin biosynthesis. The Diels–Alder cyclization to the decalin scaffold occurs at the triene-containing hexaketide stage. | ||
Therefore, the conjugated bis-olefin built by the triketide stage of chain elongation persists and subsequent failure to act at the hexaketide stage then creates a triolefinic intermediate. It is this triene that undergoes a Diels–Alder cyclization.21 LovC then acts reductively again at the heptaketide and octaketide stages. The Diels–Alder cyclization is enabled by the selective inactivity of LovC and the consequent persistence of the three olefins at the hexaketide stage. The selectivity of in trans recognition by LovC of the growing ketide chain tethered through the pantetheinyl arm to the carrier protein domain of LovB is the key feature that enables the cyclization to the decalin core scaffold of the cholesterol-lowering lovastatin.
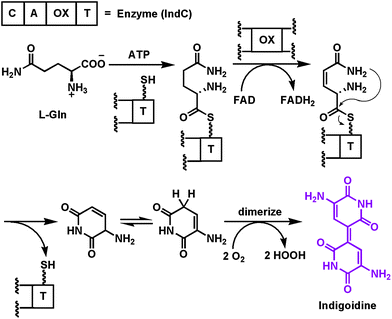 | ||
| Fig. 6 Activation and desaturation of a tethered glutamine by the oxidase domain of the NRPS module that makes the purple pigment indigoidine. | ||
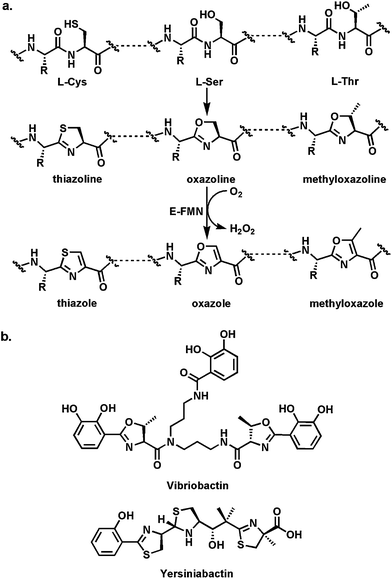 | ||
| Fig. 7 Thiazoline and oxazoline formation: (a) schematic for cyclodehydration of Cys and Ser residues in nascent proteins to thiazoline and oxazoline rings; (b) the V. cholerae siderophore vibriobactin with methyloxazoline rings; the Y. pestis siderophore yersiniabactin with thiazoline and thiazole rings. The dihydroaromatic thiazolines and methyoxazolines are part of the ferric iron chelation set. | ||
These thiazoline/oxazoline rings can also be converted to the heteroaromatic thiazoles, oxazoles, and methyloxazoles by action of flavoprotein desaturases (Fig. 7a). In the nonribosomal peptide synthetase context, as in the example of IndC above, a flavin-dependent oxidase (Ox) domain is inserted into a loop of the adenylation domain of an NRPS module. Thus, in the assembly line for the hybrid peptide/polyketide antitumor epothilones, the EpoB subunit is a C-A-Ox-T four domain module (just like IndC above). It activates and tethers cysteine and interacts with an upstream PKS subunit EpoA bearing an acetyl-S-pantetheinyl group (Fig. 8a). The C domain of EpoB transfers the acetyl moiety to the NH2 of the tethered Cys and then cyclodehydrates it to the thiazolinyl-S-T domain species.29 Now the Ox domain acts as a typical flavoprotein desaturase, presumably abstracting the acidic Cα-H as a proton and ejecting the Cβ-H as a hydride to N5 of the bound flavin coenzyme. The result of this desaturation is the conversion of the dihydrothiazoline to the heteroaromatic planar methylthiazolyl-S-EpoB. This acyl group is then transferred downstream to the polyketide synthase machinery, ultimately building the 16-member macrocyclic framework. The methylthiazole is a key activity determinant for this antimitotic cancer drug. Analogously, the bithiazole moiety of the antitumor drug bleomycin (Fig. 8b) is assembled from Cys-Cys residues on an NRPS protein by tandem cyclodehydration and desaturation.30,31 The bithiazole has the right dimensions to be a DNA intercalator and this drug acts to cause double strand breaks via oxidative damage of DNA.32
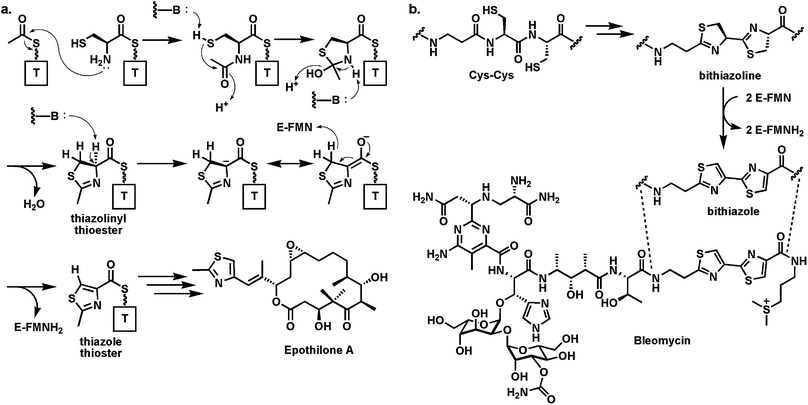 | ||
| Fig. 8 (a) Generation of methylthiazolyl-S-thiolation domain intermediate at the start of the epothilone biosynthetic assembly line from acetyl- and Cys thioesters. The FAD-dependent oxidase domain converts methylthiazoline to methylthiazole; (b) tandem conversion of adjacent Cys residues to the bithiazolyl unit in the DNA-damaging antitumor antibiotic bleomycin. | ||
Comparable logic and machinery27 is employed for posttranslational maturation of the preprotein forms of microcin B17,33 thiazolyl peptide antibiotics,34 goadsporin,35 and plantazolicin (Fig. 9).36,37 In these cases a 50–60 residue protein emerges from the bacterial ribosome as a precursor. The N-terminal 25–40 residues act as a leader peptide38 (that ultimately gets removed by proteolysis) while the C-terminal 15–40 residues undergo posttranslational maturations which include formation of thiazoles, oxazoles, and methyloxazoles. In these cases, the leader peptide is most likely an amphipathic helix that functions as a recognition element for recruitment of the cyclodehydratases and desaturases that make thiazolines and oxazolines and then convert them to the thiazole and oxazole rings. These rings replace hydrolyzable peptide backbones, rigidify the peptide scaffolds and restrict conformational flexibility, likely increasing affinity for their biological targets. The microcin B17 system is the one best characterized for the cyclodehydration and desaturation enzyme activities and also for its antibiotic action on its target DNA gyrase.39 In both contexts the flavoprotein desaturases act at the last step, and the redox change drives the morphing of amide/peptide backbones into nonhydrolyzable heterocycles.
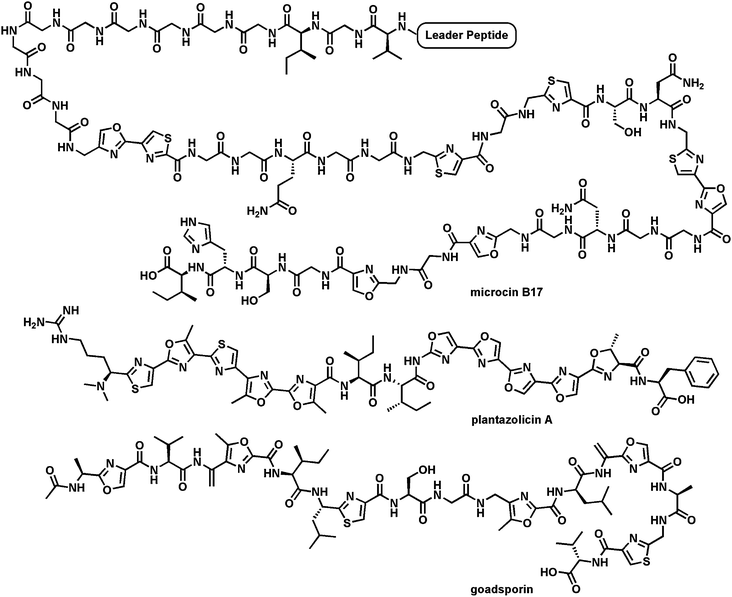 | ||
| Fig. 9 Three microbial peptides that have been posttranslationally converted into heterocyclic-containing mature products: microcin B17 targets bacterial DNA gyrase; goadsporin is a morphogen for Streptomyces development (“goads spore formation”); plantazolicin A, with two sets of five tandem heterocycles, is an antibiotic active against B. anthracis. | ||
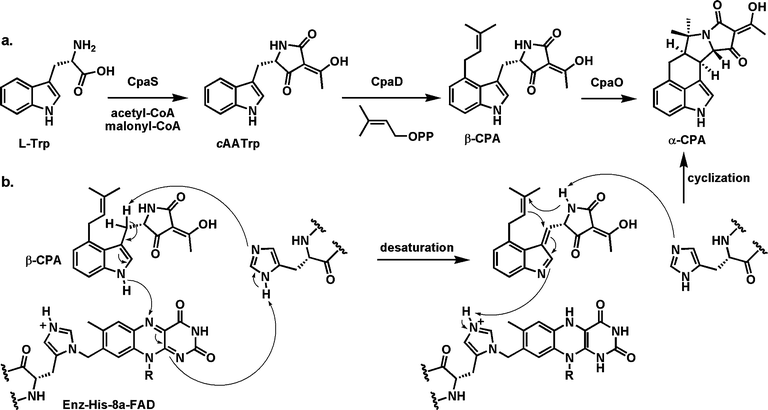 | ||
| Fig. 10 Steps in assembly of the pentacyclic fungal toxin cyclopiazonic acid A(α-CPA): (a) conversion of tryptophan to the tetramic acid cycloacetyl tryptophan (cAATrp) by action of a hybrid NRPS-PKS assembly line; (b) mechanistic proposal for the flavoenzyme CpaO-mediated-conversion of β-CPA to α-CPA with formation of the final two rings of the mature toxin. | ||
The oxidation would start with removal of one of the two prochiral hydrogens from the β-methylene of the indole moiety with electron flow into the indole and ejection of the hydrogen on the indole nitrogen as a hydride to N5 of the bound FAD. This set of steps would yield the conjugated imine as transient product along with FADH2, which gets reoxidized by O2 (see next section of flavoprotein oxygen reactions). That conjugated imine is an excellent electron sink and the double cyclization could be initiated by intramolecular attack of the weak amide nucleophile from the tetramate onto the olefin of the dimethylallyl moiety with those π-electrons in turn attacking the newly generated conjugated imine. This double cyclization cascade creates the fused pentacyclic framework of the fungal toxin.
The examples noted in this section indicate the versatility of flavin coenzymes acting formally in conjugated olefin-creating desaturations. In the specific natural product biosynthetic contexts discussed, these redox changes in nascent product frameworks create potentially reactive functionalities for Diels–Alder cyclizations, for morphing peptide backbones into five-membered ring heterocycles, and for initiating a ring-forming cascade in cyclopiazonate formation. We will note another flavoprotein-enabled Diels–Alder cyclization in the next section.
4.2 Flavin-mediated oxidations at HC–XH (X = O,N,S) centers
A second canonical set of transformations mediated by flavoenzymes, and mirroring transformations also carried out in parallel by nicotinamide-dependent enzymes, are the oxidations of sp3 carbons attached to heteroatoms: including alcohols, amines, and thiols. For our biosynthetic purposes these reductive half reactions are not particularly noteworthy mechanistically but we describe examples in natural product contexts where subsequent transformations of note are thereby enabled.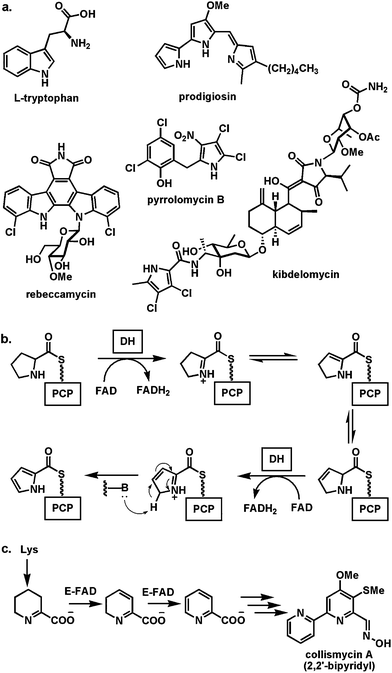 | ||
| Fig. 11 Conversion of proline units to pyrroles: (a) selected natural products where the pyrrole moieties arise from flavoenzyme desaturation of prolyl-S-thiolation domains; (b) scheme for activation and desaturation of proline monomers on NRPS modules; (c) proposal for generation of pyridine carboxylate by iterative desaturation in collismycin A biosynthesis. | ||
The conversion of proline to pyrrole carboxylate occurs while the proline is tethered in thioester to the pantetheinyl thiol arm on NRPS modules,46 in some analogy to the logic discussed in the previous sections. Proline is selected by an adenylation domain, activated as prolyl-AMP and captured by the thiol of the pantetheinyl prosthetic group of a holo T domain to yield the prolyl-S-protein (Fig. 11b). Now a free standing FAD-linked dehydrogenase converts the prolyl-S-PCP to a two electron oxidized product. This undergoes further two electron oxidation to the pyrrolyl-S-carrier protein in a process that may reflect uncatalyzed oxidative aromatization to the pyrrole. Because of its kinetic instability it is not yet clear if the two electron oxidized intermediate arises from the flavoproteins acting as amine dehydrogenases (DH), conversion of the HC–NH to the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N group via a Δ2-pyrrolinyl-S-PCP, or whether this is an α,β-desaturase, yielding the 2,3-dehydroprolyl-S-PCP as nascent product. In either case the subsequent two electron oxidation yields the pyrrole in an activated acyl linkage for subsequent transfer to a nucleophilic cosubstrate. A related double oxidation of the cyclic imine arising from Lys oxidation yields the pyridine carboxylate monomer on the way to the unusual 2,2′-bipyridyl moiety of collismycin A.47
N group via a Δ2-pyrrolinyl-S-PCP, or whether this is an α,β-desaturase, yielding the 2,3-dehydroprolyl-S-PCP as nascent product. In either case the subsequent two electron oxidation yields the pyrrole in an activated acyl linkage for subsequent transfer to a nucleophilic cosubstrate. A related double oxidation of the cyclic imine arising from Lys oxidation yields the pyridine carboxylate monomer on the way to the unusual 2,2′-bipyridyl moiety of collismycin A.47
The biosynthesis of the hexacyclic aglycone scaffold of the antitumor indolocarbazoles,48,49 exemplified by rebeccamycin and staurosporine (Fig. 12) involves an alternating set of two flavoproteins and two hemeproteins to couple two tryptophans in a process resulting in removal of 10–14 electrons as the indolocarbazole framework is assembled.50 This short pathway starts with the action of the flavoprotein RebO, followed by the hemeprotein RebD, to convert two molecules of L-Trp to chromopyrrolic acid (Fig. 12a). RebO is a conventional amine oxidase, converting each Trp to the imino acid. Condensation of one imine with a tautomeric enamine (before hydrolysis yields the inactive indolepyruvates) could yield the indicated adduct which can be captured by hemeprotein RebD acting via high valent iron intermediates to conduct one-electron chemistry, generating a benzylic radical that can lead to the C–C bond in the incipient pyrrole of chromopyrrolic acid, six electrons oxidized from the starting pair of tryptophans.51
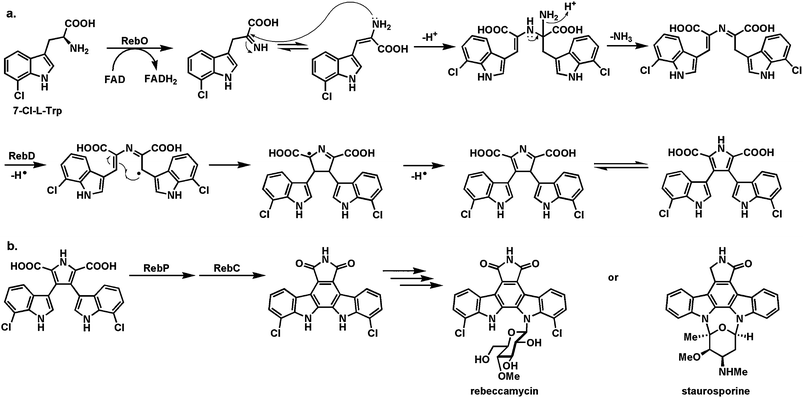 | ||
| Fig. 12 Oxidative dimerization of two tryptophans to indolocarbazole scaffolds: (a) action of RebO and RebD to generate chromopyrrolic acid: (b) action of RebP and RebC to make the final C–C bond in the indolocarbazole scaffold and control the redox state in the oxopyrrole ring. | ||
The further processing requires a reprise of the two types of flavin and heme enzymes: RebC a flavoprotein oxygenase and RebP a hemeprotein. RebP is involved in the aryl-aryl coupling to convert the chromopyrrolic acid scaffold to the indolocarbazole scaffold, while RebC is presumed to accelerate the double decarboxylation of the dicarboxypyrrole moiety and control the redox state of the final dioxopyrrole (Fig. 12b). The four enzymes, two FAD-enzymes and two heme enzymes, of the rebeccamycin pathway remove 14 electrons from the substrates/intermediates as the indolocarbazole framework is generated.
The last example of a flavoenzyme involved in a HC–NH oxidation to be featured in this section is orf Af12070, from Aspergillus fumigatus, involved in late stages of fumiquinazoline peptide alkaloid biosynthesis.52 Like the indolocarbazole biosynthetic pathway noted above, this also is a short, efficient one, with only four enzymes needed to convert anthranilate, tryptophan, and two alanines to the heptacyclic scaffold of fumiquinazoline C (FQC). The substrate for Af12070 is fumiquinazoline A (FQA) and it appears that the purified enzyme acts as an amine oxidase, oxidizing the secondary amine in the pyrazinone ring to the corresponding imine (Fig. 13). That undergoes intramolecular capture by the indicated –OH group (in turn introduced by an earlier flavoenzyme action in this pathway, noted in the oxygenase section below), generating a hemiaminal linkage in a seven membered ring and in so doing joining two tricyclic entities in the substrate to yield the remarkably complex architecture of the FQC product. The nascent imine is in equilibrium with the FQC hemiaminal because over a period of days the FQC kinetic product converts to the aminal in the thermodynamic product, fumiquinazoline D (FQD), an alternate heptacyclic framework. Af12070 is a complexity-generating redox catalyst and the HC–NH moiety oxidized is not a standard amine, but instead has elements of an amide system within the pyrazinone heterocycle.
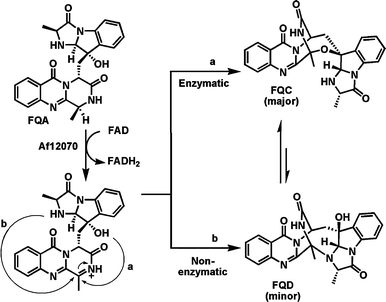 | ||
| Fig. 13 Biosynthetic pathway to fumiquinazoline C (FQC). The last step in the pathway involves oxidation of the secondary amine in fumiquinazoline A by the FAD-enzyme Af12070, followed by enzymatic intramolecular conversion of the nascent imine to the heptacyclic hemiaminal in FQC (path a) or nonenzymatic intramolecular conversion to the aminal FQD (path b). | ||
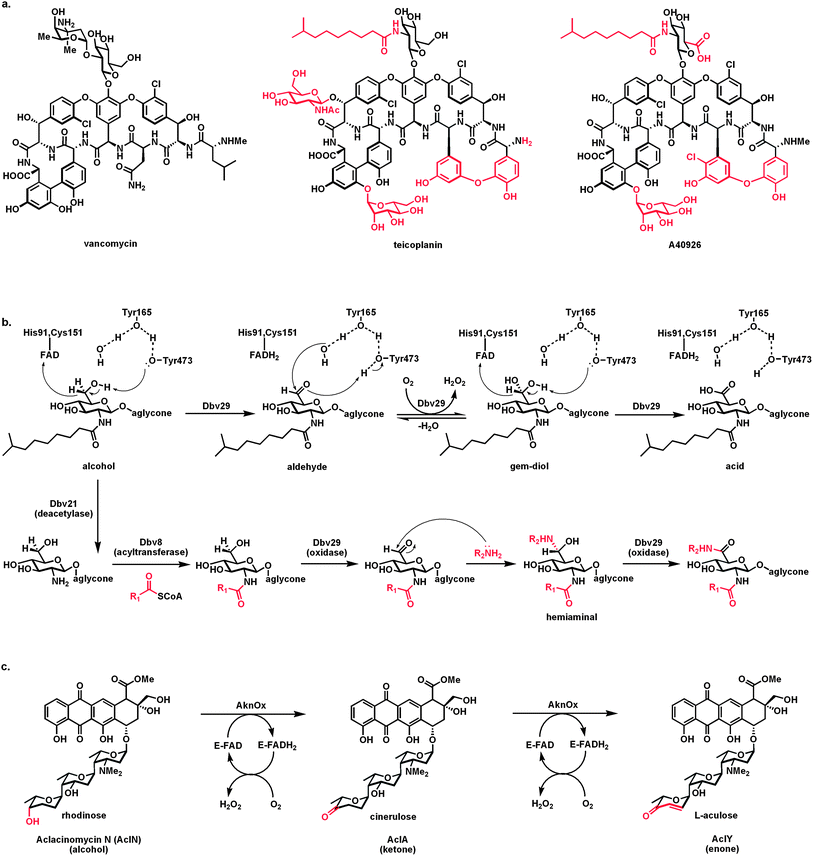 | ||
| Fig. 14 Flavoprotein alcohol oxidases carrying out net four electron oxidations: (a) structures of glycopeptide antibiotics vancomycin, teicoplanin, and A40926; (b) the flavoprotein oxidase Dbv29 oxidizes the C6-OH of the N-acetylglucosamine residue of A40926 in two discrete steps, via the aldehyde, generating the GlcNAc-6-carboxylate product; capture of the aldehyde intermediate by long chain amines and oxidation of the hemiaminal adduct produces amide variant products with two long hydrophobic substituents that confer distinct antibiotic properties; (c) Aclacinomycin oxidase acts first as an alcohol to ketone oxidase on the terminal rhodinose sugar to yield cinerulose and then carries out an α,β-desaturation to the enone functionality in the terminal sugar L-aculose. | ||
The bound aldehyde intermediate is accessible to various nucleophilic amines diffusing in from solution. The resulting hemiaminal can either be released as the imine or the hemiaminal can stay in the active site and be processed in the second redox step. In this case the product is the amide not the free acid (Fig. 14b). Turnover in the presence of amines thus gives access to glycopeptides with long chain amides at both the 2-amino and C6 (as carboxamides) of the glucosyl ring. It turns out that some of the bis-lipidated glycopeptides have desirable antibiotic properties.
An additional flavoenzyme that carries out a net four-electron oxidation on a substrate, starting with a two-electron oxidation of a sugar alcohol to the ketone, is aclacinomycin (Acl) oxidase.54 Aclacinomycins are tetracyclic aromatic polyketides with antibiotic and antitumor activity. Appended to the tetracyclic polyketide scaffold is a trisaccharide that provides binding specificity to DNA. The initial trisaccharide chain in the late stage intermediate aclacinomycin N is rhodosamine-2-deoxyfucose-rhodinose. The terminal rhodinose is a trideoxy hexose with a single alcohol substituent. The oxidase converts this –OH to the ketone, thereby generating the trideoxyketose cinerulose A (AclA; Fig. 14c). Surprisingly, the oxidase acts a second time, now not as an aldehyde oxidase but as a typical flavin-dependent desaturase, generating an olefin αβ to the newly introduced ketone to produce the αβ-enone functionality in the terminal sugar, now L-aculose. This is aclacinomycin Y, the biosynthetic endproduct. Thus the oxidase can be viewed as carrying out a hydride transfer from the rhodinose –CHOH to its covalently bound FAD in the first cycle, undergoing air reoxidation, and then in a second cycle, still with the terminal sugar, acting once more as a proton/hydride redox catalyst, but on a distinct CH2–CH2 functionality rather than a CH–OH group. No other examples of such a four-electron two functional group redox flavoenzyme are known.
A third flavoprotein alcohol oxidase of note is found in pacidamycin biosynthesis.55 It is not so much the mechanism that is of interest but the substrate is uridine and the product is uridine-5′-aldehyde (Fig. 15), a metabolite proposed several times as a necessary intermediate in peptidyl nucleoside antibiotic biogenesis,56 but until recently not known. Indeed a parallel route to this aldehyde has also been reported from UMP,57 but in that case it involves an iron-based oxygenase to yield the phosphorylated form of the gem diol as a labile form of the aldehyde, reflecting the complementary routes and roles of iron enzymes and flavoenzymes as redox catalysts. Inter alia the uridine aldehyde is then used for PLP-mediated C–C bond formations to connect amino acid moieties to the 5′-aldehydic carbon and build distinct peptidyl nucleoside scaffolds.56
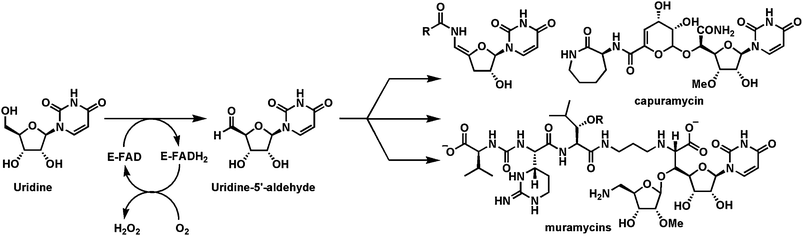 | ||
| Fig. 15 Flavoenzyme mediated conversion of uridine to uridine-5′-aldehyde, an intermediate in several peptidyl nucleoside antibiotic pathways. | ||
4.2.2.1 A Diels–Alder cyclization set up by alcohol oxidation to aldehyde. The fungus Alternaria solani, a causative agent of blight in potato and tomato, makes the polyketide solanapyrone A58 (Fig. 16) with a decalin core which has been proposed to form via a [4+2] cyclization analogous to the lovastatin scaffold construction (Fig. 5). It was presumed that a comparable PKS catalyst would be responsible for this biological Diels–Alder cyclization, but instead that PKS assembly line releases the triene prosolanapyrone. The CH3 of that metabolite is converted to an alcohol, prosolanapyrone II, by an iron-based oxygenase, setting the stage for a flavoprotein oxidoreductase Sol5. At one level this seems to be a simple alcohol to aldehyde oxidase. However, the aldehyde product is now in conjugation via the pyrone ring with the isolated olefin; that is thought to have a lowest unoccupied molecular orbital now tuned to react as a dienophile with the diene for the [4+2] cyclization to create the bicyclic decalin in solanapyrone A. Thus, the sequellae to flavin-mediated oxidation of alcohol to conjugated aldehyde are remarkable. Redox control sets the rearrangement of the diene and dienophile in motion.
![In solanapyrone biosynthesis the FAD-enzyme Sol5 oxidizes the alcohol group in prosolanapyrone II to the aldehyde which enables a [4 + 2] cyclization of the triene to the decalin ring in solanapyrone A.](/image/article/2013/NP/c2np20069d/c2np20069d-f16.gif) | ||
| Fig. 16 In solanapyrone biosynthesis the FAD-enzyme Sol5 oxidizes the alcohol group in prosolanapyrone II to the aldehyde which enables a [4 + 2] cyclization of the triene to the decalin ring in solanapyrone A. | ||
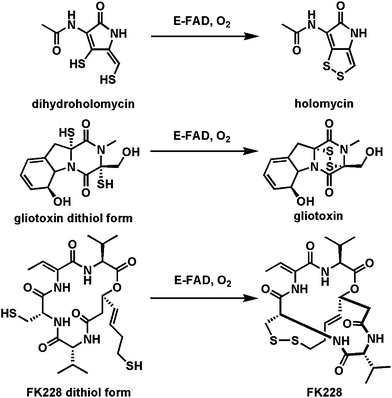 | ||
| Fig. 17 Three flavoprotein dithiol to disulfide oxidases as the last steps in the biosynthesis of holomycin, gliotoxin, and FK228. | ||
A distinct variant of thiol-oxidizing flavoproteins involve thiol oxidation of a cysteine moiety to create the thioaldehyde as an electron sink β to the carboxylate to set up a low energy decarboxylation path and create the enethiol tautomer of a vinylcysteamine63 (Fig. 18a). In the case of pantotheinoyl-cysteine decarboxylase64,65 in coenzyme A biosynthesis, the thioaldehyde is re-reduced by the FADH2 to generate the cysteamine terminus that persists in the final coenzyme A structure. In the case of posttranslational processing of the C-terminal cysteines in lantipeptide maturation, the Z-enethiol product can undergo Michael reactions with dehydroalanyl residues elsewhere in the preprotein to yield the crosslinked aminovinyl Cys residue (Fig. 18b).63
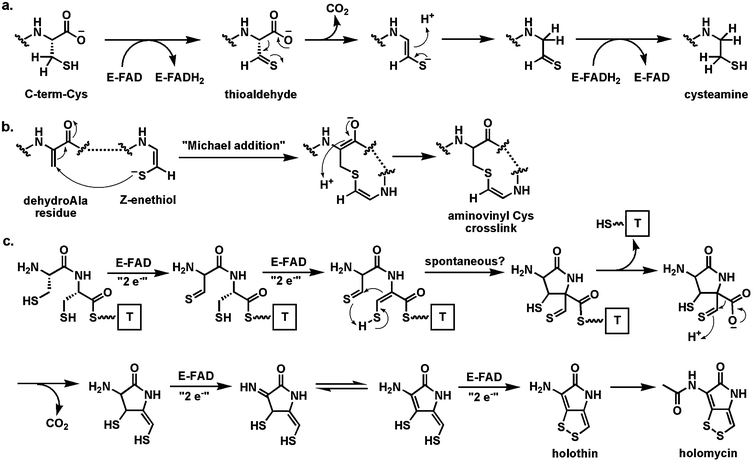 | ||
| Fig. 18 Flavoenzymes coupling thiol oxidation with decarboxylation: (a) the pantotheinyl cysteine decarboxylase in the coenzyme A biosynthetic pathway effects decarboxylation via reversible redox at the CH2–SH side chain of substrate; (b) maturation of some lantipeptides involves decarboxylation and crosslinking to yield an aminovinyl cysteine unit; (c) four flavoenzymes proposed to catalyze net eight-electron oxidation of a Cys-Cys precursor to holomycin. | ||
A homolog of this flavoenzyme class is also found in the holomycin cluster66 where it is proposed to perform a similar oxidative decarboxylation function on a Cys-Cys-dipeptide scaffold. In fact, the conversion of a Cys-Cys dipeptidyl moiety to holomycin is a formal eight-electron oxidation; there are four flavoproteins encoded in the holomycin biosynthetic gene cluster (including the dithiol oxidase HmlI noted above) suggesting flavin redox chemical versatility is employed in each of the four two-electron oxidations on the way to the mature bicyclic ene-disulfide framework of this antibiotic (Fig. 18c).
4.3 N5 of Flavin as a site of covalent adduct formation during catalytic turnover
As noted in the next section, the many dozens of flavoenzymes known to oxidize NAD(P)H in the reductive half reaction transfer a hydride to N5 of the flavin, emphasizing this end of the double imine moiety of N5-C4a-C1a-N1 as electrophilic in oxidized flavins. Two cases where carbon nucleophiles (carbanion equivalents) are shown/proposed to add covalently to N5 of the oxidized flavin coenzymes are nitroalkane oxidase and acyldihydroxyacetone-P synthase.The nitroalkane oxidase from the fungus Fusarium oxysporum forms an N5-covalent adduct with substrate in the flavin reductive half reaction.67–69 This enzyme oxidizes nitroalkanes in the presence of O2 to aldehydes and H2O2 with concomitant elimination of nitrite anion (Fig. 19). Catalysis starts with abstraction of one of the Cα-H hydrogens of the bound nitroalkane substrate as a proton because the resultant carbanion can be stabilized by delocalization into the nitro group. The carbanion then adds to N5 of FAD to yield the N5-adduct with formal transfer of two electrons such that the flavin in this adduct is in the dihydro oxidation state. The lone pair of electrons on N5 in this adduct, available because N5 is now in the electron rich dihydro redox state, act in neighboring group assistance to expel the nitrite anion. The resulting adduct to the flavin is an N5-iminium dihydroflavin. Hydrolysis via water addition yields the aldehyde and FADH2, which can be reoxidized to FAD by cosubstrate O2. The iminium adduct can be trapped by cyanide ion to yield the stable inactive covalent flavin adduct.68 This mechanism is precedent for N5 adducts to substrate during turnover, the presence of N5 product imines on pathway, and the role of the N5 lone pair as internal nucleophile.
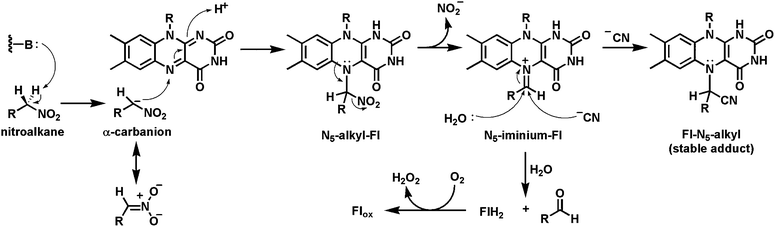 | ||
| Fig. 19 Nitroalkane oxidase catalysis involves a covalent nitroalkyl-N5 flavin adduct. | ||
A second flavoenzyme proposed to function via an iminium adduct of a dihydroflavin coenzyme to substrate during the catalytic cycle is alkyldihydroxyacetone phosphate synthase70 which converts some fraction of the diacylglycerol phospholipids in cell membranes to the monoacyl, monoalkyl ether P lipids (Fig. 20). This is a net replacement of a long chain fatty acid substituent by a long chain fatty alcohol. The catalytic cycle is proposed to start with oxidized FAD tightly bound in the active site and the removal of the diacylglycerol phospholipid (acyl DHAP) substrate hydrogen at C1 as a proton. Addition of the resultant carbanion to N5 yields the initial adduct. Participation of the N5 lone pair, just as in the nitroalkane oxidase adduct, breaks the ester bond, expelling the RCOOH product and giving an N5-flavin iminium adduct. This can be attacked by the oxygen of the fatty alcohol substrate once it has diffused into the active site and replaced the departed RCOOH. The resultant adduct is a carbinolamine and reoxidation of the flavin with C–N bond cleavage (Fig. 20) would generate a product carbanionic transition state that needs to be reprotonated to drive the release of the alkyldihydroxyacetone phosphate product (alkyl DHAP).
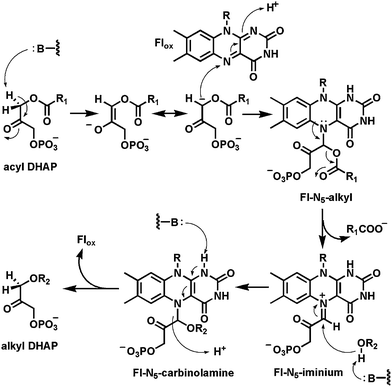 | ||
| Fig. 20 Carbanion intermediate preceeds N5 adduct formation to setup elimination of the 2-O-acyl substituent in formation of alkyl DHAP lipids. | ||
4.4 NADPH Oxidizing flavoenzymes
Most of the NADPH-oxidizing flavoenzymes found in biosynthetic pathways are involved in co-substrate oxygenation reactions. Therefore it is the reoxidative half reactions from FlH2 that are of interest for analysis of the chemical outcomes rather than the reductive half reactions which all proceed by transfer of one of the prochiral hydrogens at C4 of the dihydronicotinamide ring as a hydride to N5 of the bound oxidized flavin cofactor (Fig. 3 and 4a).However, one NADPH-oxidizing flavoenzyme variant of note is the bacterial enzyme MurB,71,72 which is the second step in assembly of the peptidoglycan scaffold in cell wall biosynthesis. The reoxidative half reaction of the FADH2 in the MurB active site involves hydride transfer from N5 to the C3′ olefinic terminus of the enol ether link in the 3′-O-pyruvyl-GlcNAc-MurNAc-UDP cosubstrate (Fig. 21). As shown the hydride transfer to C3′ and concomitant proton transfer to C2′ effects the reduction of the enol ether with chiral control to the saturated D-lactyl ether in the product UDP-muramic acid. The carboxylate of the lactate moiety is then the anchor point for addition of five amino acids, one at a time, to produce the UDP-muramyl-pentapeptide that marks completion of the cytoplasmic phase of peptidoglycan assembly.73 Reduction of the electron rich enol ether by regiospecific hydride transfer from MurB is the formal reverse of the desaturations noted in earlier section above.
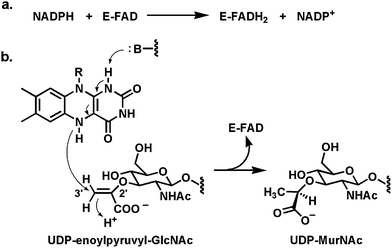 | ||
| Fig. 21 (a) The FAD-enzyme MurB uses NADPH as hydride transfer agent to generate FADH2 in the reductive half reaction; (b) in the reoxidative half reaction a hydride is transferred from N5 of FADH2 to the olefinic terminus of UDP-enolpyruvyl-GlcNAc, generating the UDP-muramic acid product. | ||
5 Biosynthetic niches for flavoprotein oxidoreductases: The reoxidative half reaction
We noted in the introductory section that dihydroflavins are spontaneously reoxidized in aerobic solutions and that the kinetically allowed pathway is by one-electron transfer, generating one-electron reduced oxygen, the superoxide anion, and the flavin semiquinone (Fig. 4b). Radical recombination leads to the bridgehead covalent Fl-4a-OOH. In flavoprotein oxidases, the intramolecular breakdown involves removal of the N5-H as a proton and elimination of the adjacent OOH−, a net production of stoichiometric amounts of hydrogen peroxide (HOOH, pKa = 10) as the oxidized flavin is regenerated. Many of the flavoproteins discussed above, with focus on their reductive half reactions, are reoxidized by this route and so function as oxidases. The bimolecular rate constant for FlH2 reaction with O2 under physiologic conditions is about 300 M−1 s−1.17 Some oxidases have kcat/Km values three orders of magnitude faster, reflecting active site microenvironmental factors that speed up reactivity with oxygen.74,75More interesting than oxidases, from the biosynthetic perspective, are flavoenzymes which redirect the default fate of the Fl-4a-OOH intermediate away from decomposition to H2O2 and towards insertion of one or, unusually, both oxygens into a cosubstrate (Fig. 22).76,77 The canonical view9,10,17 has been that such flavin monooxygenases/hydroxylases are able to transfer the distal oxygen of Fl-4a-OOH to electron rich cosubstrates, such as phenols, pyrroles, indoles, amines, and thiols, but not to less activated aromatic and aliphatic carbons. That reactivity void is filled by both mononuclear nonheme iron and heme iron (cytochrome P450) oxygenases via high valent oxo iron species.78 Several flavin dependent oxygenases are found in post-PKS assembly line tailoring of aromatic polyketides, with two examples noted below, demonstrating umpolung reactivity of the Fl-4a-OOH intermediate.
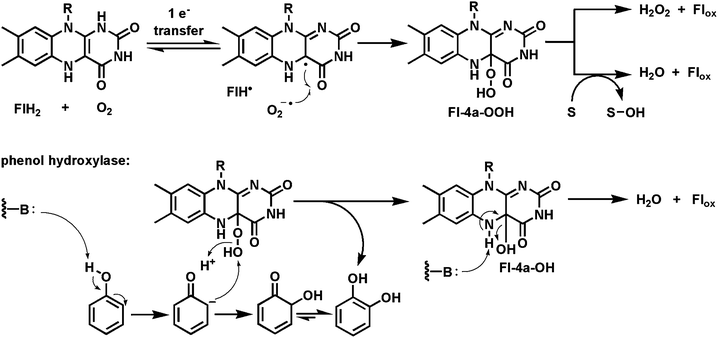 | ||
| Fig. 22 Flavoprotein monooxygenases: Transfer of the distal oxygen from Fl-4a-OOH to phenol to generate catechol. | ||
5.1 Flavoprotein hydroxylases
One example of flavoprotein hydroxylases (out of many) as tailoring enzymes is found in post assembly line modification of angucyclines (angular tetracyclic benz[a]anthracene polyketide scaffolds; Fig. 23). Early modifications involve hydroxylation of carbons 12 and 12b. PgaE hydroxylates C12, which is para to the existing phenolic-OH at C7 of the substrate.79,80 The resultant para-hydroquinone undergoes autoxidation to the quinone. A second flavoenzyme PgaM, fused to an NADH-utilizing reductase domain, acts as a hydroquinone oxidase, to produce gaudimycin C where the –OH at C12b derives from nucleophilic attack of water on an ortho-quinone methide intermediate that is then proposed to undergo oxidization by the FAD domain.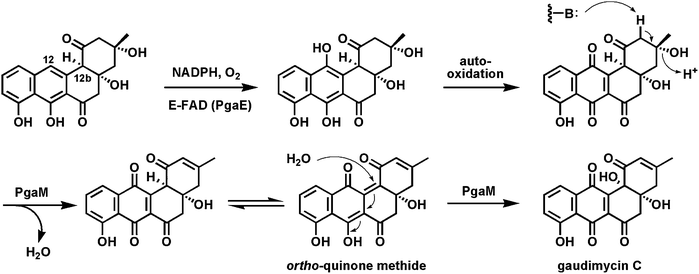 | ||
| Fig. 23 Hydroxylation of C12 in angucycline biosynthesis on the way to gaudimycin C. | ||
5.2 Flavoprotein Baeyer–Villigerases
In the canonical enzymatic reaction of phenol hydroxylation by Fl-OOH intermediates, as in the PgaE example, the weak O–O bond of the peroxide is cleaved with net delivery of an electrophilic oxygen [“OH+”] to the electron rich cosubstrate. But if the proton of the Fl-OOH can be dissociated to the Fl-OO− equivalent in a flavoenzyme active site then such a peroxy anion could behave instead as a nucleophile towards electrophilic centers (e.g ketone carbonyls). This would result in a Baeyer–Villiger type81,82 insertion of an oxygen into the ketone to produce an ester (Fig. 24a). A subclass of FAD enzymes indeed have such Baeyer–Villiger activity, as first demonstrated with simple model substrates, as in the expansion of cyclohexanone to the seven membered lactone.83–86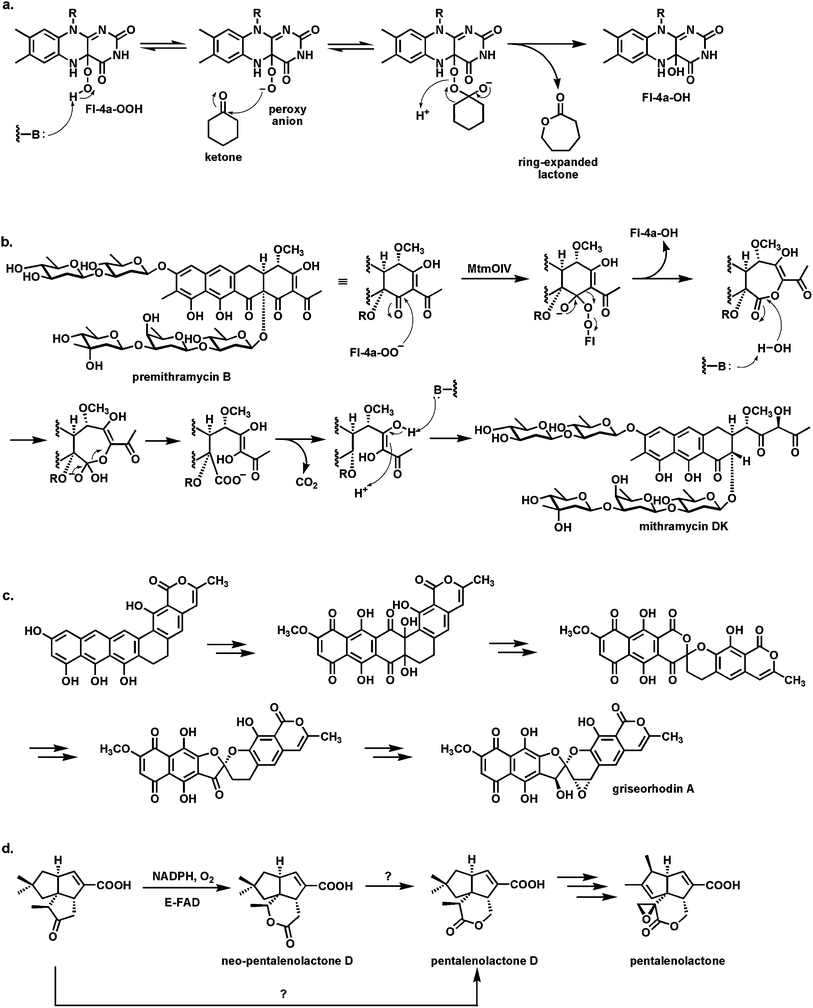 | ||
| Fig. 24 Baeyer–Villiger oxygenases act via Fl-4a-OO− as nucleophile: (a) schematic for oxygen insertion into cyclohexanone with ring expansion to the 7 member lactone; (b) conversion of the tetracyclic scaffold of premithramycin B to the tricyclic scaffold of mithramycin DK starts with a Baeyer–Villiger ring expansion of ketone to lactone, followed by lactone hydrolysis and β-keto acid decarboxylation; (c) conversion of angucycline framework to twisted spiroketal in griseorhodin involves flavoprotein catalysis and can be formulated to involve Baeyer–Villiger enzymology; (d) Baeyer–Villigerase generation of neo-pentalenolactone D. | ||
Baeyer–Villiger enzyme processing of the intermediates occurs in the aureolic family of polyketide antibiotics87 with antitumor activity, including mithramycins and chromomycins88 (Fig. 24b). The mature scaffold is tricyclic, with two oligosaccharide chains (di- and trisaccharide) decorating opposite ends of the scaffold. It is known that a tetracyclic species (e.g. premithramycin B) is precursor to the mature tricyclic framework of mithramycin DK. Cleavage of the cyclohexadione ring in the tetracyclic scaffold is accomplished by an FAD-dependent Baeyer–Villiger oxygenase. Oxidative insertion generates the seven membered ketolactone. Hydrolytic opening and equilibration with the carboxylate now uncovers a β-keto acid grouping. Facile decarboxylation completes the fragmentation and is one keto reductive step away from the mithramycin end product. Thus, the biological Baeyer–Villiger chemistry is used to set up a carbocyclic ring for an oxygen insertion, hydrolysis, decarboxylation sequence to open the tetracyclic scaffold regioselectively.
An even more remarkable cascade of carbon-carbon bond cleavages occur in the maturation of the rubromycin group of planar aromatic polyketides into the twisted spiroketal framework found for example in griseorhodin A89 (Fig. 24c). An angular hexacyclic aromatic polyketide intermediate undergoes oxidation to a quinone, rearrangement to a six-six ring spiroketal, and then contraction to a five-six ring spiroketal. Four C–C bonds are cleaved during the multistep process. At least one and possibly two of the enzymes involved are flavoproteins and both may act as Baeyer–Villiger oxygen insertion catalysts.
An FAD-dependent Baeyer–Villiger ketone to lactone ring expansion is also implicated in oxygenative tailoring of the C15 farnesyl scaffold in generation of the sesquiterpene epoxylactone pentalenolactone.90 This metabolite is produced by many Streptomyces strains and kills cells by capture of the reactive active site Cys thiolate of glyceraldehyde-3-P dehydrogenase. Amidst several maturative oxygenative transformations enacted by iron enzymes, a flavoenzyme oxygenase is proposed to convert the indicated tricyclic ketoacid to pentalenolactone D (Fig. 24d). Surprisingly, when the homologous enzyme was purified from S. avermitilis it carried out a Baeyer–Villiger ring expansion but with oxygen insertion on the other side of the ketone to give neopentalenolactone D. This is noncanonical since the more electron rich C–C bond generally migrates as the Criegee-type tetrahedral adduct breaks down in chemical and other enzymatic Baeyer–Villiger insertions.91 It may be that steric crowding dictates formation of the alternate lactone regioisomer in this case.
5.3 Flavoprotein epoxidases
Oxygen transfer to a CC double bond to create an epoxide is usually thought of as the province of P450 type heme iron monooxygenases.78 However, a famous precedent that a flavoprotein can carry out such epoxidations is the enzyme squalene epoxidase92 which epoxidizes one of the terminal double bonds in the C30 hexaene squalene to create squalene-2,3-oxide (Fig. 25a) which is then the substrate for the electrocyclization that simultaneously forms the tetracyclic framework of sterols.93 Styrene epoxidation and hypothemycin epoxidations are likewise catalyzed by flavoenzymes94 where delivery of the terminal –OH of Fl-4a-OOH as electrophilic oxygen to the π-electrons of the olefin is the proposed polarity for oxygen transfer.
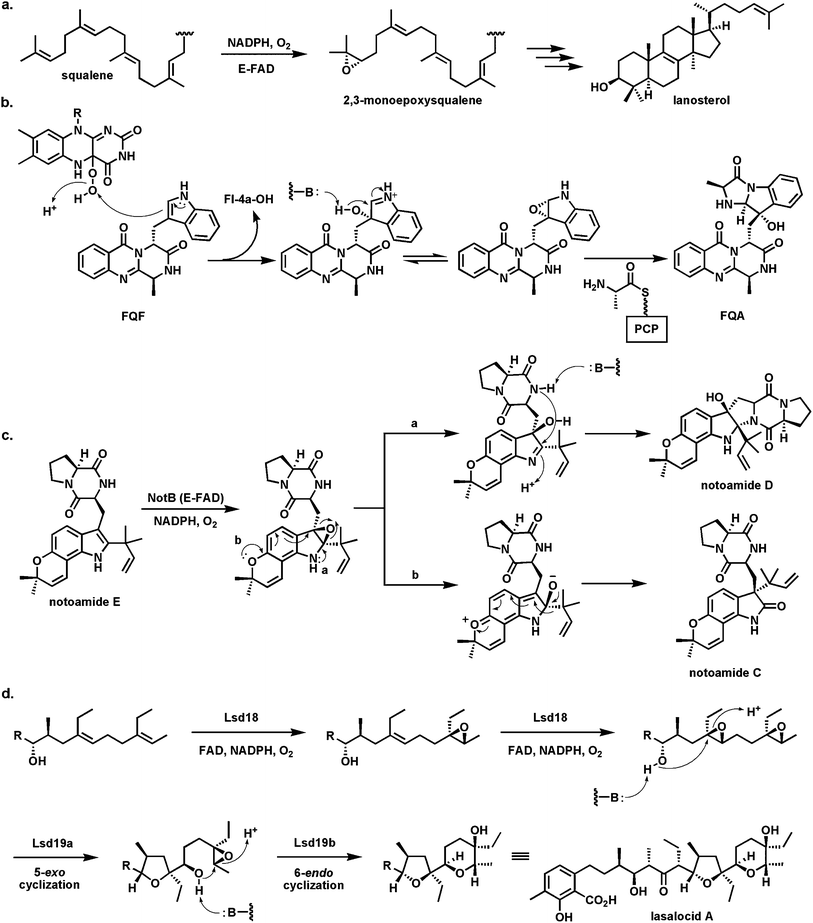 | ||
| Fig. 25 Flavoenzyme epoxidases: (a) squalene 2,3-epoxidase; (b) Af12060 converts fumiquinazoline F to fumiquinazoline A by way of an epoxyindole/hydroxyiminium ion intermediate; (c) indole epoxidation en route from notoamide E to notoamides C and D; (d) regioselective epoxidation by Lsd18 and tandem cyclization by Lsd19 in the lasalocid pathway. | ||
A recent example of another flavoenzyme with comparable epoxidase activity stems from study of the earlier steps in the fungal fumiquinazoline biosynthetic pathway. The FAD-enzyme Af1206095 acts on fumiquinazoline F (FQF) in the presence of cosubstrates NADPH and O2 to generate an unstable oxygenated intermediate that is then processed by an alanine-activating NRPS module Af12050 to produce fumiquinazoline A (FQA). The net result of action by these two enzymes is an annulation of the indole by the incoming alanine to produce a tricyclic imidazolindolone moiety, with a hydroxyl substituent at what had been the β-carbon of the indole (Fig. 25b). Af12060 is proposed to start the process by transfer of an oxygen atom across the double bond in the pyrrole ring of the indole. This can be formulated as transfer of an “OH+” equivalent from the FAD-4a-OOH to the electron rich Cβ of the indole acting as nucleophile. The resultant product may be a hybrid between the epoxyindole and the hydroxyiminium contributor. This is the species that can be captured by the alanyl-thioester installed on the carrier protein domain of Af12060.
A related family of tremorgenic fungal peptidyl alkaloids appear to be assembled by this indole epoxidation/annulation strategy.96 The Af12060 enzyme represents the first example of a characterized flavin-dependent “indole epoxidase”catalyst. Paired with Af12070 noted above52 as the amine/amide oxidase converting FQA to heptacyclic FQC, the two flavoenzymes in the short (four enzymes total) fumiquinazoline biosynthetic pathway illustrate how FAD redox chemistry can be utilized in biological framing of complex scaffolds.
Analogously, NotB in the notoamide pathway is proposed to generate an indole epoxide intermediate from notoamide A (Fig. 25c). Two fates are proposed. One involves intramolecular conversion to the hydroxyiminium species with subsequent capture by the amide of the diketopiperazine moiety to produce notoamide D. The alternative alkoxyiminium tautomer could be quenched by intramolecular migration of the prenyl group on the way to notoamide C.97
The last epoxidase example (Fig. 25d) occurs in the lasalocid pathway where Lsd18 acts twice in succession to build the bis-epoxide that is then substrate for epoxide hydrolase action that creates the tetrahydrofuran and tetrahydropyran rings of lasalocid with regio-and stereochemical control.98,99
5.4 A flavoenzyme oxidative path to amide hydrolysis
The degradation of uracil to 3-hydroxypropionate, 2 NH3, and CO2 (Fig. 26) by an E. coli strain has been shown to require the rut genes.100 Two of the required proteins are an NADH/FMN reductase, RutF, and a partner protein, RutA, which can use the provided FMNH2, generate the FMN-4a-OOH, and use that to cleave uracil to Z-3-ureidoacrylate.101 This pair is representative of a subgroup of flavin-utilizing enzymes split into two partners.17 The NAD(P)H-FMN reductases generate FMNH2 by typical hydride transfer but are unusual in allowing rapid release of the dihydroflavin from the reductase active site. Now the FMNH2 must diffuse to the active site of the partner protein which captures it by one-electron redox manifold with O2 to yield the FMN-4a-OOH when the specific oxygen transfer work of the partner protein begins. The FMNH2 while diffusing between the partner proteins is running an autoxidation gauntlet in solution and in principle represents an inefficient way to get dihydroflavin equivalents to the oxygenative active site.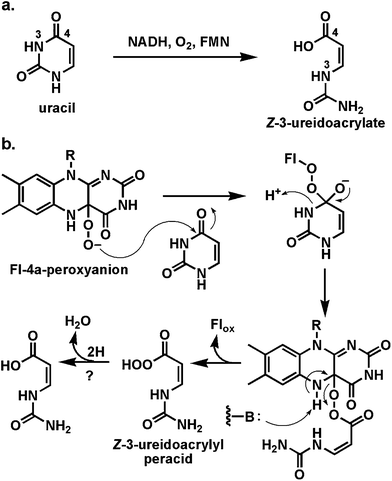 | ||
| Fig. 26 Oxidative cleavage of uracil to Z-3-ureidoacrylate: proposed involvement of Fl-4a-peroxyanion in ring-opening step. | ||
In a formal sense the unraveling of uracil by this two enzyme partnership is a hydrolytic cleavage of the N3-C4 amide bond. However, the reaction path is not hydrolytic but instead oxidative. The proposal is that the FMN-4a-OOH acts as nucleophile towards the uracil C4-carbonyl (akin to the reactivity in the Baeyer–Villiger reactions above) to yield a tetrahedral adduct.101 Expulsion of N3 from this adduct cleaves the N3-C4 amide bond and leaves a flavin-acrylate perester. Reoxidation to FMN would release the peracid form of the product. An as yet uncharacterized reduction of the ureidoacrylate peracid to the acid product would complete the reaction. The authors note chemical precedent for oxidative cleavage of amide bonds but this is the first documented biological example. It is a testimony to the utility of the Fl-4a-OOH for nucleophilic oxidative chemistry.
5.5 Using flavin-4a-OOH to create an electrophilic biological chlorinating agent
More than 4000 natural products are known that contain carbon–halogen bonds, the great majority being carbon–chlorine bonds, generated during biosynthetic processes.102 The enzymatic machinery utilized for such halogenations is matched to the electronic demands of substrate. Three distinct types of cofactors are employed by halogenases103,104 acting in biosynthetic pathways: (1) vanadium in haloperoxidases; (2) nonheme mononuclear iron in α-ketoglutarate-dependent halogenases; and (3) flavin coenzymes. The vanadium and iron-dependent halogenases are beyond the scope of this discussion but there are recent reviews delineating structures, mechanisms, and types of carbon centers that are halogenated by such catalysts.103,104The flavin dependent halogenases can be thought of as variants of flavin hydroxylases. The substrate range is comparable to flavin-mediated hydroxylations.103 Electron rich phenols, pyrroles, and indoles incorporate one or more halogens regiospecifically and suggest, as in the case of transfer of an electrophilic “OH+” by the oxygenases, that an electrophilic “Cl+” equivalent is generated and transferred in the active site of the flavin-dependent halogenases (Fig. 27a). Typically these are two component enzyme systems:105 an FMN/FAD reductase uses NAD(P)H to generate the FlH2 which diffuses away from the reductase and to the active site of the apo form of the halogenase (analogy to RutF and RutA above). The FlH2 then reacts with O2 in the halogenase active site in two one-electron steps to generate the canonical Fl-4a-OOH. Now chloride ion is a cosubstrate, bound between two tryptophan side chains in the active site106,107 and attacks the distal oxygen of the flavin peroxide, cleaving the weak O–O bond, leaving the Fl-4a-OH and generating hypochlorous acid, HOCl, as the nascent product. This constitutes transfer of “OH+” to the attacking Cl−, in keeping with the polarity of the Fl-OOH.103 Intriguingly, once that has happened, the chlorine in HOCl is instead a “Cl+” equivalent, consistent with the subsequent reactivity towards electron rich phenols and pyrroles in the substrates that get chlorinated.
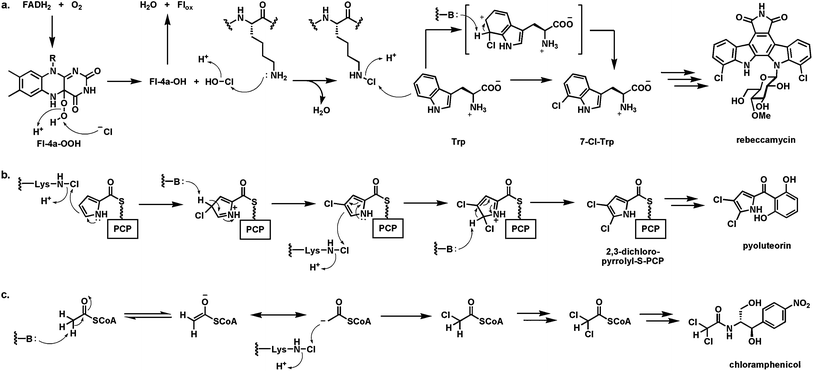 | ||
| Fig. 27 Halogenases utilizing FADH2 and O2 to generate a “Cl+” chlorinating species for electron rich cosubstrates: (a) generation of nascent HOCl in halogenase active sites and proposed conversion to a Lys-N6-Cl chloramine as proximal halogenating species by tryptophan 7-halogenase; (b) sequential chlorination of pyrrolyl-S-carrier protein at Cβ and Cα during pyoluteorin biosynthesis; (c) tandem chlorination on Cβ of acetyl CoA to generate the dichloroacetyl group during chloramphenicol assembly. | ||
There is indirect evidence in the tryptophan 7-halogenase that starts rebeccamycin biosynthesis (vide supra) that the nascent HOCl, in diffusing down an active site tunnel to get to the bound tryptophan substrate, encounters the ε-NH2 of a Lys side chain conserved in these dihydroflavin-dependent halogenases.108 Thus, formation of a Lys-NH-Cl chloramine intermediate may occur and be the proximal chlorinating agent, still polarized to deliver a “Cl+” equivalent, as shown in Fig. 27a for regiospecific chlorination at C7 of the indole side chain. In biosynthesis of the hexadepsipeptide antifungal agent kutzneride109 there are a pair of FlH2-dependent halogenases that act in tandem to generate the 7-Cl-Trp and then the 6,7-dichloro-Trp residue that gets incorporated into the hexadepsipeptide antifungal kutzneride scaffold.110 Analogously, a FlH2-utilizing halogenase chlorinates both the α- and β-carbons of a pyrrolyl-S-pantetheinyl carrier protein,111 whose formation from the prolyl-S-PCP precursor by a flavoprotein oxidase we noted earlier (Fig. 27b).46 In chloramphenicol biosynthesis two tandem chlorinations are carried out by the flavin-dependent halogenase ClmS,112 but now on the same carbon, converting an acetyl to a dichloroacetyl group. ClmS is proposed to act on acetyl CoA converting it to dichloroacetyl CoA as the proximal acylating reagent in the pathway (Fig. 27c). Utilization of acetyl CoA as halogenation substrate would allow the readily accessible thioester enolate to act as a nucleophile towards the ClmS chloramine intermediates as a donor of “Cl+”.
5.6 Biological oxidative Favorskii rearrangement during enterocin assembly
The bacteriostatic agent enterocin is an octaketide from Streptomyces maritimus that has undergone extensive rearrangement during biosynthetic maturation. The EncM flavoenzyme appears to be the major change agent, acting on a polyketidyl-S-ACP substrate that has undergone prior reduction of one of the eight ketones to the C9-OH.113 This kind of regiospecific reduction of one specific ketone in a polyketonic chain is typical as a set point for determining cyclization connectivity in tetracyclic aromatic polyketides.114 In this system, that –OH group becomes a crucial internal nucleophile during the EncM-mediated scaffold rearrangements (Fig. 28). It is thought115 that EncM has monooxygenase activity, with the Fl-4a-OOH transferring an “OH+” equivalent to an enolate to yield a hydroxy C12 species, sandwiched between the C11 and C13 ketones. Further oxidation of that alcohol (a second hydroxylation to the gem diol, or an alcohol to ketone type of dehydrogenation) is proposed to yield the 11,12,13 triketo intermediate.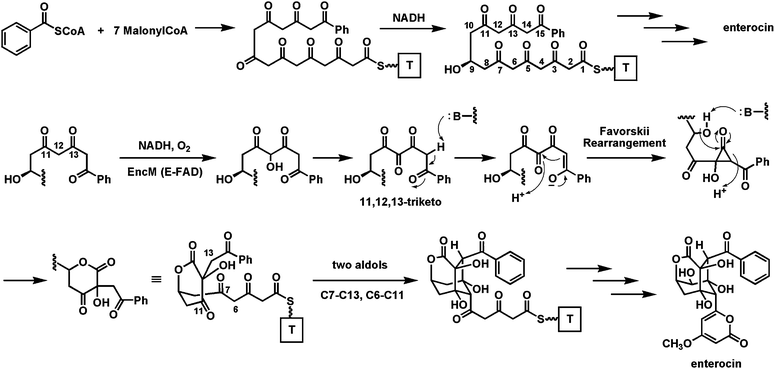 | ||
| Fig. 28 The role of EncM in enterocin biosynthesis: oxidation of a polyketonic intermediate to an 11,12,13-triketo species that undergoes a Favorskii rearrangement and then two regiospecific aldol condensations. | ||
Now a Favorskii-type rearrangement116 is envisioned. In the thioester enolate tautomer C14 is nucleophilic and could attack the C12 keto to yield a hydroxycyclopropanone species. In turn this could then be captured by the C9-OH as nucleophile attacking the strained C13 carbonyl within the cyclopropanone. Lactone formation would be coupled to C13–C14 bond cleavage. This alters the backbone chain connectivity and sets the central hydroxy-keto-lactone framework in enterocin. This intermediate, while still bound in the EncM active site, could then undergo two directed aldol condensations (C6–C11 and C7–C13 in the pre-enterocin framework) to construct the remaining two six membered rings of the enterocin tricyclic core. Presumably the folded acyclic polyketidyl chain conformer that is stabilized in the EncM active site is crucial not only for the original oxygenation/oxidation to yield the triketo intermediate, but also to enable the proposed Favorskii rearrangement and the subsequent double aldol condensations. The cascade of chemistry that is unleashed after the flavin coenzyme-mediated redox steps is reminiscent of the post-redox cyclization to the decalin framework in the solanapyrone synthase active site.58
5.7 Morphing FMN into dimethylbenzimidazole: A flavin “destructase”
Vitamin B12 binds a redox active cobalt atom in the equatorial plane of the tetrapyrrolic corrin macrocycle. The fifth and six ligands are in axial positions. The bottom axial ligand is a nitrogen atom from a tethered dimethylbenzimidazole ribonucleotide moiety (Fig. 29a). The top axial ligand in the vitamin can be water or cyanide and is replaced with an adenosyl moiety or a methyl group in the coenzymatically active adenosyl B12 forms that participate in various mutase reactions and in methionine synthesis. B12 is produced only by microbes and so is essential for humans.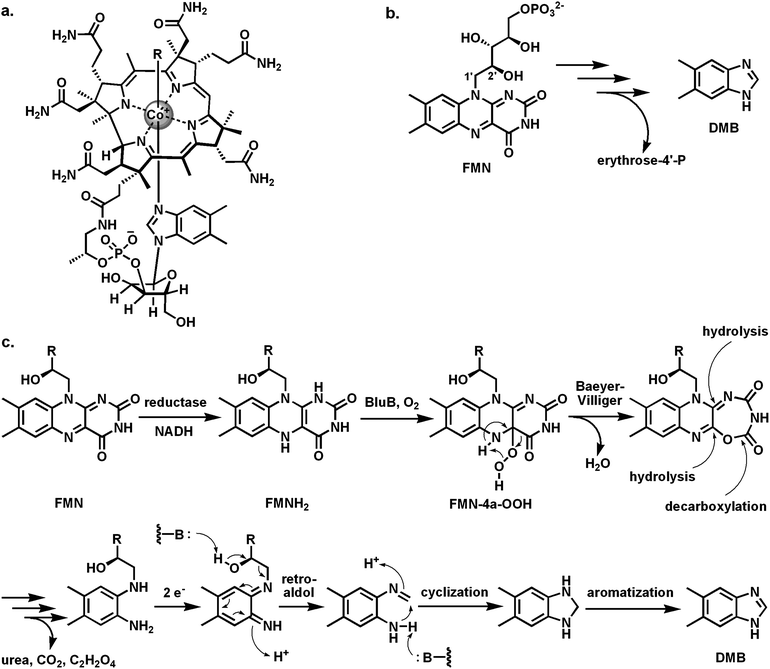 | ||
| Fig. 29 Creating the dimethylbenzimidazole ligand for B12 from FMN: (a) vitamin B12 with the dimethylbenzimidazole (DMB) as bottom axial ligand to the cobalt atom; (b) BluB acts as a flavin “destructase”, generating DMB and erythrose-4-P from FMN; (c) possible mechanism involving a Fl-4a-OOH and an internal Baeyer–Villiger reaction on the pyrimidine ring of FMN. | ||
The last piece of B12 biosynthesis to be uncovered was the mechanism of formation of the dimethylbenzimidazole (DMB) bicyclic ring system. It had been known for years that FMN is the precursor for DMB and that C1′ of the ribityl chain of FMN becomes the C2 of the imidazole moiety of DMN, but the responsible enzyme, BluB, has only recently been identified in a DMB auxotroph of Sinorhizobium meliloti.117 BluB turns out to be the flavin “destructase”, cleaving the ribityl chain between C1′ and C2′ to release erythrose-4′-phosphate and to contract the tricyclic isoalloxazine to the bicyclic DMB (Fig. 29b). In keeping with the theme of two component flavoenzymes noted in previous sections (e.g. the halogenases and the Rut enzymes), a separate NADH-dependent reductase generates FMNH2 which diffuses to the active site of BluB that then reacts FMNH2 with O2.
The mechanism of BluB as flavin “destructase” has not been resolved but it is reasonable to postulate the canonical FMN-4a-OOH as an early intermediate.118 It is possible that this undergoes an intramolecular Baeyer–Villiger type oxygen insertion into its pyrimidine ring to yield the 7-member lactone (Fig. 29c). This sets up that ring for a series of hydrolytic steps that would take apart both the ring-expanded pyrimidine and the central pyrazine: loss of oxalic acid, urea, and CO2 could generate the indicated diaminobenzene. Oxidation and retroaldol cleavage would precede cyclization and aromatizing oxidation to dimethylbenzimidazole.
The examples in this Section 5 deal with the different reaction manifolds open to Fl-4a-OOH intermediates and the control of their reactivity in enzyme microenvironments. The utilization of flavin hydroperoxides, as both electrophilic and nucleophilic oxygenation reagents on a diverse range of cosubstrates to effect some remarkable chemical transformations, explains the many crossroads of natural product tailoring and maturation occupied by flavoenzymes.
6 Nucleophilic and general base catalysis for FlH2 in flavoenzymes
In Section 5 we focused on the one-electron manifold of reduced flavins and the two step reactions that lead to the covalent Fl-4a-OOH intermediate. The other well known 4a adducts in flavoenzyme chemistry are the Fl-4a-thiol adducts that form during the catalytic cycles of dithiol oxidases/disulfide reductases such as the well studied glutathione reductase,119 lipoamide dehydrogenase,120 mercuric ion reductases,121,122 and plant phototropins.123 All of these FAD enzymes in their resting state have an active site disulfide proximal to the bound flavin coenzyme isoalloxazine ring system. Once the FADH2 has been generated in the reductive half reaction, the reoxidative half reaction involves adduct formation in a two-electron manifold with C4a of the dihydroFMN/FAD acting as nucleophile (Fig. 3). The 4a-thiol-flavin adduct is then subjected to the indicated participation of the second thiol to regenerate the resting oxidation state of this enzyme subclass, with FAD and active site disulfide reformed.N5 of Flavins is the other port of entry and exit of electrons as repeatedly noted above in the transfer of hydride ions to and from nicotinamide coenzymes as substrates and also in the covalent adducts formed during nitroalkane oxidation and alkyl ether phospholipid biosynthesis. Recent evidence has accrued that N5 in FADH2 and FMNH2 can also act as a general acid/base catalyst for proton transfer, without reoxidation or net change in the FlH2 oxidation state.
6.1 N5 of FMNH2 as a proton transfer catalyst
The case for FlH2 as a proton transfer catalyst has been made for the enzyme IDI-2,124 catalyzing the interconversion of Δ2- and Δ3-isopentenyl diphosphates (Fig. 30), generating the pool of isomers needed for all isoprenoid pathways. In certain pathogenic bacteria the enzyme specifically requires the labile FMNH2 and there is no obvious or net redox change during this isomerization. The pathway is an alternative to the more widespread cofactor-independent isopentenyl diphosphate epimerase. Use of FMN analogs with different redox potentials has shown that the electronic properties of the dihydroflavin are consequential for catalysis.125 A recent proposal for the mechanism126 is that N5 of the dihydroflavin might act as both acid and base to facilitate proton transfers between C2 and C4 of a carbocation intermediate generated from either Δ2- or Δ3-IDP substrate.127 A diprotonated cationic N5 form of the FMNH2 has been proposed. This is the first suggestion that dihydroflavin N5 can act as a proton transfer catalyst to carbon centers of bound substrates.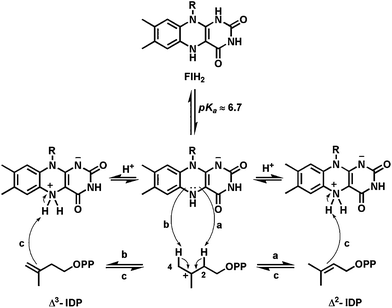 | ||
| Fig. 30 Proposed role of FlH2 in the type II isopentenyl diphosphate isomerase: N5 as proton transfer catalysts for the 1,3-allylic isomerization. | ||
6.2 N5 of dihydroflavin as nucleophile to initiate catalysis
A second example of N5 flavin-substrate adducts, this time starting from the dihydro rather than the oxidized state of the flavin coenzyme noted in Section 4.3, is found in the enzyme UDP-galactopyranose mutase128,129 found in Mycobacterium tuberculosis and also in Aspergillus fungal strains. The enzyme converts the predominant form of galactose (gal), the pyranose cyclic hemiacetal (galp), to the minor furanose hemiketal (galf; Fig. 31). The galactofuranose units are key building blocks for the arabinogalactan oligosaccharide chains in mycobacterial cell walls and are required for viability of that pathogen. The enzyme absolutely requires the reduced FADH2 although there is no net redox change during catalysis.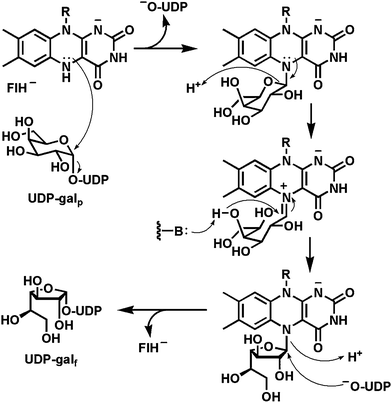 | ||
| Fig. 31 Proposed role of FlH2 in the UDP-galactopyranose mutase reaction: Covalent FlH2-N5-galactose-C1 adduct as reaction intermediate. | ||
Indeed, the current mechanistic paradigm features no change in FADH2 redox state.130–132 The catalytic cycle is proposed to initiate with attack of N5 of FADH2 on the C1′ of the galactosyl moiety of UDP-galp, displacing UDP as a good leaving group and generating the galp in amine linkage to N5 of the dihydroflavin. Use of the N5 lone pair of electrons, in direct analogy to the nitroalkane oxidase precedent,67 would generate the iminium adduct as the C1′–O5 bond of the galactose is cleaved, generating the acylic form of the sugar. This could reclose to the amino adduct by back attack of the C5′–O or instead close by attack of the C4′–O which generates the galf form of the amino adduct. Now, recapture of the galactosyl group by the UDP initially expelled at the start of the cycle, but still held in the enzyme active site, will generate the product UDP-galf. Two of the virtues of interconversion of galp and galf starting and ending at the nucleotide diphosphosugar level are that (a) UDP is a good leaving group to lower the energy barrier for attack by N5 of FADH2 and (b) then the product has the galf activated at C1′ for subsequent galactosyl transfer and oligomerization.
7 Concluding remarks
Flavin coenzymes are immensely versatile heterocycles that enable many kinds of chemical transformations as complexity is built into biosynthetic scaffolds. The tricyclic isoalloxazine with its fused pyrazine-pyrimidine rings can access more diverse reaction manifolds than any other organic or metal-centered coenzyme. This includes both two- and one-electron transfer pathways that position flavins at all the metabolic redox crossroads where flux steps down from two-electron only (e.g. NAD(P)H) to one-electron only (e.g. FeIII) redox partners. Flavins are responsive to cosubstrate demands, as exemplified by their ability to participate in transfer of hydrogens with two electrons (H−), one electron (H˙), or no electrons (H+). FAD and FMN are electrophilic in the oxidized state and nucleophilic in the reduced state. Both N5 and bridgehead C4a are ports of entry and exit of electrons, as evidenced by covalent adducts at those positions to substrates.The oxygen reactivity in the FlH2 reoxidative half reaction via one-electron transfer and radical recombination to the Fl-4a-OOH opens up biosynthetically useful sets of oxygen transfers. While transfer of an electrophilic “OH+” equivalent to nucleophilic cosubstrates is the predominant oxygen transfer mode, the behavior of the Fl-4a-peroxyanion as nucleophile towards electrophilic centers in cosubstrates is the basis of Baeyer–Villiger oxygenative ring expansions of ketones to lactones and conversions of aldehydes to esters. The discovery of a large class of flavoenzymes with halogenase activity highlights the ability of the Fl-OOH intermediates to transfer “OH+” to chloride anions and thereby generate “Cl+” equivalents as nascent HOCl and then likely enzyme-chloramines as proximal chlorinating agents.
Flavoenzymes are key complexity-generating enzymes in natural product maturations, setting up [4+2] cyclizations by distinct routes in lovastatin and solanapyrone assembly. They are active, virtuoso catalysts in the creation of tricyclic and then heptacyclic peptide alkaloid scaffolds in fumiquinazolines C and D, of the pentacyclic and heptacyclic scaffolds in chromopyrrolic acid and the subsequent indolocarbazole frameworks of rebeccamycin and staurosporine. They engineer oxygenative opening of tetracyclic precursors in angucycline maturation. EncM catalyzes a four-ring circus with a Favorskii rearrangement at the heart of enterocin assembly. The UDP-gal mutase mediates a remarkable ring contraction in producing galf monomers for mycobacterial cell wall assembly.
The bis-nitrogen heterocyclic portion of the isoalloxazine core in the flavin coenzymes is without peer in the scope of biological redox catalysis enabling control of framework architecture and functional group disposition of a huge range of natural products. As more microbial genome sequences turn up predicted flavoprotein orfs in biosynthetic gene clusters, additional new biosynthetic chemistry is likely to emerge.
8 Acknowledgements
Work in the Walsh laboratory has been supported by NIH grants GM20011 and GM49338. We sincerely thank Jared B. Parker for critical reading of this manuscript and valuable feedback. We gratefully acknowledge the work done by all of our colleagues cited in this review.9 References
- M. A. Fischbach and C. T. Walsh, Chem. Rev., 2006, 106, 3468–3496 CrossRef CAS.
- C. Hertweck, Angew. Chem., Int. Ed., 2009, 48, 4688–4716 CrossRef CAS.
- E. S. Sattely, M. A. Fischbach and C. T. Walsh, Nat. Prod. Rep., 2008, 25, 757–793 RSC.
- C. T. Walsh, Acc. Chem. Res., 2008, 41, 4–10 CrossRef CAS.
- F. Kopp, C. Mahlert, J. Grünewald and M. A. Marahiel, J. Am. Chem. Soc., 2006, 128, 16478–16479 CrossRef CAS.
- J. A. Read and C. T. Walsh, J. Am. Chem. Soc., 2007, 129, 15762–15763 CrossRef CAS.
- C. Walsh, Enzymatic Reaction Mechanisms, W.H. Freeman Inc, San Francisco, 1979 Search PubMed.
- J. Valton, C. Mathevon, M. Fontecave, V. Nivière and D. P. Ballou, J. Biol. Chem., 2008, 283, 10287–10296 CrossRef CAS.
- V. Massey, J. Biol. Chem., 1994, 269, 22459–22462 CAS.
- C. Walsh, Acc. Chem. Res., 1980, 13, 148–155 CrossRef CAS.
- S. O. Mansoorabadi, C. J. Thibodeaux and H.-W. Liu, J. Org. Chem., 2007, 72, 6329–6342 CrossRef CAS.
- S. Ghisla and V. Massey, Eur. J. Biochem., 1989, 181, 1–17 CAS.
- D. J. T. Porter, J. G. Voet and H. J. Bright, J. Biol. Chem., 1973, 248, 4400–4416 Search PubMed.
- C. Thorpe and C. H. Williams, Jr, J. Biol. Chem., 1976, 251, 7726–7728 Search PubMed.
- M. Mewies, W. S. McIntire and N. S. Scrutton, Protein Sci., 1998, 7, 7–20 Search PubMed.
- M.-T. Lai and H.-W. Liu, J. Am. Chem. Soc., 1992, 114, 3160–3162 CrossRef CAS.
- B. A. Palfey and C. A. McDonald, Arch. Biochem. Biophys., 2010, 493, 26–36 Search PubMed.
- D. I. Chan and H. J. Vogel, Biochem. J., 2010, 430, 1–19 CrossRef CAS.
- S. Smith and S.-C. Tsai, Nat. Prod. Rep., 2007, 24, 1041–1072 RSC.
- S. M. Ma, J. W.-H. Li, J. W. Choi, H. Zhou, K. K. M. Lee, V. A. Moorthie, X. Xie, J. T. Kealey, N. A. Da Silva, J. C. Vederas and Y. Tang, Science, 2009, 326, 589–592 CrossRef CAS.
- C. D. Campbell and J. C. Vederas, Biopolymers, 2010, 93, 755–763 CrossRef CAS.
- S. B. Bumpus, N. A. Magarvey, N. L. Kelleher, C. T. Walsh and C. T. Calderone, J. Am. Chem. Soc., 2008, 130, 11614–11616 CrossRef CAS.
- S. Reverchon, C. Rouanet, D. Expert and W. Nasser, J. Bacteriol., 2002, 184, 654–665 CAS.
- H. Takahashi, T. Kumagai, K. Kitani, M. Mori, Y. Matoba and M. Sugiyama, J. Biol. Chem., 2007, 282, 9073–9081 CrossRef CAS.
- J. G. Owen, K. J. Robins, N. S. Parachin and D. F. Ackerley, Environ. Microbiol., 2012, 14, 1198–1209 Search PubMed.
- R. SinhaRoy, J. Milne, P. Belshaw, A. Gehring and C. Walsh, Nat. Prod. Rep., 1999, 16, 249–263 RSC.
- C. T. Walsh, S. J. Malcolmson and T. S. Young, ACS Chem. Biol., 2012, 7, 429–442 Search PubMed.
- J. H. Crosa and C. T. Walsh, Microbiol. Mol. Biol. Rev., 2002, 66, 223–249 CrossRef CAS.
- H. Chen, S. O'Conner, D. E. Cane and C. T. Walsh, Chem. Biol., 2001, 8, 899–912 CrossRef CAS.
- T. L. Schneider, B. Shen and C. T. Walsh, Biochemistry, 2003, 42, 9722–9730 CrossRef CAS.
- L. Du, M. Chen, Y. Zhang and B. Shen, Biochemistry, 2003, 42, 9731–9740 CrossRef CAS.
- J. Chen and J. Stubbe, Nat. Rev. Cancer, 2005, 5, 102–112 CrossRef CAS.
- Y.-M. Li, J. C. Milne, L. L. Madison, R. Kolter and C. T. Walsh, Science, 1996, 274, 1188–1193 CrossRef CAS.
- C. T. Walsh, M. G. Acker and A. A. Bowers, J. Biol. Chem., 2010, 285, 27525–27531 CrossRef CAS.
- H. Onaka, M. Nakaho, K. Hayashi, Y. Igarashi and T. Furumai, Microbiology, 2005, 151, 3923–3933 CrossRef CAS.
- K. J. Molohon, J. O. Melby, J. Lee, B. S. Evans, K. L. Dunbar, S. B. Bumpus, N. L. Kelleher and D. A. Mitchell, ACS Chem. Biol., 2011, 6, 1307–1313 CrossRef CAS.
- B. Kalyon, S. E. Helaly, R. Scholz, J. Nachtigall, J. Vater, R. Borriss and R. D. Süssmuth, Org. Lett., 2011, 13, 2996–2999 CrossRef CAS.
- T. J. Oman and W. A. van der Donk, Nat. Chem. Biol., 2010, 6, 9–18 CrossRef CAS.
- J. G. Heddle, S. J. Blance, D. B. Zamble, F. Hollfelder, D. A. Miller, L. M. Wentzell, C. T. Walsh and A. Maxwell, J. Mol. Biol., 2001, 307, 1223–1234 Search PubMed.
- N. W. Seidler, I. Jona, M. Vegh and A. Martonosi, J. Biol. Chem., 1989, 264, 17816–17823.
- X. Liu and C. T. Walsh, Biochemistry, 2009, 48, 8746–8757 CrossRef CAS.
- X. Liu and C. T. Walsh, Biochemistry, 2009, 48, 11032–11044 Search PubMed.
- W. C. Kenney, D. E. Edmondson, T. P. Singer, D. J. Steenkamp and J. C. Schabort, FEBS Lett., 1974, 41, 111–114 Search PubMed.
- C. T. Walsh, S. Garneau-Tsodikova and A. Howard-Jones, Nat. Prod. Rep., 2006, 23, 517–531 RSC.
- J. W. Phillips, M. A. Goetz, S. K. Smith, D. L. Zink, J. Polishook, R. Onishi, S. Salowe, J. Wiltsie, J. Allocco, J. Sigmund, K. Dorso, S. Lee, S. Skwish, M. de la Cruz, J. Martin, F. Vicente, O. Genilloud, J. Lu, R. E. Painter, K. Young, K. Overbye, R. G. K. Donald and S. B. Singh, Chem. Biol., 2011, 18, 955–965 Search PubMed.
- S. Garneau-Tsodikova, P. C. Dorrestein, N. L. Kelleher and C. T. Walsh, J. Am. Chem. Soc., 2006, 128, 12600–12601 CrossRef CAS.
- I. Garcia, N. M. Vior, A. F. Braña, J. González-Sabin, J. Rohr, F. Moris, C. Méndez and J. A. Salas, Chem. Biol., 2012, 19, 399–413 Search PubMed.
- C. Sánchez, C. Méndez and J. A. Salas, Nat. Prod. Rep., 2006, 23, 1007–1045 RSC.
- C. Sánchez, I. A. Butovich, A. F. Braña, J. Rohr, C. Méndez and J. A. Salas, Chem. Biol., 2002, 9, 519–531 CrossRef CAS.
- A. R. Howard-Jones and C. T. Walsh, J. Am. Chem. Soc., 2006, 128, 12289–12298 CrossRef CAS.
- A. R. Howard-Jones and C. T. Walsh, Biochemistry, 2005, 44, 15652–15663 CrossRef CAS.
- B. D. Ames, S. W. Haynes, X. Gao, B. S. Evans, N. L. Kelleher, Y. Tang and C. T. Walsh, Biochemistry, 2011, 50, 8756–8769 CrossRef CAS.
- Y.-C. Liu, Y.-S. Li, S.-Y. Lyu, L.-J. Hsu, Y.-H. Chen, Y.-T. Huang, H.-C. Chan, C.-J. Huang, G.-H. Chen, C.-C. Chou, M.-D. Tsai and T.-L. Li, Nat. Chem. Biol., 2011, 7, 304–309 CrossRef CAS.
- I. Alexeev, A. Sultana, P. Mäntsälä, J. Niemi and G. Schneider, Proc. Natl. Acad. Sci. U. S. A., 2007, 104, 6170–6175 CrossRef CAS.
- A. E. Ragab, S. Grüschow, D. R. Tromans and R. J. Goss, J. Am. Chem. Soc., 2011, 133, 15288–15291 Search PubMed.
- C. T. Walsh and W. Zhang, ACS Chem. Biol., 2011, 6, 1000–1007 Search PubMed.
- Z. Yang, X. Chi, M. Funabashi, S. Baba, K. Nonaka, P. Pahari, J. Unrine, J. M. Jacobsen, G. I. Elliott, J. Rohr and S. G. Van Lanen, J. Biol. Chem., 2011, 286, 7885–7892 Search PubMed.
- K. Kasahara, T. Miyamoto, T. Fujimoto, H. Oguri, T. Tokiwano, H. Oikawa, Y. Ebizuka and I. Fujii, ChemBioChem, 2010, 11, 1245–1252 CrossRef CAS.
- C. Davis, S. Carberry, M. Schrettl, I. Singh, J. C. Stephens, S. M. Barry, K. Kavanagh, G. L. Challis, D. Brougham and S. Doyle, Chem. Biol., 2011, 18, 542–552 CrossRef CAS.
- M. Schrettl, S. Carberry, K. Kavanagh, H. Haas, G. W. Jones, J. O'Brien, A. Nolan, J. Stephens, O. Fenelon and S. Doyle, PLoS Pathog., 2010, 6, e1000952 Search PubMed.
- B. Li and C. T. Walsh, Biochemistry, 2011, 50, 4615–4622 CrossRef CAS.
- C. Wang, S. R. Wesener, H. Zhang and Y.-Q. Cheng, Chem. Biol., 2009, 16, 585–593 Search PubMed.
- C. S. Sit, S. Yoganathan and J. C. Vederas, Acc. Chem. Res., 2011, 44, 261–268 CrossRef CAS.
- E. Strauss, C. Kinsland, Y. Ge, F. W. McLafferty and T. P. Begley, J. Biol. Chem., 2001, 276, 13513–13516 CAS.
- E. Strauss, H. Zhai, L. A. Brand, F. W. McLafferty and T. P. Begley, Biochemistry, 2004, 43, 15520–15533 CrossRef CAS.
- B. Li and C. T. Walsh, Proc. Natl. Acad. Sci. U. S. A., 2010, 107, 19731–19735 CrossRef CAS.
- P. F. Fitzpatrick, A. M. Orville, A. Nagpal and M. P. Valley, Arch. Biochem. Biophys., 2005, 433, 157–165 CrossRef CAS.
- A. Nagpal, M. P. Valley, P. F. Fitzpatrick and A. M. Orville, Biochemistry, 2006, 45, 1138–1150 Search PubMed.
- A. Héroux, D. M. Bozinovski, M. P. Valley, P. F. Fitzpatrick and A. M. Orville, Biochemistry, 2009, 48, 3407–3416 Search PubMed.
- A. Razeto, F. Mattiroli, E. Carpanelli, A. Aliverti, V. Pandini, A. Coda and A. Mattevi, Structure, 2007, 15, 683–692 CrossRef CAS.
- T. E. Benson, C. T. Walsh and J. M. Hogle, Structure, 1996, 4, 47–54 CrossRef CAS.
- W. J. Lees, T. E. Benson, J. M. Hogle and C. T. Walsh, Biochemistry, 1996, 35, 1342–1351 Search PubMed.
- C. T. Walsh, T. E. Benson, D. H. Kim and W. J. Lees, Chem. Biol., 1996, 3, 83–91 Search PubMed.
- G. T. Lountos, R. Jiang, W. B. Wellborn, T. L. Thaler, A. S. Bommarius and A. M. Orville, Biochemistry, 2006, 45, 9648–9659 Search PubMed.
- A. M. Orville, G. T. Lountos, S. Finnegan, G. Gadda and R. Prabhakar, Biochemistry, 2009, 48, 720–728 Search PubMed.
- B. Entsch, D. P. Ballou and V. Massey, J. Biol. Chem., 1976, 251, 2550–2563 CAS.
- B. Entsch, D. P. Ballou, M. Husain and V. Massey, J. Biol. Chem., 1976, 251, 7367–7379 Search PubMed.
- Cytochrom P450, ed. P. R. Ortiz de Montellano, Kluwer Academic/Plenum Publishers, New York, 3rd edn, 2005 Search PubMed.
- P. Kallio, Z. Liu, P. Mäntsälä, J. Niemi and M. Metsä-Ketelä, Chem. Biol., 2008, 15, 157–166 CrossRef CAS.
- P. Kallio, P. Patrikainen, J.-P. Suomela, P. Mäntsälä, M. Metsä-Ketelä and J. Niemi, Biochemistry, 2011, 50, 5535–5543 Search PubMed.
- C. T. Walsh and Y.-C. J. Chen, Angew. Chem., Int. Ed. Engl., 1988, 27, 333–343 CrossRef.
- R. Orru, H. M. Dudek, C. Martinoli, D. E. Torres Pazmiño, A. Royant, M. Weik, M. W. Fraaije and A. Mattevi, J. Biol. Chem., 2011, 286, 29284–29291 CrossRef CAS.
- N. A. Donoghue, D. B. Norris and P. W. Trudgill, Eur. J. Biochem., 1976, 63, 175–192 CAS.
- C. C. Ryerson, D. P. Ballou and C. Walsh, Biochemistry, 1982, 21, 2644–2655 CrossRef CAS.
- B. P. Branchaud and C. T. Walsh, J. Am. Chem. Soc., 1985, 107, 2153–2161 CrossRef CAS.
- D. Sheng, D. P. Ballou and V. Massey, Biochemistry, 2001, 40, 11156–11167 CrossRef CAS.
- M. K. Kharel, P. Pahari, M. D. Shepherd, N. Tibrewal, S. E. Nybo, K. A. Shaaban and J. Rohr, Nat. Prod. Rep., 2012, 29, 264–325 RSC.
- M. P. Beam, M. A. Bosserman, N. Noinaj, M. Wehenkel and J. Rohr, Biochemistry, 2009, 48, 4476–4487 CrossRef CAS.
- Z. Yunt, K. Reinhardt, A. Li, M. Engeser, H.-M. Dahse, M. Gütschow, T. Bruhn, G. Bringmann and J. Piel, J. Am. Chem. Soc., 2009, 131, 2297–2305 CrossRef CAS.
- J. Jiang, C. N. Tetzlaff, S. Takamatsu, M. Iwatsuki, M. Komatsu, H. Ikeda and D. E. Cane, Biochemistry, 2009, 48, 6431–6440 CrossRef CAS.
- M. D. Mihovilovic and P. Kapitán, Tetrahedron Lett., 2004, 45, 2751–2754 CrossRef CAS.
- A. Nagumo, T. Kamei, J. Sakakibara and T. Ono, J. Lipid Res., 1995, 36, 1489–1497 Search PubMed.
- M. W. Huff and D. E. Telford, Trends Pharmacol. Sci., 2005, 26, 335–340 Search PubMed.
- C. J. Thibodeaux, W. C. Chang and H. W. Liu, Chem Rev, 2011 Search PubMed.
- B. D. Ames, X. Liu and C. T. Walsh, Biochemistry, 2010, 49, 8564–8576 CrossRef CAS.
- X. Gao, Y.-H. Chooi, B. D. Ames, P. Wang, C. T. Walsh and Y. Tang, J. Am. Chem. Soc., 2011, 133, 2729–2741 CrossRef CAS.
- S. Li, J. M. Finefield, J. D. Sunderhaus, T. J. McAfoos, R. M. Williams and D. H. Sherman, J. Am. Chem. Soc., 2012, 134, 788–791 CrossRef CAS.
- A. Minami, M. Shimaya, G. Suzuki, A. Migita, S. S. Shinde, K. Sato, K. Watanabe, T. Tamura, H. Oguri and H. Oikawa, J. Am. Chem. Soc., 2012, 134, 7246–7249 CrossRef CAS.
- D. E. Cane, W. D. Celmer and J. W. Westley, J. Am. Chem. Soc., 1983, 105, 3594–3600 CrossRef CAS.
- K. D. Loh, P. Gyaneshwar, E. Markenscoff Papadimitriou, R. Fong, K.-S. Kim, R. Parales, Z. Zhou, W. Inwood and S. Kustu, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 5114–5119 Search PubMed.
- T. Mukherjee, Y. Zhang, S. Abdelwahed, S. E. Ealick and T. P. Begley, J. Am. Chem. Soc., 2010, 132, 5550–5551 Search PubMed.
- G. W. Gribble, J. Chem. Educ., 2004, 81, 1441–1449 CrossRef CAS.
- F. H. Vaillancourt, E. Yeh, D. A. Vosburg, S. Garneau-Tsodikova and C. T. Walsh, Chem. Rev., 2006, 106, 3364–3378 CrossRef.
- C. S. Neumann, D. G. Fujimori and C. T. Walsh, Chem. Biol., 2008, 15, 99–109 CrossRef CAS.
- E. Yeh, S. Garneau and C. T. Walsh, Proc. Natl. Acad. Sci. U. S. A., 2005, 102, 3960–3965 CrossRef CAS.
- C. Dong, S. Flecks, S. Unversucht, C. Haupt, K.-H. van Pee and J. H. Naismith, Science, 2005, 309, 2216–2219 CrossRef CAS.
- X. Zhu, W. De Laurentis, K. Leang, J. Herrmann, K. Ihlefeld, K.-H. van Pée and J. H. Naismith, J. Mol. Biol., 2009, 391, 74–85 Search PubMed.
- E. Yeh, L. C. Blasiak, A. Koglin, C. L. Drennan and C. T. Walsh, Biochemistry, 2007, 46, 1284–1292 CrossRef CAS.
- D. G. Fujimori, S. Hrvatin, C. S. Neumann, M. Strieker, M. A. Marahiel and C. T. Walsh, Proc. Natl. Acad. Sci. U. S. A., 2007, 104, 16498–16503 CrossRef.
- J. R. Heemstra, Jr. and C. T. Walsh, J. Am. Chem. Soc., 2008, 130, 14024–14025 Search PubMed.
- P. C. Dorrestein, E. Yeh, S. Garneau-Tsodikova, N. L. Kelleher and C. T. Walsh, Proc. Natl. Acad. Sci. U. S. A., 2005, 102, 13843–13848 CrossRef CAS.
- K. Podzelinska, R. Latimer, A. Bhattacharya, L. C. Vining, D. L. Zechel and Z. Jia, J. Mol. Biol., 2010, 397, 316–331 CrossRef CAS.
- L. Xiang, J. A. Kalaitzis and B. S. Moore, Proc. Natl. Acad. Sci. U. S. A., 2004, 101, 15609–15614 CrossRef CAS.
- C. Hertweck, A. Luzhetskyy, Y. Rebets and A. Bechthold, Nat. Prod. Rep., 2007, 24, 162–190 RSC.
- Q. Cheng, L. Xiang, M. Izumikawa, D. Meluzzi and B. S. Moore, Nat. Chem. Biol., 2007, 3, 557–558 CrossRef CAS.
- A. Favorskii, J. Russ. Phys. Chem. Soc., 1905, 37, 643 Search PubMed.
- M. E. Taga, N. A. Larsen, A. R. Howard-Jones, C. T. Walsh and G. C. Walker, Nature, 2007, 446, 449–453 CrossRef CAS.
- T. P. Begley, A. Chatterjee, J. W. Hanes, A. Hazra and S. E. Ealick, Curr. Opin. Chem. Biol., 2008, 12, 118–125 Search PubMed.
- G. E. Schulz, R. H. Schirmer, W. Sachsenheimer and E. F. Pai, Nature, 1978, 273, 120–124 CrossRef.
- C. Thorpe and C. H. Williams, Jr, J. Biol. Chem., 1976, 251, 3553–3557 Search PubMed.
- B. Fox and C. T. Walsh, J. Biol. Chem., 1982, 257, 2498–2503 Search PubMed.
- B. S. Fox and C. T. Walsh, Biochemistry, 1983, 22, 4082–4088 Search PubMed.
- W. R. Briggs, C. F. Beck, A. R. Cashmore, J. M. Christie, J. Hughes, J. A. Jarillo, T. Kagawa, H. Kanegae, E. Liscum, A. Nagatani, K. Okada, M. Salomon, W. Rüdiger, T. Sakai, M. Takano, M. Wada and J. C. Watson, Plant Cell, 2001, 13, 993–997 CrossRef CAS.
- K. Kaneda, T. Kuzuyama, M. Takagi, Y. Hayakawa and H. Seto, Proc. Natl. Acad. Sci. U. S. A., 2001, 98, 932–937 CrossRef CAS.
- C. J. Thibodeaux, W.-C. Chang and H.-W. Liu, J. Am. Chem. Soc., 2010, 132, 9994–9996 Search PubMed.
- C. J. Thibodeaux, S. O. Mansoorabadi, W. Kittleman, W.-C. Chang and H.-W. Liu, Biochemistry, 2008, 47, 2547–2558 Search PubMed.
- J. Calveras, C. J. Thibodeaux, S. O. Mansoorabadi and H.-W. Liu, ChemBioChem, 2012, 13, 42–46 Search PubMed.
- P. M. Nassau, S. L. Martin, R. E. Brown, A. Weston, D. Monsey, M. R. McNeil and K. Duncan, J. Bacteriol., 1996, 178, 1047–1052 CAS.
- F. Pan, M. Jackson, Y. Ma and M. McNeil, J. Bacteriol., 2001, 183, 3991–3998 CrossRef CAS.
- M. Soltero-Higgin, E. E. Carlson, T. D. Gruber and L. L. Kiessling, Nat. Struct. Mol. Biol., 2004, 11, 539–543 CrossRef CAS.
- T. D. Gruber, W. M. Westler, L. L. Kiessling and K. T. Forest, Biochemistry, 2009, 48, 9171–9173 CrossRef CAS.
- H. G. Sun, M. W. Ruszczycky, W.-C. Chang, C. J. Thibodeaux and H.-W. Liu, J. Biol. Chem., 2012, 287, 4602–4608 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2013 |