Recent advances in the efficient reduction of graphene oxide and its application as energy storage electrode materials
Tapas Kuilaa, Ananta Kumar Mishrab, Partha Khanraa, Nam Hoon Kimc and Joong Hee Lee*abc
aWCU Programme, Department of BIN Fusion Technology, Chonbuk National University, Jeonju, Jeonbuk 561-756, Republic of Korea. E-mail: tkuila@gmail.com; parthakhanra@gmail.com; jhl@jbnu.ac.kr; Fax: +82 832702341; Tel: +82 832702342
bDepartment of Polymer and Nano Engineering, Chonbuk National University, Jeonju, Jeonbuk 561-756, Republic of Korea. E-mail: spuananta@gmail.com
cDepartment of Hydrogen and Fuel Cell Engineering, Chonbuk National University, Jeonju, Jeonbuk 561-756, Republic of Korea. E-mail: namhk99@naver.com
First published on 7th November 2012
Abstract
Efficient reduction of graphene oxide (GO) by chemical, thermal, electrochemical, and photo-irradiation techniques has been reviewed. Particular emphasis has been directed towards the proposed reduction mechanisms of GO by different reducing agents and techniques. The advantages of using different kinds of reducing agents on the basis of their availability, cost-effectiveness, toxicity, and easy product isolation processes have also been studied extensively. We provide a detailed description of the improvement in physiochemical properties of reduced GO (RGO) compared to pure GO. For example, the electrical conductivity and electrochemical performance of electrochemically obtained RGO are much better than those of chemically or thermally RGO materials. We provide examples of how RGO has been used as supercapacitor electrode materials. Specific capacitance of GO increases after reduction and the value has been reported to be 100–300 F g−1. We conclude by proposing new environmentally friendly types of reducing agents that can efficiently remove oxygen functionalities from the surface of GO.
 Tapas Kuila | Dr Tapas Kuila has been working as a Research Professor in the Department of BIN Fusion Technology, Chonbuk National University since February, 2012. He joined the Department as Postdoctoral Fellow in November, 2010. He received his PhD in Chemistry from Indian Institute of Technology, Kharagpur, India. He has completed MSc and BSc from Vidyasagar University, West Bengal, India. Presently, he is working on the surface modification of chemically derived graphene and its application in supercapacitor devices. He is also working on the green reduction of graphene oxide and graphene-based composites. |
 Ananta Kumar Mishra | Dr Ananta Kumar Mishra completed his MSc and MPhil in Organic Chemistry from Sambalpur University, India. He received his PhD degree in Polymer Science from Indian Institute of Technology, Kharagpur, India, in 2010. He has been pursuing his Post-Doctoral Research in Chonbuk National University, South Korea, since November 2010. Currently, he is working on the hybrid membranes for proton exchange membrane fuel cells and fabrication of graphene-based hydrogen barrier film through layer-by-layer assembly techniques. |
 Partha Khanra | Mr Partha Khanra has completed a MSc in Physics from Devi Ahilya Vishwavidyalaya, Indore, India. Currently he is continuing his PhD at Chonbuk National University, South Korea. He is working on the chemical and electrochemical synthesis of monolayer functionalized graphene. Both the chemically and electrochemically derived graphene finds application in supercapacitor devices. |
 Nam Hoon Kim | Mr Nam Hoon Kim is perusing his PhD on graphene based composite materials at Chonbuk National University, South Korea. He has completed Bachelor and Master degrees in Polymer Science from Chonbuk National University. He is working on the simultaneous reduction and functionalization of graphene oxide to increase the electrical conductivity of graphene for composite application. |
 Joong Hee Lee | Professor Joong Hee Lee has more than 25 years of research experience in the fabrication of polymer and polymer composites for electrical and electronic applications. He has received a PhD in Polymer Science from the University of Minnesota, USA. He has received a Gold Medal in Korea Patent Award and Best Project Award. His current research interests are focused on the preparation and surface modification of graphene. He is also working on graphene-based supercapacitors, biosensors, and composites. Part of his research group is engaged in the development of highly conducting composite bipolar plates and hybrid membranes for proton exchange membrane fuel cells. |
1 Introduction
The unique physiochemical and electrical characteristics of graphene make it an ideal choice for use in electronic devices such as touch panels, p–n junction materials, flexible thin-film transistors, hydrogen storage materials and supercapacitor electrode materials.1–8 To meet demand, large-scale production of graphene is required. Several methods such as chemical vapour deposition (CVD), plasma-enhanced CVD, epitaxial growth on electrically insulating surfaces, electric arc discharge, and solution-based chemical oxidation–reduction have been developed to produce graphene.9–18 Among these reported techniques, the most promising method for the large-scale production of graphene is the chemical oxidation of graphite, conversion of the resulting graphite oxide to graphene oxide (GO), and the subsequent reduction of GO. The surface of GO bears different oxygen-containing functional groups such as carboxyl, hydroxyl, epoxy, and keto groups, which make GO hydrophilic and insulating in nature.19,20 For application of GO in supercapacitor devices, it is essential that the GO be reduced to ensure electronic conjugation. A supercapacitor is also called an electrochemical capacitor which can store energy by means of electrical double-layer capacitors (EDLC) and pseudocapacitors.4 The supercapacitor shows high power density and long-term cyclic stability than those of the conventional capacitors. The EDLC stores charge non-faradically through reversible adsorption of ions at the electrode–electrolyte interface. On the contrary, pseudocapacitance arises for the faradic reactions occurring due to the remaining oxygen functionalities of RGO. The presence of oxygen functionalities also facilitates better interaction with the electrolyte.4 Therefore, controlled reduction of GO using strong or mild reducing agents is always preferred. Different kinds of chemical reducing agents have been used to reduce GO to graphene. In most of the cases, strong reducing agents such as hydrazine monohydrate, hydroquinone, sodium borohydride, hydroxylamine, phenylenediamine, and hydrohalic acid have been used as reducing agents.21–27 However, these reducing chemicals may damage the environment or be cost-prohibitive when used for the large-scale production of graphene. Moreover, graphene layers tend to agglomerate and form graphitic structures when strong reducing agents are used. Recently, reduction of GO using metals (Fe, Al, Zn) has attracted significant interest.28–30 However, metal particles may remain as impurities after the metal/hydrochloric acid reduction of GO. To overcome these shortcomings, non-toxic reducing agents such as L-ascorbic acid, vitamin-C, D-glucose, wild carrot root, alcohols, acetic acid, dextran, and tea solutions have been used for the reduction of GO.31–36 These non-toxic reducing agents can efficiently reduce GO to graphene and the physicochemical properties of the reduced GO (RGO) are comparable to those of hydrazine–RGO. Thermal and electrochemical techniques have also been developed to reduce GO.37–42 These approaches can successfully remove oxygen functionalities from the surface of GO. However, thermal reduction of GO in the presence of hydrogen gas requires high temperatures and special reactors, while electrochemical reduction is energy-consuming and requires expensive platinum electrodes. Therefore, new strategies for the large-scale production of RGO under mild and environmentally benign conditions are of great interest.Numerous groups have investigated the reduction of GO by chemical, thermal, and electrochemical methods for supercapacitor application. In this article, we review recent trends in the reduction of GO and provide a short review on the preparation of GO by chemical methods. We also highlight the structure–property relationships of GO and discuss why it is essential to reduce GO for supercapacitor application. All these methods reduce GO successfully and produce graphene with excellent physicochemical properties. However, these methods have limitations with respect to toxicity, cost-effectiveness, ease of availability, and experimental complexity. We conclude by proposing future cost effective and ‘green’ methodologies for the reduction of GO to graphene for application in various electrochemical devices.
2 Preparation of GO
Natural flake graphite and exfoliated (expandable) graphite are generally used to prepare graphite oxide. Several methods for the preparation of graphite oxide have been reported.43–53 Hummers' original method has been modified in several ways to increase the degree of oxidation of graphite. Pre-oxidized graphite has also been used to prepare graphite oxide by a modified Hummers' method.44 The Staudenmaier method is also an effective way to produce chemically oxidized graphite. A few other oxidants such as Jones' reagent (H2CrO4/H2SO4), Brodie's reagent, and peroxides have been used to oxidize graphite.43,49,50 In this section, we discuss the preparation of graphite oxide using different chemical methods.| KMnO4 + 3H2SO4 → K+ + MnO3− + H3+ + 3HSO4− |
| MnO3− + MnO4− → Mn2O7 |
Alternatively, potassium dichromate (K2Cr2O7) can be used instead of KMnO4 to oxidize graphite. This reagent is known as Jones' reagent and is regarded as a relatively inexpensive method for the conversion of graphite to graphite oxide. In this case, chromic acid (H2CrO4) is the oxidizing agent, and is generated in situ by the reaction of K2Cr2O7 and H2SO4. The conversion proceeds through an intermediate, dichromic acid (H2Cr2O7), as shown in the following reaction diagram:
| K2Cr2O7 + H2SO4 → H2Cr2O7 ↔ H2CrO4 |
Recently, there has been increased interest in using pre-oxidized graphite to increase the extent of oxidation. In this method, pre-oxidation of graphite (2.0 g) is carried out in the presence of a concentrated H2SO4 (20 ml) solution in which K2S2O8 (1.0 g) and P2O5 (1.0 g) are completely dissolved at 80 °C.44 The mixture, in a beaker, is kept at 80 °C for 5 h using an oil bath, after which the mixture is cooled down and diluted with 1 liter of distilled water. The final product is filtered and washed with distilled water followed by vacuum drying at 75 °C. The dry pre-oxidized graphite powder obtained is then oxidized by Hummers' method. The resulting graphite oxide contains more than 30% of oxygen (atomic%) as compared to graphite oxide obtained by the oxidation of pure graphite.
The Staudenmaier method is also regarded as an effective method to oxidize graphite and the extent of oxidation is comparable to that obtained by Hummers' method. However, this method is not popular due to the difficulty in controlling the reaction temperature.54 In a typical preparation procedure, a reaction flask containing a magnetic stir bar is charged with sulfuric acid (87.5 ml) and nitric acid (45 ml) and cooled by immersion in an ice bath. The acid mixture is stirred and allowed to cool for 15 min, and graphite (5 g) is added under vigorous stirring to avoid agglomeration. After the graphite powder is well dispersed, potassium chlorate (55 g) is added slowly over 15 min to avoid sudden increases in temperature. The reaction flask is loosely capped to allow evolution of gas from the reaction mixture and stirred for 96 h at room temperature. The only drawback of this method is the use of explosive potassium chlorate, which makes it very difficult to control the temperature of the reaction.
Graphite oxide can be prepared directly in a one-step process where graphite powder and peroxides are mixed and ground to fine powder using a mortar and pestle.43 This powder is heated in a small beaker at 110 °C. Peroxides are strong oxidizers and may explode when heated in a closed container. However, this is not an effective method for the conversion of graphite to graphite oxide.
All of the above-mentioned methods are used to prepare multi-layers of graphite oxides. To obtain a single layer of GO, graphite oxide is mechanically exfoliated by ultra-sonication in water. Single layers of GO are separated by centrifugation of the total graphite oxide dispersion in water. Un-exfoliated graphite oxide is removed as deposited material at the bottom of the centrifuge tube. The GO is collected as a stable dispersion in water, and the water is removed by freeze-drying. The conversion of graphite oxide to GO depends on the degree of oxidation of the graphite, the dispersion medium, the ultrasonic frequency, and the sonication time, among other factors. It has been found that the degree of exfoliation increases as the extent of oxidation, ultrasonic frequency, and sonication time increase.
3 Reduction of GO
Graphite oxide consists of layered GO sheets that contain many oxygen functional groups (e.g. hydroxyl, epoxide, carbonyl, and carboxyl groups). These functional groups significantly alter van der Waals interactions between the layers and endow GO with strong hydrophilicity.19,20 Based on the different models proposed by Hoffmann, Ruess, Nakajima–Matsuo, and Lerf–Klinowski (shown in Fig. 1), GO contains oxygen functionalities on the basal planes and edges of sheets.55–57 The presence of these functional groups destroys the planar sp2 carbons of graphite and converts them to sp3 carbons.19,20 Therefore, the π–π electronic conjugation of graphite is destroyed in GO, resulting in a significant decrease in electrical conductivity. It has been reported that GO is insulating in nature. In contrast, the electrical conductivity of pure graphite has been reported to be 2500 ± 15 S m−1 at room temperature.58,59 Therefore, GO cannot be used as a conductive material for electrical and electrochemical devices without further processing. In addition, the presence of oxygen functional groups makes GO thermally unstable when it undergoes pyrolysis at elevated temperatures.60–62 Notably, it has been demonstrated that the electrical conductivity of GO (and presumably its thermal stability as well) can be restored close to the level of graphite by chemical reduction.63–65 The electrical conductivity of RGO has been reported to be in the range of 200–42![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 S m−1 depending on the type of reducing agent, the reduction time, temperature, annealing time, and annealing temperature, etc.60,66–68 The specific capacitance of RGO has been found to be significantly higher than that of GO. The cyclic stability of GO can also be increased after the reduction. However, the transparency and dispersibility of RGO are significantly poorer than those of pure GO. Oxygen-containing functional groups are removed during reduction, resulting in a decrease in the dispersibility of GO in different solvents. The restoration of π-electronic conjugation and the change in color from brown to black are the main reasons for the observed decrease in transparency.
000 S m−1 depending on the type of reducing agent, the reduction time, temperature, annealing time, and annealing temperature, etc.60,66–68 The specific capacitance of RGO has been found to be significantly higher than that of GO. The cyclic stability of GO can also be increased after the reduction. However, the transparency and dispersibility of RGO are significantly poorer than those of pure GO. Oxygen-containing functional groups are removed during reduction, resulting in a decrease in the dispersibility of GO in different solvents. The restoration of π-electronic conjugation and the change in color from brown to black are the main reasons for the observed decrease in transparency.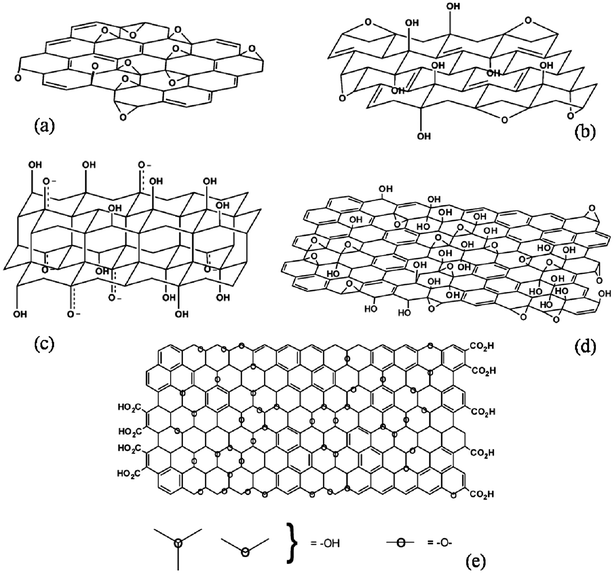 | ||
| Fig. 1 (a) Hofman, (b) Ruess, (c) Nakajima–Matsuo, and (d and e) Lerf–Klinowski model structures of GO showing the presence of oxygen functionalities above and below the basal plane. (Reproduced with permission from ref. 55–57.) | ||
Stankovich et al. were the first authors to report the preparation of electrically conducting graphene–polystyrene (PS) composites with a percolation threshold of 0.1 vol% graphene. These authors reduced the GO–PS colloidal suspension using dimethylhydrazine at 80 °C for 24 h.67,69 The excellent electrical conductivity of the resulting composite spurred great interest in developing methods to obtain electrically conductive graphene via chemical, electrochemical, or thermal reduction of GO. In this section, we discuss different approaches to sequentially reduce GO to graphene.
3.1 Chemical reduction
![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C in as-synthesized GO was observed at ∼230 nm; this peak red-shifted to ∼260 nm upon reduction.
C in as-synthesized GO was observed at ∼230 nm; this peak red-shifted to ∼260 nm upon reduction. | ||
| Fig. 2 Proposed reaction mechanisms for the reduction of oxygen functionalities (epoxy and keto carbonyl) of GO using hydrazine monohydrate. (Reproduced with permission from ref. 70.) | ||
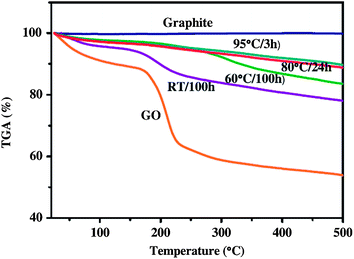 | ||
| Fig. 3 Thermogravimetric analysis (TGA) curves of graphite, GO, and RGO. (Reproduced with permission from ref. 67.) | ||
The most widely used reducing agent to prepare RGO sheets is hydrazine monohydrate.60–75 However, agglomeration of graphene sheets during reduction remains a major problem. Li et al. prepared processable aqueous dispersions of graphene by reducing GO without using polymeric or surfactant stabilizers.76 However, in most of the cases, processable graphene was prepared by surface modification of GO using an organic stabilizer followed by hydrazine reduction. The presence of hydrophilic or hydrophobic functionalities on the foreign stabilizer prevents restacking during reduction. Ongoing research on graphene revealed that primary alkyl amines, organic isocyanates, polymeric surfactants, and aromatic polyatomic molecules can be used to modify the surface of GO. These stabilizing molecules (macromolecules) either formed direct bonds (covalent interactions) with GO or adsorbed on the surface of GO and helped it to be dispersed in different solvents. Kuila et al. demonstrated the reduction of 6-amino-4-hydroxy-2-naphthalenesulfonic acid (ANS)-functionalized GO using hydrazine monohydrate.51 XPS elemental analysis revealed that the ANS functionalized graphene contained 4.2% oxygen (atomic%), confirming successful reduction, and a satisfactory electrical conductivity value (145 S m−1) was obtained at room temperature.
The reduction of GO or functionalized GO can also be carried out in the presence of a polymer when preparing graphene-based composite materials. This type of reduction helps to produce homogeneously dispersed graphene–polymer composites. Stankovich et al. were the first to propose this technique to prepare phenyl isocyanate-modified GO–PS composites in a dimethylformamide (DMF) solvent.69 Reduction was carried out in situ and excess hydrazine was removed by washing in methanol. Hu et al. prepared graphene–PS composites using an in situ emulsion polymerization method.77 Kuila et al. prepared graphene–polymethyl methacrylate (PMMA) composites using a similar method.78 In both the cases, polymerization of monomers (styrene or methyl methacrylate) was carried out in the presence of a water dispersion of GO. On completion of polymerization, hydrazine monohydrate was used to reduce the polymer (PS or PMMA)-wrapped GO. The reduction of GO in the presence of PS or PMMA was confirmed by Fourier transform infrared spectroscopy (FT-IR) and Raman spectra analysis.77,78 Nanocomposites of polyurethane (PU) with functionalized graphene sheets (FGS) were prepared by different techniques.79–82 The FGS were finely dispersed in the PU matrices as evidenced from the TEM analysis. Thermal stability and modulus values in the composites were improved significantly. Most notably the nanocomposite containing 2 parts of FGS per 100 parts of PU had an electrical conductivity of 10−4 S cm−1, a 107 times increase over that of pristine PU. This is attributed to the large surface area, high aspect ratio and good interfacial adhesion between FGS and PU. Lee et al. prepared the nanocomposite films of FGS–poly(ethylene oxide) (PEO) and GO–epoxy by solution mixing.83,84 Differential scanning calorimetry analysis suggested that FGS had a positive impact on the nucleating effect of PEO crystallization. The dynamic mechanical properties of the PEO–FGS composites were significantly higher than that of pure PEO. The FGS also efficiently improved the electrical conductivity of PEO. It is seen that the electrical conductivity was increased by more than 103-fold compared to that of pristine PEO with the addition of 2 parts of FGS per 100 parts of PEO.
Bourlinos et al. described the reduction of GO and the formation of graphitic structures using hydroquinone and sodium borohydride (NaBH4) as reducing agents before the discovery of graphene in 2004.63 They found that the intensity of the XRD peak at 2θ ∼ 24° increased and that of the XRD peak at 2θ ∼ 11.2° decreased as the reduction time of GO was increased. Wang et al. showed that hydroquinone is an efficient reducing agent for the reduction of GO.85 Although the full reduction mechanism of GO is not yet known, the reduction process must involve the removal of oxygen functional groups. The protons generated by the dissociation of hydroquinone in water can protonate the oxygen functionalities of GO and help to eliminate these groups at elevated temperatures. The appearance of a 002 reflection peak at 2θ ∼ 25° indicated the stacking of graphene sheets upon reduction. The intensity of the D band increased and that of the G band decreased in hydroquinone–RGO, confirming the reduction of GO. Shin et al. studied the effect of NaBH4 reduction on the electrical conductivity of RGO.86 They found that the efficiency of reduction increased on increasing the concentrations of NaBH4. The reduction of GO to graphene was confirmed by observing the change in the peak position from 230 to 260 nm in the UV-vis spectra of GO and RGO. The removal of oxygen functionalities occurred through the formation of intermediate, boron oxide complexes, which in turn increased the interlayer spacing of GO. The intermediate complexes were then removed by the gradual removal of carbonyl and hydroxyl groups, causing the interlayer distances to contract. It has been found that the sheet resistance of NaBH4–RGO is much lower than that of hydrazine-reduced films. This is likely due to the formation of C–N bonds during hydrazine reduction.
Zhou et al. described a rapid and cost-effective method for GO reduction using hydroxylamine as a reducing agent.87 The reduction of GO with hydroxylamine can occur rapidly under mild conditions, and the as-produced graphene sheets showed high electrical conductivity, a fair crystalline state, and good aqueous dispersibility without the need to use stabilizing reagents. A proposed mechanism for the removal of epoxide and hydroxyl groups from GO by hydroxylamine is shown in Fig. 4. However, the exact mechanism of GO reduction using hydroxylamine is still unclear. According to Zhou et al., hydroxylamine molecules attacked the carbon atom of the epoxide groups, and converted into hydroxyl groups by ring opening reactions.87 After releasing one water molecule, an N-hydroxylaziridine intermediate was formed. The hydroxylamine molecules can also attack the β carbon of the hydroxyl group in GO and transfer a proton to the hydroxyl group to release H2O with the formation of a similar N-hydroxylaziridine intermediate. The N-hydroxylaziridine intermediate will quickly convert into a conjugated vinyl (ethenyl) group and release unstable nitrogen oxide (NOx) as well as H2O. The removal of oxygen functionalities was confirmed by XPS analysis of pure GO and RGO, as shown in Fig. 5. The electrical conductivity values of hydroxylamine RGO were almost similar to those of the hydrazine–RGO.
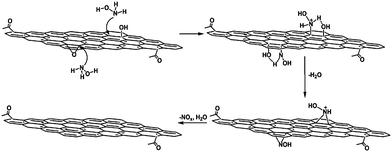 | ||
| Fig. 4 Schematic of the proposed mechanism for the reduction of GO by hydroxylamine. (Reproduced with permission from ref. 87.) | ||
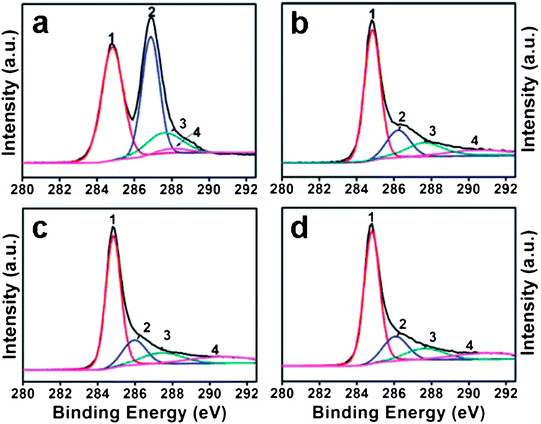 | ||
| Fig. 5 C 1s XPS spectra of GO before (a) and after reduction for 30 min (b), 1 h (c), and 12 h (d). Peaks 1, 2, 3, and 4 correspond to CC/C–C in aromatic rings, C–O (epoxy and alkoxy), CO, and COOH groups, respectively. (Reproduced with permission from ref. 87.) | ||
Simultaneous reduction and surface modification of GO using p-phenylene diamine (PPD) as a reducing and surface-modifying agent was reported.88 The mechanism of GO reduction is presumably quite similar to that of hydrazine- and hydroxylamine-based GO reduction. The oxidation product of PPD (OPPD) was adsorbed on the surface of graphene through π–π interactions and played an important role in the dispersion of graphene in different solvents. It was suggested that the positively charged OPPD could prevent the aggregation of graphene layers by electrostatic repulsive forces.
Zhou et al. and Sun et al. noted that sodium hydrosulfite was an effective reducing agent for GO reduction.58,89 These authors used XPS, TGA, XRD, Raman spectroscopy, solid-state 13C NMR spectroscopy and electrical conductivity to confirm the successful reduction of GO to graphene. However, the exact mechanism of GO reduction using sodium hydrosulfite is still an open question. GO might be reduced due to the low electrode potential of Na2S2O4 (E0 SO32−/S2O24− = −1.12 V). The epoxide and hydroxyl groups of GO were attacked by a nucleophile with a back-side SN2 nucleophilic reaction, resulting in the formation of an intermediate. Then, a thermal elimination reaction was used to obtain RGO and the reducing agent (S2O42−) was oxidized to sulfite (SO32−). The electrical conductivity of the sodium hydrosulfite–RGO was 1377 S m−1, which is about seven orders of magnitude higher than that of GO (3 × 10−4 S m−1) and comparable to that of pristine graphite (2500 ± 15 S m−1). Sun et al. showed that the absorption capability of GO can be enhanced through in situ reduction with sodium hydrosulfite as the reducing agent.89 The adsorption capacity of pure GO towards acridine orange is almost doubled after reduction with sodium hydrosulfite. This is due to the conversion of carboxyl groups into hydroxyl groups after the reduction of GO.
Recently, simultaneous reduction and functionalization of GO using a single chemical have been reported. Chemicals with reducing and functionalizing ability can be used to obtain processable colloidal dispersions of RGO, which can be used in a wide range of applications. Lin et al. described the reduction of GO in a DMF–water solvent at temperatures of 100 and 150 °C.90,91 The exact mechanism for this type of reduction is yet to be determined. It has been proposed that upon the “collision” of oxygen-containing functional groups within the basal plane of GO, CO2 or CO gas is released. Solvothermally obtained RGO (STRG) has been shown to exhibit a specific capacitance of up to 276 F g−1 at a discharge current density of 0.1 A g−1 in a 1 M H2SO4 electrolyte.91 Pham et al. reported a simple and effective method for reducing and functionalizing GO into chemically converted graphene by solvothermal reduction of GO suspension in N-methyl-2-pyrrolidone (NMP).66 Reduction of GO occurred through a free radical mechanism. The free radicals generated from NMP functionalized the GO sheets during solvothermal reduction in air. It has been found that the dispersibility of STRG was excellent in various organic solvents, while slightly functionalized STRG showed excellent electrical conductivity. A freestanding STRG paper reduced for 1 h exhibited electrical conductivities as high as 21600 S m−1, while the dispersibility of STRG that had been reduced for 5 h was as high as 1.4 mg ml−1. Efficient synthesis of processable graphene sheets using pyrrole as a reducing and surface modifying agent has also been reported.92 Reduction occurred through the exchange of electrons between pyrrole monomers and GO dispersed in water. During the reduction process, the pyrrole monomers reduced GO to RGO sheets and were themselves oxidized to form an oxidized product of pyrroles (OPy). OPy, which are possibly oligomers of pyrrole, facilitated the dispersion of graphene in organic solvents (ethanol, isopropanol, DMF, DMSO, NMP, THF, and acetone) through their adsorption to GO by π–π interactions. The formation of crystalline RGO sheets as compared to amorphous GO was demonstrated by transmission electron microscopy (TEM) analysis (Fig. 6). Simultaneous surface functionalization and reduction of GO with octadecylamine (ODA) can also be used to obtain RGO sheets.93 FT-IR spectra (Fig. 7) shows that the carboxyl functionality at 1710 cm−1 is absent in the ODA functionalized RGO. The appearance of a new band at 1564 cm−1 indicates the formation of –C–NH–C– bonds due to the reactions between epoxide and amine groups.
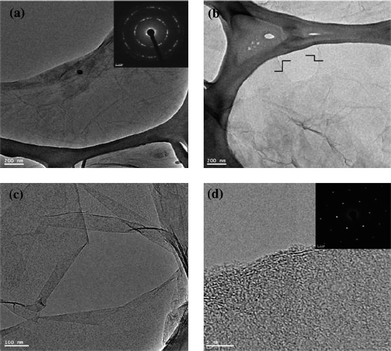 | ||
| Fig. 6 TEM images of (a) GO (inset shows SAED of GO), (b) low-magnification image of RGO, (c) scrolled graphene sheet and (d) HRTEM image of the RGO edge. The inset shows the typical SAED pattern of RGO. (Reproduced with permission from ref. 92.) | ||
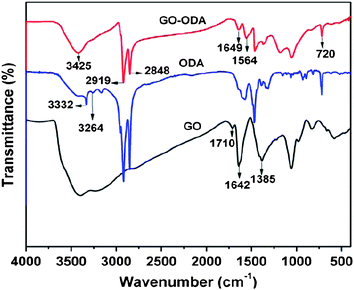 | ||
| Fig. 7 FT-IR spectra of GO, ODA and GO–ODA (20 h) (adapted from ref. 93). | ||
Fan et al. were the first to describe an environmentally friendly and efficient route for the reduction of GO by aluminum powder (Al)/hydrochloric acid (HCl) within 30 min.28 The bulk conductivity of the RGO was 2000 S m−1 at room temperature. They also studied the reduction of GO using iron (Fe)/HCl at room temperature.29 The appearance of a broad peak (Fig. 8) centered at 24.5° confirmed the stacking of graphene sheets during reduction. In comparison to Fe and Al, zinc (Zn) powder has been used extensively for the reduction of GO to graphene.30,94,95 According to these authors, reduction was completed within 1 minute after ultrasonication. The atomic ratios of carbon to oxygen in GO increased from 2.58 to 33.5 after reduction, as determined by XPS elemental analysis. The RGO had a conductivity of 15000 S m−1. The reaction between metals (Al, Fe, and Zn) and GO in an acidic solution can be understood in terms of the reduction potentials of two half cell reactions as follows:
| M (Al, Fe, Zn) → Mn+ + ne−, for Fe and Zn, n = 2, and for Al, n = 3 |
| GO + aH+ + be− → RGO + cH2O |
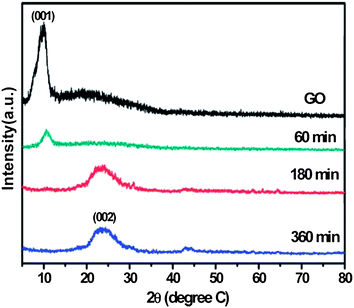 | ||
| Fig. 8 X-ray diffraction patterns of GO before and after reduction via Fe for different reduction times. (Reproduced with permission from ref. 29.) | ||
The standard reduction potentials of Al3+/Al, Fe2+/Fe, and Zn2+/Zn vs. SHE (standard electrode potential) are +1.66, +0.44, and +0.76 V, respectively. In contrast, the electrode potential of GO reduction is about −0.40 V vs. SHE in a solution with a pH ∼ 4.96 Therefore, the efficiency of GO reduction decreases in the following order: Al > Zn > Fe. The main drawback associated with metal reduction of GO is the need to separate the metal particles from the RGO to avoid metal impurities.
Pei et al. developed a simple but highly effective process to reduce GO using hydrohalic acids.97 The possible reaction mechanism is presented in Fig. 9. In the first step, the acidic reducing agents can catalyze the ring-opening reaction of epoxy groups and convert them into hydroxyl groups. In the final stage, hydroxyl groups were substituted by halogen atoms. The substituted halogen atoms can be easily eliminated from the carbon lattice to produce graphene. GO films reduced for 1 h at 100 °C in 55% hydroiodic (HI) acid had an electrical conductivity as high as 29800 S m−1 and a C/O ratio above 12, both of which are much higher than those of films reduced by other chemical methods. The use of an acetic acid–HI mixture for the efficient reduction of GO was also reported.33 The C/O atomic ratio in the resulting RGO was 6.6 and the electrical conductivity of the RGO pellet was recorded as 30400 S m−1. This novel approach can be used to fabricate and process various graphene papers and thin films to develop new flexible substrates, particularly for low-temperature processes. Chen et al. demonstrated the efficient reduction of GO using hydrobromic acid (HBr).27 The RGO could be dispersed in water and 2–3 graphene layers were observed in TEM images. The maximum specific capacitances of the RGO dispersed in water and ionic liquid were 348 and 158 F g−1, respectively, at a current density of 0.2 A g−1. Hydriodic acid (HI) in the presence of trifluoroacetic acid was also found to be an efficient reducing agent.26 Reduction using HI occurred at temperatures below 0 °C, resulting in highly graphitized RGO sheets. The resulting graphene sheets were highly deoxygenated and formed a well-crystallized graphitic material without any nitrogen and sulfur impurities. RGO–polymer composite materials that require subzero temperatures are expected to enter commercial production in the near future.
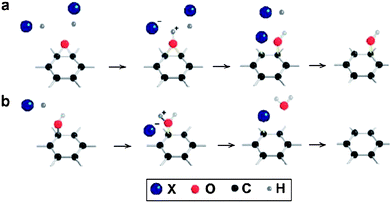 | ||
| Fig. 9 Possible reaction mechanism of GO reduction by hydrohalic acids: (a) ring-opening reaction of an epoxy group and (b) halogenation substitution reaction of a hydroxyl group. The substituted halogen atoms are expected to be easy to eliminate from the carbon lattice (X = iodine or bromine). (Reproduced with permission from ref. 97.) | ||
Hydrazine, hydrazine monohydrate, dimethylhydrazine, sodium borohydride, hydroquinone, hydroxylamine, p-phenylene diamine, and sodium hydrosulfide have been shown to efficiently reduce GO to graphene. Unfortunately, these reducing agents introduce additional functional groups onto the surface of RGO during chemical reduction, thereby increasing sheet resistance due to the scattering of electrons.98 Moreover, all these chemical reducing agents are highly poisonous and explosive. Therefore, strict precautions must be taken when using large quantities of these chemicals and their use must be minimized to protect the environment. Consequently, new approaches for effectively converting GO into stable graphene sheets under mild conditions need to be explored.
Similar to the reduction of GO in acidic solutions, alkaline solutions are also very efficient for the deoxygenation of GO. The addition of sodium hydroxide (NaOH) or potassium hydroxide (KOH) to a GO dispersion results in the conversion of the yellow-brown dispersion of GO to a homogeneous black dispersion.100 Although the proposed mechanism is unclear, it has been shown that dispersed GO undergoes faster reduction as the concentration of NaOH or KOH increases.
Dreyer et al. showed that a variety of commercially available alcohols (methyl alcohol, ethyl alcohol, isopropyl alcohol, and benzyl alcohol) can efficiently reduce GO to graphene.101 The resulting RGO exhibited high C:O ratios (up to 30:1, as determined by elemental combustion analysis), high conductivities (up to 4600 S m−1), and good specific capacitances (up to 35 F g−1) when tested as electrode materials in ultracapacitors. Su et al. reported an efficient technique for the removal of oxygen functionalities from the surface of GO using ethanol vapors.102 The RGO films had highly graphitic structures, excellent electrical conductivity, good transparency (>96%), and a high effective field-effect hole mobility (210 cm2 V−1).
Chen et al. and Pham et al. published the first environment-friendly method to reduce GO using amino acids.103,104 A possible mechanism for the chemical reduction of GO by L-cysteine is shown in Fig. 10. L-Cysteine is an amino acid that contains a thiol group. This thiol group is susceptible to oxidization upon which it releases a proton, thereby functioning as a nucleophile.105 The binding energy of the protons to oxygen-containing groups, such as the hydroxyl and epoxide groups of GO, is very high, resulting in the formation of H2O molecules.31 Finally, the L-cysteine is oxidized into L-cystine, leading to the formation of RGO sheets. The electrical conductivity of L-cysteine–RGO is about 106 times higher than that of pure GO. The reduction of GO by L-glutathione also proceeds through similar pathways. L-Glutathione–RGO can be dispersed in distilled water and several polar aprotic solvents, including THF, DMF, and DMSO, at a typical concentration of 0.5 mg ml−1 after ultrasonication for 30 min. The stabilization of graphene suspensions may originate from the oxidized products of L-cysteine. Bose et al. showed a simple, efficient, and environmentally friendly approach towards the reduction of GO using glycine as both a chemical functionalizer and a reducing agent.106 The reduction occurred under mild conditions and the as-prepared graphene sheet showed good crystalline behavior. The removal of oxygen functionalities from GO was confirmed by XPS and Raman spectra analysis. The RGO formed stable dispersion in water when a large amount of glycine was used.
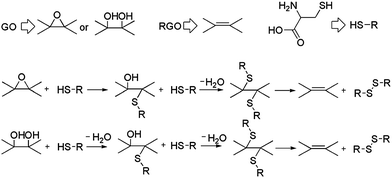 | ||
| Fig. 10 Possible mechanism of reduction of GO with L-cysteine. (Reproduced with permission from ref. 103.) | ||
Jiang et al. prepared nitrogen doped RGO by the reaction of concentrated ammonia and GO under hydrothermal conditions.107 The effects of temperature on the surface chemistry, morphology and structure of N-doped graphene sheets were investigated. Elemental mapping images (Fig. 11) indicated the presence of N, C and O elements in the RGO. It revealed that the distribution of N atoms in the RGO sheet was highly homogeneous suggesting the successful formation of N-doped graphene. Li et al. showed that thermal annealing (at 300 °C) of GO in the presence of ammonia (NH3) gas could be used to generate nitrogen-doped graphene sheets.108 The oxygen functionalities of GO are thought to be responsible for this kind of reduction. Electrical conductivity measurements of individual GO sheets demonstrated that GO annealed in NH3 exhibited higher conductivity than that annealed in H2. A novel approach for the bulk production of lithium-intercalated graphene sheets through the reduction of GO in liquid NH3 and lithium metal was reported.109 The proposed mechanism for the reduction of GO by Li/NH3 is shown in Fig. 12. Electrochemical studies of Li–RGO electrodes showed a significant enhancement in the specific capacitance of lithium batteries over commercially available graphite electrodes. Graphene sheets were prepared from GO using urea as an expansion–reduction agent.110 Thermal decomposition of urea produces ammonia gas, and the use of NH3 as a reducing gas is not new. According to Wakeland et al., the as-produced NH3 has two functions. First, the local ‘shock’ created by the rapid expansion of the gas separated the layers of the solid GO and increased the amount of surface exposed. Second, the reducing species generated by the thermal decomposition of expansion-reducing solids combined with the oxygen functionalities of GO, resulting in the formation of stable gaseous molecules and leaving RGO sheets on the crucible.
 | ||
| Fig. 11 (a) The BF-STEM images of the N-doped graphene and the corresponding EDS maps, (b) C, (c) N, and (d) O. (Reproduced with permission from ref. 107.) | ||
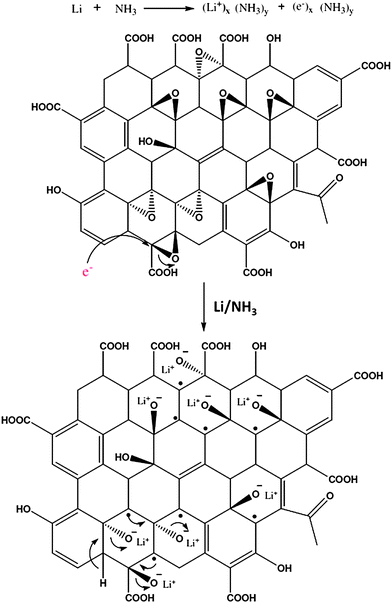 | ||
| Fig. 12 Reduction mechanism of GO by lithium metal in liquid ammonia. (Reproduced with permission from ref. 109.) | ||
RGO–nanocrystalline platinum hybrid materials were synthesized by simultaneous co-reduction of GO and chloroplatinic acid with sodium citrate in water at 80 °C (pH 7 and 10).111 This RGO served as an excellent support for dispersing and stabilizing Pt nanoparticles. The resultant hybrid materials exhibited high thermal stability, and are soluble in nonpolar organic solvents with the assistance of oleylamine.
Salas et al. and Wang et al. reported the reduction of GO by the respiration of environmental microbes in the genus Shewanella.112,113 Reduction was facilitated by electron transfer mechanisms at the Shewanella–GO interface, where at least two distinct extracellular electron transfer pathways have been proposed to occur, namely, direct charge transfer from the cell surface, and self-secreted soluble redox electron mediators. The electrochemical properties of the microbially reduced graphene (MRG) were comparable to those of hydrazine–RGO. The specific capacitance of MRG was 117 F g−1 at a charging current density of 1 A g−1, and decreased slightly to 91 F g−1 at a current density of 5 A g−1. Reduction of GO using wild carrot root was also found to be a fruitful method for obtaining graphene sheets.34 Endophytic microorganisms in carrots removed the oxygen functionalities from the surface of GO and restored electronic conjugation in graphene.
Zhu et al. reported the preparation of graphene sheets by adding reducing sugars, such as glucose, fructose, and sucrose to a GO dispersion in water.32 They explained the reduction of GO and its stabilization in water as follows. First, glucose is oxidized to aldonic acid by GO in the presence of an ammonia solution. The aldonic acid is converted into lactone. This oxidized product of glucose contains a large number of hydroxyl groups and carboxyl groups. The RGO may also have some remaining oxygen functionalities, such as hydroxyl and carboxylic groups. Thus, the hydroxyl and carboxyl groups in the oxidized products of glucose form hydrogen bonds with the residual oxygen functionalities of the RGO surfaces. The resulting RGO showed good electrocatalytic activity toward catecholamines (dopamine, epinephrine, and norepinephrine). Kim et al. described the reduction of GO using dextran as a multifunctional reducing agent.35 The reduction mechanism of GO by dextran might be similar to that of glucose, where glucose is oxidized to aldonic acid by GO in the presence of ammonia, and then further converted into lactone which interacts with GO via hydrogen bonding. The RGOs were found to be highly dispersible in water due to the adsorption of dextran molecules through hydrogen bonding.
Dopamine-induced reduction and functionalization of GO was reported by Xu and colleagues. At a weak alkaline pH, dopamine underwent self-polymerization to produce polydopamine (PDA), which was adsorbed onto the substrate materials.114 Fast reduction, self-polymerization, and adhesion properties of dopamine allow its use as a simultaneous reducing agent for GO and as a capping agent to stabilize and decorate the resulting RGO surfaces for further functionalization. PDA-capped RGO formed a homogeneous dispersion in water at a concentration of 0.05 mg ml−1. Kaminska et al. used a dopamine derivative for the simultaneous reduction and noncovalent functionalization of GO using azide terminated dopamine.115 The azide functional groups of dopamine readily reacted with alkynyl terminated molecules via a “click chemistry” approach using the copper(I)-catalyzed Huisgen 1,3-dipolar cycloaddition.
Wang et al. prepared water-dispersible graphene via GO reduction in green tea solution.36 It is well known that green tea contains aromatic polyphenolic compounds that act as both reducing agents and stabilizers; reduction of GO in green tea resulted in RGO with good dispersibility in both aqueous and organic solvents. This is a low-cost, non-toxic, easily scalable method that is highly effective at removing the functional groups of GO.
Lei et al. reported eco-friendly reduction of GO using tannic acid (TA).116 TA can act as both a reducing and a surface-modifying agent. The TA–RGO was demonstrated to disperse in ethanol, DMF, and DMSO at a concentration of 0.5 mg ml−1. The electrical conductivity of the TA–RGO was 656.7 S m−1, a value comparable to that achieved using other chemical reducing agents.
Khanra et al. showed that baker's yeast can be used for the simultaneous bio-functionalization and reduction of GO.117 Baker's yeast containing nicotinamide adenine dinucleotide phosphate (NADPH) is the main reducing and functionalizing agent. The amine functional groups of NADPH can easily couple with the epoxy functionalities of GO and form a stable water dispersion of RGO obtained by yeast reduction (YR-GO). The maximum dispersibility of YR-GO in water was recorded as 2.5 mg ml−1 at room temperature. The main advantages of this technique over the traditional chemical reduction are environmentally friendly approach and easy product isolation process. Simultaneous reduction and functionalization of GO was performed using amino-terminated poly(amidoamine) (PAMAM) dendrimers.118 The PAMAM modified RGO catalyzed the Knoevenagel condensation of benzaldehyde and dimethyl acrylate with yields up to 99%, and thus was a very active organocatalyst. Zhang et al. showed polyelectrolyte induced reduction of GO to obtain water dispersible graphene sheets.119 Controlled experiment suggested that there was no conversion of GO to graphene in the absence of the polyelectrolyte, poly(diallyldimethylammonium chloride) (PDDA). It was found that the PDDA served the roles of both a reducing and a stabilizing agent for preparing metal nanoparticle decorated graphene. It is anticipated that the N+ groups of PDDA attacked the oxygen atom of epoxide groups in GO inducing ring opening reaction as shown in Fig. 13. The initial derivative produced by the epoxide ring opening reacted further via the elimination of nitroso groups to form a CC bond. On the other hand, the hydroxyl groups were removed by a thermal dehydroxylation process.
 | ||
| Fig. 13 Proposed reaction pathway for the deoxygenation of exfoliated GO in a PDDA solution to obtain graphene nanosheets (G), where “R–N+” was used to designate PDDA. (Reproduced with permission from ref. 119.) | ||
All the methods discussed above are environment friendly methods for reducing GO. The RGO obtained using these methods were demonstrated to have good electrical conductivity, specific capacitance values, and good dispersibility in different organic solvents. However, 100% removal of oxygen functionalities from the surface of GO is not possible using mild reducing agents. Therefore, thermal and electrochemical methods for the large-scale production of RGO have been proposed.
3.2 Thermal reduction
Thermal reduction is a well-known method for the removal of oxygen functionalities from the surface of GO.120–123 In this method, oxygen functionalities are removed in the form of water, carbon dioxide, and carbon monoxide. The sample is placed in a pre-heated furnace at 1000–1100 °C for 30–45 s in the absence of air.54 The generated gases expand the layers of graphite oxide (or GO) and reduce these materials to graphene.Schniepp et al. reported thermal reduction of GO in a specially designed furnace.54 In brief, they inserted GO in an alumina boat into a 25 mm inner diameter, 1.3 m long quartz tube that was sealed at one end. The other end of the quartz tube was closed using a rubber stopper. An argon (Ar) inlet and thermocouple were then inserted through the rubber stopper. The sample was flushed with Ar for 10 min, and then the quartz tube was quickly inserted into a Lindberg tube furnace preheated to 1050 °C. These authors reported that the carbon content in the RGO increased with increasing furnace temperature.
McAllister et al. studied the exfoliation of GO and its reduction mechanism using TGA-FT-IR.123 According to their study, thermal decomposition of GO obeyed second order rate kinetics and loss can be attributed to the decomposition of oxygen-containing functional groups. The thermal energy evolved during the decomposition of GO heats the sample locally, allowing faster reaction rates and higher internal temperatures, ensuring sufficient rapid pressure buildup for uniform exfoliation. The resultant RGO samples were highly aggregated and dispersed easily in solvents. The dispersed materials consisted of at least 80% single graphene sheets.
Mattevi et al. showed that annealing of GO at 450 °C or above is equivalent to chemical reduction via hydrazine monohydrate at 80 °C followed by heating at 200 °C.124 XPS elemental analysis showed that the RGO contains ∼8% of oxygen.125 Thermal treatment of a GO paper at 700 °C under Ar or hydrogen atmosphere can efficiently remove the oxygen-containing functional groups and restore the sp2 network structure. The resulting RGO exhibited electrical conductivities as high as 8100 S m−1 which is five orders of magnitude higher than that of the original GO paper. The RGO obtained by thermal exfoliation and reduction can be used as active electrodes in the positive half-cell of a vanadium redox flow battery.126 It has been found that the RGO obtained at 1000 °C exhibited an outstanding electrochemical performance in terms of peak current densities and reversibility. This excellent behavior is attributed to the restoration of sp2 networks after thermal treatment, which implies the formation of a graphene with a high electrical conductivity and large surface area. Moreover, the remaining oxygen containing functional groups acted as active sites towards the vanadium redox reactions. Extensive research was carried out on the thermal reduction of GO, RGO characterization and its application but the exact mechanism is still unclear. Therefore, to investigate the thermal reduction mechanism, Acik et al. took in situ transmission infrared absorption spectroscopy measurements of GO films upon thermal annealing at 60–850 °C under vacuum (10−3 to 10−4 Torr).127 The proposed mechanism for thermal reduction of GO proceeds through radical reactions as shown in Fig. 14. Based on this mechanism, thermal reduction of GO occurred in three steps: initiation, propagation, and termination of free radicals. During thermal degradation, hydroxyl, hydronium, and hydroperoxyl radicals formed at the sheet edges; this determined the efficiency of the radical reactions. In the first reaction, reactive hydroxyl radicals attacked the hydroxyl groups undergoing radical propagation, leading to the formation of carbonyls. Carboxylates are known to decompose via decarboxylation, forming CO2 radicals. The unpaired electrons of the CO2 radicals then migrated through the carbon framework, resulting in the formation of covalent bonds after finding other unpaired electrons. The reactive free hydroxyl radicals are also likely to convert 1,2-diols at the etch holes into carbonyls. These radicals then propagated by reaction with the oxygen groups of GO or the carbon dangling bonds at defective sites. The newly formed species after these reactions determined the amount of oxygen that remained in the RGO structure after annealing.
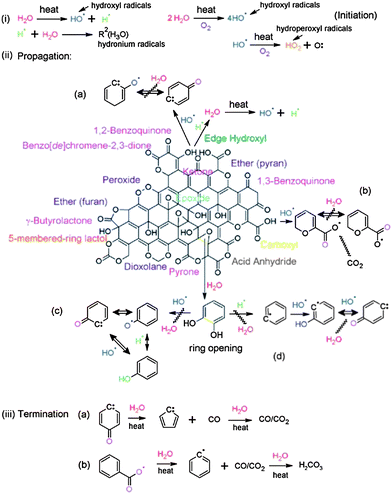 | ||
| Fig. 14 Schematic of thermal reduction mechanisms for GO reduction indicating the decomposition of oxygen species through radical reactions starting with (i) hydrolysis via combustion reactions (formation of hydroxyl, hydronium, and hydroperoxyl radicals), (ii) propagation via decarboxylation or attack on hydroxyls as well as ring-opening of epoxides, and (iii) termination into benzyl/phenyl radicals with CO/CO2 production. (Reproduced with permission from ref. 127.) | ||
Recently Park et al. described thermochemical reduction of GO for the preparation of highly conducting functionalized graphene.68 The aryl diazonium salt of 4-iodoaniline was grafted on the surface of GO to prepare phenyl functionalized GO (I-Ph-GO). Thermal annealing of I-Ph-GO at 300 °C for 6 h produces functionalized graphene (R-Ph-GO). The schematic for the preparation of thermochemically reduced graphene is shown in Fig. 15. The electrical conductivity of the R-Ph-GO increased with increasing annealing time and the maximum conductivity was found to be 42000 at 6 h annealing time. The enhanced conductivity of R-I-Ph-GO is attributed to the reduction of GO by the release of HI from I-Ph-GO during thermal annealing.
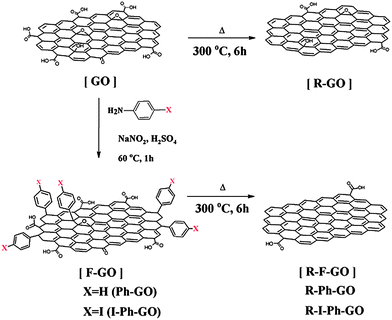 | ||
| Fig. 15 Schematic of functionalized GO (F-GO) and reduced F-GO (RFGO). Phenyl functionalized GO (Ph-GO) and GO can be thermally reduced. 4-Iodophenyl functionalized GO (I-Ph-GO) can be thermochemically reduced at 300 °C. (Reproduced with permission from ref. 68.) | ||
Thermal reduction is an easy, cost-effective and environmentally friendly fabrication route for flexible conducting graphene papers of great application potential as flexible electrodes in fields, such as energy storage/harvest (supercapacitors, batteries) and sensors. However, this method requires a complicated experimental set-up and consumes a huge amount of energy.
3.3 Electrochemical reduction
In comparison to chemical and thermal reduction, electrochemical reduction of GO is ‘green’ and fast, and the reduced material is contaminant-free. Guo et al. reported a simple approach for the large-scale synthesis of high quality graphene sheets through electrochemical reduction of an exfoliated GO precursor material at a cathodic potential of 1.5 V.128 The electrical conductivity of electrochemically obtained RGO (ERGO) was higher than that of CRGO. This may be due to the residual defects in CRGO, which could be minimized during electrochemical reduction. Shao et al. performed electrochemical reduction of GO by extended cyclic voltammetry (CV, −1.0 to 1.0 V vs. reversible hydrogen electrode, 50 mV s−1) in a 0.1 M Na2SO4 solution in a standard three-electrode cell with Hg/Hg2SO4 and Pt foil as the reference and counter electrodes, respectively (all electrode potentials are scaled to RHE except when stated otherwise).129 The specific capacitance of these ERGO sheets was 164 F g−1, which is much higher than that of CRGO. ERGO had greatly enhanced catalytic activity with regard to the electrochemical reduction of O2 and H2O2. Harima et al. demonstrated the electrochemical reduction of GO casted on the conductive substrates (fluorine-doped-tin-oxide) in the presence of organic solvents (acetonitrile, propylene carbonate, DMF, DMSO).130 The electricity required for the complete reduction was 2.0 coulombs. The resulting ERGO had an electrical conductivity of about 300 S m−1 and a specific capacitance of 147.2 F g−1 in propylene carbonate. An et al. described an effective method for the simultaneous electrochemical reduction and electrophoretic deposition (EPD) of RGO on various substrates including Cu, Ni, Al, stainless steel, and p-type Si.131 GO sheets in an aqueous dispersion migrated towards the anode, were deposited there and reduced to RGO when DC voltage was applied. The deposition rate depends on the concentration of GO dispersion, applied DC voltage and the conductivity of the substrate (anode plate). It has been found that a smooth film of RGO was obtained under the following experimental conditions: 1.5 mg ml−1 GO concentration in water, 10 V applied DC voltage and less than 30 s reduction time. However, the efficiency of reduction was less than the RGO obtained by hydrazine reduction as observed from the XPS elemental analysis. Electrochemical reduction of oriented graphene layers was proposed by Ramesha and Sampath.132 GO can be assembled on the conducting gold substrate by a layer-by-layer assembly technique through electrostatic interaction. Reduction of assembled GO could be performed electrochemically using applied DC bias by scanning the potential from 0 to −1 V vs. a saturated calomel electrode in an aqueous electrolyte. The electrochemical reduction of assembled GO may open up another way to obtain highly conducting graphene electrodes for nano-electronic devices.Fu et al. prepared graphene–Au composites by electrochemical co-reduction of GO and Au(III) in ionic liquids.39 They studied I–t performances to confirm the simultaneous reduction of GO and Au(III). The composite-modified electrodes exhibited enhanced electrochemical activity and can potentially be used in biosensors. The electrochemical reduction of GO prepared from exfoliated graphite in an acetamide–urea–ammonium nitrate ternary eutectic melt was reported.40 Electrochemical reduction of an oriented GO film was carried out at room temperature using a ternary molten electrolyte. Wang et al. described a method for the electrochemical reduction of 3-aminopropyltriethoxysilane functionalized GO.133 The functionalized GO is adsorbed on the surface of glassy carbon electrode and reduced to functionalized graphene in a 0.5 M NaCl solution saturated with N2. The functionalized graphene was further immobilized with glucose oxidase and used to detect glucose.
Electrochemical reduction of GO is very fast and avoids the use of harmful chemical reducing agents and hence, further purification of RGO is not required. The defect concentration in the ERGO is almost negligible suggesting a bright possibility of application in the areas of supercapacitor devices, touch screens and flexible electronics. Moreover, power consumption for reduction is also negligible as compared to chemical or thermal reduction. However, large scale production of RGO is the main disadvantage for commercial application. The degree of reduction is also not comparable to that of the chemical or thermal reduction method.
3.4 Microwave and photoreduction
In addition to the conventional reduction techniques described earlier, various other methods were used to deoxygenate GO. Cote et al. reported a chemical-free flash reduction process in which a photographic camera flash triggers instantaneous deoxygenation of GO by photothermal heating.134 This method decreased the sheet resistance from 2 × 108 Ω sq−1 (for pure GO) to 9.5 kΩ sq−1 (for the RGO). The hydrophilicity of the flashed area was also significantly decreased than that of pure GO. In comparison to chemical, thermal, and electrochemical reduction, flash reduction is rapid, chemical-free, and energy efficient. This technique holds great promise for patterning GO films for device and composite applications. Ng et al. demonstrated the feasibility of using a bismuth vanadate (BiVO4) visible light photocatalyst to reduce GO photocatalytically.135 Using the BiVO4–RGO system, these authors reported the generation of a higher photocurrent density in response to visible light irradiation than that generated by the TiO2 system in response to UV light irradiation. These authors attributed this finding to the longer electron lifetime of excited BiVO4 compared to TiO2, as the electrons are injected into RGO instantly at the site of generation, minimizing charge recombination. Chen et al. reported the rapid and mild thermal reduction of GO by microwaves.136 Reduction was carried out in a mixed solution of N,N-dimethylacetamide and water (DMAc–H2O). The mixed solution functioned as both a solvent for the produced graphene and a medium to control the temperature of the reactive system up to 165 °C. GO can be reduced within few minutes using microwaves, with the efficiency of reduction dependent on the duration of exposure to microwaves. The RGO obtained by the microwave induced reduction dispersed well in DMAc and formed a stable dispersion. Furthermore, the electrical conductivity of the RGO was 104 times higher than that of pure GO. Yao et al. demonstrated that GO can be directly converted to graphene sheets by applying electric current through the GO film deposited on a conductive substrate.137 The reduction occurred through the following mechanism: GO + H+ + e− = RGO + H2O. It is well known that GO contains ionizable carboxyl functionalities on its surfaces. Therefore, the GO paper itself can supply the necessary H+ ions for the reduction. It has been found that completely dried GO disfavors the reduction process. Go'mez-Navarro et al. prepared RGO on copper or gold grids by dipping the grids into the GO dispersion for 8–10 minutes and subsequently chemically reduced under hydrogen plasma.138 They have determined the atomic structure of RGO by TEM analysis. The largest portion of the RGO layer is comprised of clean well crystallized graphene areas as shown in Fig. 16. The TEM images also revealed the presence of a remarkable amount of topological defects after reduction. It was found that almost 60% of the surface of RGO remained undisturbed. Akhavan and Ghaderi prepared RGO films deposited on the TiO2 substrate by the photocatalytic irradiation.139 The concentration of the C–OH, CO, and OC–OH bonds decreased to 73%, 85%, and 72%, respectively, on increasing the irradiation time to 4 h. This is attributed to the effective reduction of the GO platelets to graphene. Moreover, on increasing the irradiation time, the antibacterial activity of the RGO–TiO2 thin film increased under solar light irradiation. The antibacterial activity of the RGO–TiO2 thin film was ∼7.5 times higher than that of the GO–TiO2 thin films. Williams et al. and Guardia et al. investigated UV-assisted photocatalytic reduction of GO.140,141 The change in the absorption spectrum following the UV-irradiation over a 15 min period is shown in Fig. 17. The change in color of the sample from dark brown to black can be seen as the reduction of GO proceeds. However, no significant changes in the absorption could be seen when TiO2 was not used. The TiO2 accumulated electrons and interacted with the GO in order to reduce certain functional groups. The following mechanism has been proposed for the reduction of GO to graphene.| TiO2 + hν → TiO2 (h + e) → TiO2 (e) + C2H4OH + H+ |
| TiO2 (e) + GO → TiO2 + RGO |
 | ||
| Fig. 16 Atomic resolution, aberration-corrected TEM image of a single layer reduced-GO membrane. (a) Original image and (b) with color added to highlight the different features. (Reproduced with permission from ref. 138.) | ||
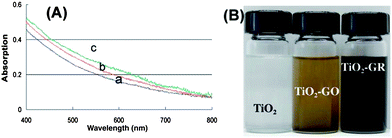 | ||
| Fig. 17 (A) The absorption spectra of GO and TiO2 suspension in ethanol (a) before UV-irradiation, (b) after 5 min, and (c) after 15 min of UV irradiation. (B) The change in color of a 10 mM solution of TiO2 nanoparticles with 0.5 mg ml−1 GO before and after UV irradiation for 2 h. A suspension of 10 mM TiO2 nanoparticles is also shown for comparison. (Reproduced with permission from ref. 140.) | ||
The RGO dispersed easily in a range of polar solvents. The main advantage of photocatalytic reduction is that it can be triggered on demand by tuning the UV-irradiation. Nanosecond pulse LASER analysis suggests that this method not only provides an on-demand UV assisted reduction technique but also opens up new ways to obtain photoactive graphene–semiconductor composites.
4 RGO-based supercapacitor
The insulating nature of GO is not useful for supercapacitor application because of a very low specific capacitance value.142–152 In contrast, the specific capacitance of GO increases after reduction and the value has been reported to be 100–300 F g−1. In the previous sections we have discussed the reduction of GO using different kinds of reducing agents to find out the environmentally friendly reduction route. The current section describes the usefulness of RGO as energy storage electrode materials and its application in supercapacitor devices.The unique features of RGO, including high electrical conductivity and porous macrostructure, are credited for improved cyclic stability and capacitance performance. Owing to its large surface area, RGO can store electrons in its π–π network and discharges stored electrons non-faradically.141 The pseudocapacitance arises due to the reversible redox reactions between the residual oxygen functionalities of RGO and H+ ions of the electrolyte. Lin et al. observed the faradic reaction of RGO in a 1 M H2SO4 electrolyte but there was no redox reaction in a 1 M Na2SO4 solution.91 The effect of reduction time on the supercapacitive performance of hydrazine–RGO has been investigated in detail.143 The C and N contents in the RGO increase with increasing reduction time at the expense of specific surface area. This is attributed to the removal of hydrophilic oxygen functionalities from the surface of RGO during reduction resulting in extensive agglomeration via π–π interactions. The RGO obtained by 30 min reduction shows highest specific capacitance of 192 F g−1 in a 6 M KOH electrolyte. Zhao et al. investigated the effect of thermal reduction temperature on the specific capacitance of RGO.144 It has been found that the specific capacitance of RGO annealed at 200 °C is highest and decreases with increasing annealing temperature. Oxygen containing functional groups played an important role in the charge–discharge processes, as they served as a passage for the ions to the internal electrode–electrolyte interface. Fig. 18 shows the variation of specific capacitance of different RGOs at different current densities. Ye et al. also reported that the RGO obtained by annealing at 200 °C and ambient pressure showed good electrochemical performance, with a maximum specific capacitance of 315 F g−1.145 The RGO-based supercapacitors in two-electrode systems were designed in 1-butyl-3-methylimidazolium hexafluorophosphate (BMIPF6) and 1-butyl-3-methylimidazolium tetrafluoroborate (BMIBF4), respectively.146 The retention in specific capacitance is 141% of the initial value after 3000 cycles. An environment-friendly method to produce water-dispersible RGO by using glutathione as a reducing and stabilization agent has been developed.147Fig. 19(a–f) shows CV, charge–discharge, specific capacitance retention and Nyquist plots of the RGO electrode. The CV curves maintain their rectangle shape very well at a scan rate of 10–500 mV s−1. The specific capacitance of the RGO electrode was determined from galvanostatic charge–discharge tests and is noted to be as high as 238 F g−1 at a current density of 0.1 A g−1. Mhamane et al. showed the reduction of GO using triethylene glycol and its usefulness as energy storage electrode materials.148 Lei et al. reported that urea reduced GO shows a gravimetric capacitance of 255 F g−1 and a volumetric capacitance of 196 F cm−3.149 The superior capacitive performance of RGO suggested that urea is a good reducing agent for the mass production of graphene-based materials for energy storage devices. Zhu et al. used RGO to fabricate super-capacitor devices.150 The maximum specific capacitance for the activated graphene was 166 F g−1 at a current density of 2.8 A g−1. Li et al. designed supercapacitor electrode materials by mixing few-layer-graphene, carbon black and poly(tetrafluoroethylene) in a mass ratio of 80:15:5 and dispersed in ethanol.151 The conductivity of the RGO is measured to be ∼3.2 × 104 S m−1, which is comparable to that of pristine graphite. The CV performance and galvanostatic charge–discharge measurement was performed in a 1 M Na2SO4 solution. The charge–discharge curves are linear in the total range of potential with constant slopes, showing a nearly perfect capacitive behavior. The specific capacitance of the designed electrode can reach upto 180 F g−1 which suggests that the RGO can be used as next-generation electrode materials for green energy storage systems due to its higher electrochemical performance. Jiang et al. investigated the capacitive behavior of N-doped graphene obtained by the reduction of GO with ammonia.107 Nitrogen doped graphene possesses greater gravimetric capacitances (144 F g−1 at 0.2 A g−1) than that of pure graphene. This is attributed to the pseudocapacitive effect based on the N-doping for graphene. After 500 cycles, the capacitance decreases to only 10% of initial capacitance, exhibiting excellent cycle stability. Yoo et al. reported an in-plane fabrication approach for an ultrathin capacitor comprised of pristine graphene and RGO.152 A schematic depiction of the stacked geometry used in the fabrication of these supercapacitor devices is provided in Fig. 20. The novel, ultrathin design allowed for the formation of an efficient electrical double layer and most importantly, for the utilization of the maximum electrochemical surface area. These solid-state supercapacitors are prototypes for a broad range of thin-film based energy storage devices. Stoller et al. studied the specific capacitance of chemically modified graphene (CMG).153 Three different electrolytes commonly used in commercial EDLCs were used to check the performance of the CMG-based ultracapacitor cells. Galvanostatic charge–discharge of CMG was performed at constant currents of 10 and 20 mA. Specific capacitance was determined from galvanostatic charge–discharge using C = I/(dV/dt) with dV/dt calculated from the slope of the discharge curves. A specific capacitance of 135 F g−1 in a 5.0 M aqueous KOH solution was recorded. In contrast, the specific capacitance in 1 M tetraethylammonium tetrafluoroborate was found to be 99 F g−1. The specific capacitance, energy density and power density of hydrazine reduced GO154 were found to be 205 F g−1, 28.5 W h kg−1 and 10 kW kg−1. The retention in specific capacitance was ∼90% after 1200 charge–discharge cycles. Du et al. investigated the specific capacitance behavior of graphene sheets derived by the chemical reduction of GO.155 The electrochemical performance of EDLC was affected by the specific surface area, pore characteristics, layer stacking and oxygen-functionalities of electrode materials. The high level of oxidation of graphite led to a large specific surface area and more graphene edges, which are favorable for the enhancement of capacitive performance. The maximum specific capacitance of 150 F g−1 at a current density of 0.1 A g−1 was recorded. Zhu et al. studied the capacitive behavior of RGO obtained by microwave assisted reduction of GO.156 The simple preparation of RGO could provide a promising route for the scalable and cost-effective production of processable graphene materials. The specific capacitance of the RGO materials was found to be 191 F g−1 in a KOH electrolyte.
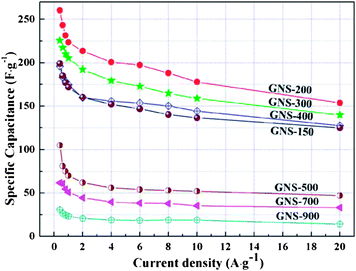 | ||
| Fig. 18 Rate capacitances of the samples in 6 M KOH with different current densities. GNS is used to designate thermally reduced RGO and the numbers followed by a hyphen (-) suggest the annealing temperature. (Reproduced with permission from ref. 144.) | ||
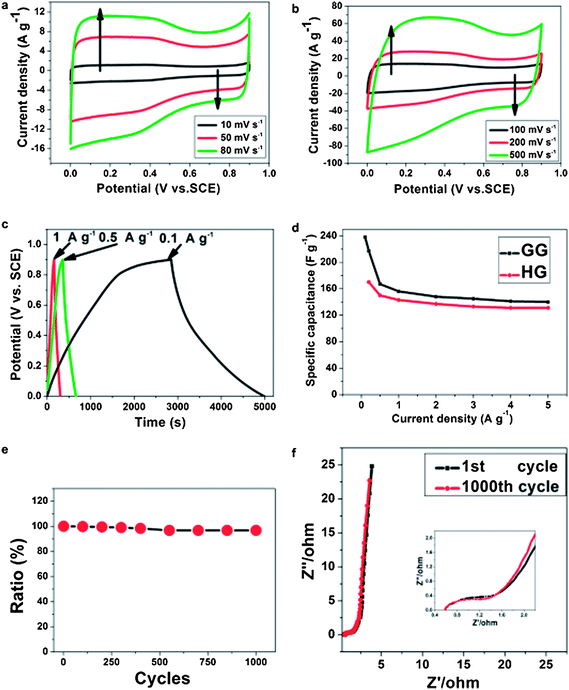 | ||
| Fig. 19 (a and b) CV curves of GG at various scan rates with potential from 0 V to 0.9 V (vs. SCE) in 1 M H2SO4; (c) galvanostatic charge–discharge curves of graphene sheets (GG) at different current densities of 0.1, 0.5 and 1 A g−1; (d) the specific capacitances of GG and graphene by hydrazine reduction (HG) at different current densities; (e) capacitance retention ratio of GG from the 1st to 1000th cycle at the current density of 2 A g−1; and (f) the Nyquist impedance plots of GG in the frequency range of 100 kHz to 0.1 Hz (main chart) and high frequency area (inset image) after the 1st cycle and 1000th cycle m2. (Reproduced with permission from ref. 147.) | ||
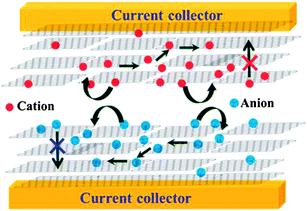 | ||
| Fig. 20 Schematic of the stacked geometry used for the fabrication of supercapacitor devices. (Reproduced with permission from ref. 152.) | ||
5 Conclusions and future research directions
We have highlighted recent advances in the reduction of GO by chemical, thermal, electrochemical, and photo-irradiation techniques. For each approach, we provided possible mechanisms for the removal of oxygen functionalities from the surface of GO. Chemical reduction of GO involves two main types of reducing agents: toxic chemicals, which are very strong reducing agents, or eco-friendly reducing agents. The use of strong reducing agents such as hydrazine, sodium borohydride, hydroxylamine, and para-phenylenediamine should be minimized due to their adverse environmental impacts. In general, eco-friendly reducing agents are mild agents and these agents are derived from natural fruits and vegetables. In some cases, eco-friendly reducing agents have been found to be more effective than strong reducing agents. Thermal reduction of GO requires a very high temperature (∼1000 °C), and can eliminate the oxygen groups from GO surfaces. We also described electrochemical reduction of GO, although the exact mechanism underlying electrochemical reduction is still unclear. The RGO obtained by chemical, thermal, or electrochemical methods has significantly improved electrical and electrochemical properties compared to pure GO. The electrical conductivity and electrochemical stability of ERGO are higher than those of chemically or thermally obtained RGO. Although we have discussed the reduction of GO by different reducing agents and techniques in detail, exact mechanisms for each and every case are yet to be determined. RGO holds great promise for use in supercapacitor devices due to its high specific capacitance value.The ‘green’ reduction of GO by biological reducing agents is another exciting future direction. Different kinds of biological reducing agents such as sp. AS2.2241,157 plant tissues,158 coconut water (Cocos nucifera L.),159,160 grapes,161,162 red algae (Cyanidioschyzon merolae 10D and Cyanidium caldarium)163 and soaked Phaseolus aureus164 were used in the reduction of aromatic and aliphatic organic ketones and esters. It is expected that these kinds of reducing agents will be a good choice for the reduction of GO to graphene. Therefore, by utilizing abundant and sustainable natural sources, biocatalysts that are selective, simple to use, non-toxic, and ultra-low cost can be developed with the ultimate goal of the large-scale production of RGO.
Acknowledgements
This study was supported by the Converging Research Center Program (2012K001428), the Human Resource Training Project for Regional Innovation, and the World Class University (WCU) program (R31-20029) funded by the Ministry of Education, Science and Technology (MEST) and National Research Foundation (NRF) of Korea. It was also supported by the Research Professor Program of Chonbuk National University.References
- K. S. Novoselov, A. K. Geim, S. V. Morozov, D. Jiang, Y. Zhang, S. V. Dubonos, I. V. Grigorieva and A. A. Firsov, Science, 2004, 306, 666–669 CrossRef CAS.
- A. K. Geim, Science, 2009, 324, 1530–1534 CrossRef CAS.
- Y. B. Zhang, Y. W. Tan, H. L. Stormer and P. Kim, Nature, 2005, 438, 201–204 CrossRef CAS.
- J. J. Yoo, K. Balakrishnan, J. Huang, V. Meunier, B. G. Sumpter, A. Srivastava, M. Conway, A. L. M. Reddy, J. Yu, R. Vajta and P. M. Ajayan, Nano Lett., 2011, 11, 1423–1427 CrossRef CAS.
- G. Eda, G. Fanchini and M. Chhowalla, Nat. Nanotechnol., 2008, 3, 270–274 CrossRef CAS.
- M. Terrones, A. R. Botello-Méndez, J. Campos-Delgado, F. López-Urías, Y. I. Vega-Cantú, F. J. Rodríguez-Macías, A. L. Elias, E. Muñoz-Sandova, A. G. Cano-Márquez, J. Charlier and H. Terrones, Nano Today, 2010, 5, 351–372 CrossRef.
- F. Schedin, A. K. Geim, S. V. Morozov, E. W. Hill, P. Blake, M. I. Katsnelson and K. S. Novoselov, Nat. Mater., 2007, 6, 652–655 CrossRef CAS.
- S. Chae, F. Gunes, K. K. Kim, E. S. Kim, G. H. Han, S. M. Kim, H. J. Shin, S. M. Yoon, J. Y. Choi, M. H. Park, C. W. Yang, D. Pribat and Y. H. Lee, Adv. Mater., 2009, 21, 2328–2333 CrossRef CAS.
- X. Li, W. Cai, J. An, S. Kim, J. Nah, D. Yang, R. Piner, A. Velamakanni, I. Jung, E. Tutuc, S. K. Banerjee, L. Colombo and R. S. Ruoff, Science, 2009, 324, 1312–1314 CrossRef CAS.
- K. S. Kim, Y. Zhao, H. Jang, Y. S. Lee, J. M. Kim, K. S. Kim, J. H. Ahn, P. Kim, J. Y. Choi and B. H. Hong, Nature, 2009, 457, 706–710 CrossRef CAS.
- Z. Wang, N. Li and Z. Gu, Nanotechnology, 2010, 21, 175602 CrossRef.
- M. Keidar, A. Shashurin, O. Volotskova, Y. Raitses and I. I. Beilis, Phys. Plasmas, 2010, 17, 057101 CrossRef.
- Z. Yan, Z. Peng, Z. Sun, J. Yao, Y. Zhu, Z. Liu, P. M. Ajayan and J. M. Tour, ACS Nano, 2011, 5, 8187–8192 CrossRef CAS.
- A. Malesevic, R. Vitchev, K. Schouteden, A. Volodin, L. Zhang, G. V. Teneloo, A. Vanhulsei and C. V. Haesendonck, Nanotechnology, 2008, 19, 305604 CrossRef.
- S. Park and R. S. Ruoff, Nat. Nanotechnol., 2009, 4, 217–224 CrossRef CAS.
- I. Vlassiouk, M. Regmi, P. Fulvio, S. Dai, P. Datskos and G. Eres, ACS Nano, 2011, 5, 6069–6076 CrossRef CAS.
- G. Ruan, Z. Sun, Z. Peng and J. M. Tour, ACS Nano, 2011, 5, 7601–7607 CrossRef CAS.
- T. Kuila, S. Bose, A. K. Mishra, P. Khanra, N. H. Kim and J. H. Lee, Prog. Mater. Sci., 2012, 57, 1061–1105 CrossRef CAS.
- J. I. Paredes, S. Villar-Rodil, A. Martínez-Alonso and J. M. D. Tascón, Langmuir, 2008, 24, 10560–10564 CrossRef CAS.
- R. D. Dreyer, S. Park, C. W. Bielawski and R. S. Ruoff, Chem. Soc. Rev., 2010, 39, 228–240 RSC.
- M. C. Kim, G. S. Hwang and R. S. Ruoff, J. Chem. Phys., 2009, 131, 064704 CrossRef.
- X. Dong, C. Y. Su, W. Zhang, J. Zhao, Q. Ling, W. Huang, P. Chen and L. Li, Phys. Chem. Chem. Phys., 2010, 12, 2164–2169 RSC.
- Y. Li, L. Tang and J. Li, Electrochem. Commun., 2009, 11, 846–849 CrossRef CAS.
- L. A. Mashat, K. Shin, K. Kalantar-zadeh, J. D. Plessis, S. H. Han, R. W. Kojima, R. B. Kaner, D. Li, X. Gou, S. J. Ippolito and W. Wlodarski, J. Phys. Chem. C, 2010, 114, 16168–16173 Search PubMed.
- S. Mao, K. Yu, S. Cui, Z. Bo, G. Lu and J. Chen, Nanoscale, 2011, 3, 2849–2853 RSC.
- P. Cui, J. Lee, E. Hwang and H. Lee, Chem. Commun., 2011, 47, 12370–12372 RSC.
- Y. Chen, X. Zhang, D. Zhang, P. Yu and Y. Ma, Carbon, 2011, 49, 573–580 CrossRef CAS.
- Z. Fan, K. Wang, T. Wei, J. Yan, L. Song and B. Shao, Carbon, 2010, 48, 1686–1689 CrossRef CAS.
- Z. J. Fan, W. Kai, J. Yan, T. Wei, L. J. Zhi, J. Feng, Y. Ren, L. P. Song and F. Wei, ACS Nano, 2011, 5, 191–198 CrossRef CAS.
- X. Mei and J. Ouyang, Carbon, 2011, 49, 5389–5397 CrossRef CAS.
- J. Zhang, H. Yang, G. Shen, P. Cheng, J. Zhang and S. Guo, Chem. Commun., 2010, 46, 1112–1114 RSC.
- C. Zhu, S. Guo, Y. Fang and S. Dong, ACS Nano, 2010, 4, 2429–2437 CrossRef CAS.
- I. K. Moon, J. Lee, R. S. Ruoff and H. Lee, Nat. Commun., 2010, 1, 73–76 CrossRef.
- T. Kuila, S. Bose, P. Khnara, A. K. Mishra, N. H. Kim and J. H. Lee, Carbon, 2012, 50, 914–921 CrossRef CAS.
- Y. K. Kim, M. H. Kim and D. H. Min, Chem. Commun., 2011, 47, 3195–3317 RSC.
- Y. Wang, Z. Shi and J. Yin, ACS Appl. Mater. Interfaces, 2011, 3, 1127–1133 CAS.
- B. Qi, H. Liu, X. Bo, H. Yang and G. Liping, Chem. Eng. J., 2011, 171, 340–344 CrossRef CAS.
- J. Ping, Y. Wang, K. Fan, J. Wu and Y. Ying, Biosens. Bioelectron., 2011, 28, 204–209 CrossRef CAS.
- C. Fu, Y. Kuang, Z. Huang, X. Wang, N. Du, J. Chen and H. Zhou, Chem. Phys. Lett., 2010, 499, 250–253 CrossRef CAS.
- V. S. Dilimon and S. Sampath, Thin Solid Films, 2011, 519, 2323–2327 CrossRef CAS.
- Z. Wei, D. Wang, S. Kim, S. Y. Kim, Y. Hu, M. K. Yakes, A. R. Laracuente, Z. Dai, S. R. Marder, C. Berger, W. P. King, W. A. de Heer, P. E. Sheehan and E. Riedo, Science, 2010, 328, 1373–1376 CrossRef CAS.
- X. Wang, L. Zhi and K. Mullen, Nano Lett., 2008, 8, 323–327 CrossRef CAS.
- J. Shen, Y. Hu, M. Shi, X. Lu, C. Qin, C. Li and M. Ye, Chem. Mater., 2009, 21, 3514–3520 CrossRef CAS.
- B. G. Choi, H. Park, T. J. Park, M. H. Yang, J. S. Kim, S. Y. Jang, N. S. Heo, S. Y. Lee, J. Kong and W. H. Hong, ACS Nano, 2010, 4, 2910–2918 CrossRef CAS.
- W. S. Hummers and R. E. Offeman, J. Am. Chem. Soc., 1958, 80, 1339 CrossRef CAS.
- L. Staudenmaier, Ber. Dtsch. Chem. Ges., 1899, 31, 1394–1399 CrossRef.
- T. Nakajima and Y. Matsuo, Carbon, 1994, 32, 469–475 CrossRef CAS.
- J. Li, H. Shi, N. Li, M. Li and J. Li, Cent. Eur. J. Chem., 2010, 8, 783–788 CrossRef CAS.
- Y. Zhu, S. Murali, W. Cai, X. Li, J. W. Suk, J. R. Potts and R. S. Ruoff, Adv. Mater., 2010, 22, 3906–3924 CrossRef CAS.
- Y. Zhang, L. Ren, S. Wang, A. Marathe, J. Chaudhuri and G. Li, J. Mater. Chem., 2011, 21, 5386–5391 RSC.
- T. Kuila, P. Khanra, S. Bose, N. H. Kim, B. C. Ku, B. Moon and J. H. Lee, Nanotechnology, 2011, 22, 305710 CrossRef.
- G. Wang, X. Shen, B. Wang, J. Yao and J. Park, Carbon, 2009, 47, 1359–1364 CrossRef CAS.
- J. Geng and H. T. Jung, J. Phys. Chem. C, 2010, 114, 8227–8234 CAS.
- H. C. Schniepp, J. L. Li, M. J. McAllister, H. Sai, M. H. Alonso, D. H. Adamson, R. K. Prud'home, R. Car, D. A. Savilley and I. A. Aksay, J. Phys. Chem. B, 2006, 110, 8535–8539 CrossRef CAS.
- A. Lerf, H. He, M. Forster and J. Klinowski, J. Phys. Chem. B, 1998, 102, 4477–4482 CrossRef CAS.
- T. Szabo, O. Berkesi, P. Forgo, K. Josepovits, Y. Sanakis, D. Petridis and I. Dekany, Chem. Mater., 2006, 18, 2740–2749 CrossRef CAS.
- H. He, J. Klinowski, M. Forster and A. Lerf, Chem. Phys. Lett., 1998, 287, 53–56 CrossRef CAS.
- T. Zhou, F. Chen, K. Liu, H. Deng, Q. Zhang, J. Feng and Q. Fu, Nanotechnology, 2011, 22, 045704 CrossRef.
- T. Kuila, S. Bhadra, D. Yao, N. H. Kim, S. Bose and J. H. Lee, Prog. Polym. Sci., 2010, 35, 1350–1375 CrossRef.
- S. Stankovich, D. A. Dikin, R. D. Piner, K. A. Kohlhaas, A. Kleinhammes, Y. Jia, Y. Wu, S. T. Nguyen and R. S. Ruoff, Carbon, 2007, 45, 1558–1565 CrossRef CAS.
- H. P. Boehm, A. Clauss, G. Fischer and U. Hofmann, Surface properties of extremely thin graphite lamellae, Proceedings of the Fifth Conference on Carbon, Pergamon Press, 1962, pp. 73–80 Search PubMed.
- H. P. Boehm, A. Clauss, G. O. Fischer and U. Hofmann, Z. Anorg Allg Chem., 1962, 316, 119–127 CrossRef CAS.
- A. B. Bourlinos, D. Gournis, D. Petridis, T. Szabo, A. Szeri and I. Dekany, Langmuir, 2003, 19, 6050–6055 CrossRef CAS.
- P. Xiao, M. Xiao, P. Liu and K. Gong, Carbon, 2000, 38, 626–628 CrossRef CAS.
- N. A. Kotov, I. Dekany and J. H. Fendler, Adv. Mater., 1996, 8, 637–641 CrossRef CAS.
- V. H. Pham, T. V. Cuong, S. H. Hur, E. Oh, E. J. Kim, E. W. Shin and J. S. Chung, J. Mater. Chem., 2011, 21, 3371–3377 RSC.
- P. G. Ren, D. X. Yan, X. Ji, T. Chen and Z. M. Li, Nanotechnology, 2011, 22, 055705 CrossRef.
- O. K. Park, M. G. Hahm, S. Lee, H. I. Joh, S. I. Na, R. Vajtai, J. H. Lee and B. C. Ku, Nano Lett., 2012, 12, 1789–1793 CrossRef CAS.
- S. Stankovich, D. A. Dikin, G. H. B. Dommett, K. M. Kohlhaas, E. J. Zimney, E. A. Stach, R. D. Piner, S. T. Nguyen and R. S. Ruoff, Nature, 2006, 442, 282–286 CrossRef CAS.
- X. Gao, J. Jang and S. Nagase, J. Phys. Chem. C, 2010, 114, 832–842 CAS.
- H. A. Becerril, J. Mao, Z. Liu, R. M. Stoltenberg, Z. Bao and Y. Chen, ACS Nano, 2008, 2, 463–470 CrossRef CAS.
- J. T. Robinson, F. K. Perkins, E. S. Snow, Z. Wei and P. E. Sheehan, Nano Lett., 2008, 8, 3137–3140 CrossRef CAS.
- S. Pei and H.-M. Cheng, Carbon, 2012, 50, 3210–3228 CrossRef CAS.
- K. P. Loh, Q. Bao, G. Eda and M. Chhowalla, Nat. Chem., 2010, 2, 1015–1024 CrossRef CAS.
- J. T. Robinson, M. Zalalutdinov, J. W. Baldwin, E. S. Snow, Z. Wei, P. Sheehan and B. H. Houston, Nano Lett., 2008, 8, 3441–3445 CrossRef CAS.
- D. Li, M. B. Muller, S. Gijle, R. B. Kaner and G. G. Wallace, Nat. Nanotechnol., 2008, 3, 101–105 CrossRef CAS.
- H. Hu, X. Wang, J. Wang, L. Wan, F. Liu, H. Zheng, R. Chen and C. Xu, Chem. Phys. Lett., 2010, 484, 247–253 CrossRef CAS.
- T. Kuila, S. Bose, P. Khanra, N. H. Kim, K. Y. Rhee and J. H. Lee, Composites, Part A, 2011, 42, 1856–1861 CrossRef.
- A. V. Raghu, Y. R. Lee, H. M. Jeong and C. M. Shin, Macromol. Chem. Phys., 2008, 209, 2487–2493 CrossRef CAS.
- D. A. Nguyen, Y. R. Lee, A. V. Raghu, H. M. Jeong, C. M. Shin and B. K. Kim, Polym. Int., 2009, 58, 412–417 CrossRef CAS.
- Y. R. Lee, A. V. Raghu, H. M. Jeong and B. K. Kim, Macromol. Chem. Phys., 2009, 210, 1247–1254 CrossRef CAS.
- N. D. Anh, A. V. Raghu, H. M. Jeong and C. M. Shin, Polym. Polym. Compos., 2010, 18, 351–358 Search PubMed.
- H. B. Lee, A. V. Raghu, K. S. Yoon and H. M. Jeong, J. Macromol. Sci., Part B: Phys., 2010, 49, 802–809 CrossRef CAS.
- Y. R. Lee, S. C. Kim, H. Lee, H. M. Jeong, A. V. raghu, K. R. Reddy and B. K. Kim, Macromol. Res., 2011, 19, 66–71 CrossRef CAS.
- G. Wang, J. Yang, J. Park, X. Gou, B. Wang, H. Liu and J. Yao, J. Phys. Chem. C, 2008, 112, 8192–8195 CAS.
- H. J. Shin, K. K. Kim, A. Benayad, S. M. Yoon, H. K. Park, I. S. Jung, M. H. Jin, H. K. Jeong, J. M. Kim, J. Y. Choi and Y. H. Lee, Adv. Funct. Mater., 2009, 19, 1987–1992 CrossRef CAS.
- X. Zhou, J. Zhang, H. Wu, H. Yang, J. Zhang and S. Guo, J. Phys. Chem. C, 2011, 115, 11957–11961 CAS.
- Y. Chen, X. Zhang, P. Yu and Y. Ma, Chem. Commun., 2009, 4527–4529 RSC.
- L. Sun, H. Yu and B. Fugetsu, J. Hazard. Mater., 2010, 203, 101–110 CrossRef.
- Z. Lin, Y. Yao, Z. Li, Y. Liu, Z. Li and C. P. Wong, J. Phys. Chem. C, 2010, 114, 14819–14825 CAS.
- Z. Lin, Y. Liu, Y. Yao, O. J. Hildreth, Z. Li, K. Moon and C. P. Wong, J. Phys. Chem. C, 2011, 115, 7120–7125 CAS.
- C. A. Amarnath, C. E. Hong, N. H. Kim, B. C. Ku, T. Kuila and J. H. Lee, Carbon, 2011, 49, 3497–3502 CrossRef CAS.
- W. Li, X. Z. Tang, H. B. Zhang, Z. G. Jiang, Z. Z. Yu, X. S. Du and Y. W. Mai, Carbon, 2011, 49, 4724–4730 CrossRef CAS.
- Y. Liu, Y. Li, M. Zhong, Y. Yang, Y. Wen and M. Wang, J. Mater. Chem., 2011, 21, 15449–15455 RSC.
- R. S. Dey, S. Hajra, R. K. Sahu, C. R. Raj and M. K. Panigrahi, Chem. Commun., 2012, 48, 1787–1789 RSC.
- M. Zhou, Y. Wang, Y. Zhai, J. Zhai, W. Ren, F. Wang and S. Dong, Chem.–Eur. J., 2009, 15, 6116–6120 CrossRef CAS.
- S. Pei, J. Zhao, J. Du, W. Ren and H. M. Cheng, Carbon, 2010, 48, 4466–4474 CrossRef CAS.
- J. H. Park, W. C. Mitchel, H. E. Smith, L. Grazulis and K. G. Eyink, Carbon, 2010, 48, 1670–1673 CrossRef CAS.
- M. J. Fernandez-Merino, L. Guardia, J. I. Paredes, S. Villar-Rodil, P. Solis-fernandez, A. Martinez-alonso and J. M. D. Tascon, J. Phys. Chem. C, 2010, 114, 6426–6432 CAS.
- X. Fan, W. Peng, Y. Li, X. Li, S. Wang, G. Zhang and F. Zhang, Adv. Mater., 2008, 20, 4490–4493 CrossRef CAS.
- D. R. Dreyer, S. Murali, Y. Zhu, R. S. Ruoff and C. W. Bielawski, J. Mater. Chem., 2010, 21, 3443–3447 RSC.
- C. Y. Su, Y. Xu, W. Zhang, J. Zhao, A. Liu, X. Tang, C. H. Tsai, Y. Huang and L. J. Li, ACS Nano, 2010, 4, 5285–5292 CrossRef CAS.
- D. Chen, L. Li and L. Guo, Nanotechnology, 2010, 22, 325601 CrossRef.
- T. A. Pham, J. S. Kim, J. S. Kim and Y. T. Jeong, Colloids Surf., A, 2011, 384, 543–548 CrossRef CAS.
- K. Brocrklehurst and G. Little, Biochem. J., 1972, 128, 471–474 Search PubMed.
- S. Bose, T. Kuila, A. K. Mishra, N. H. Kim and J. H. Lee, J. Mater. Chem., 2012, 22, 9696–9703 RSC.
- B. Jiang, C. Tia, L. Wang, L. Sun, C. Chen, X. Nong, Y. Qiao and H. Fu, Appl. Surf. Sci., 2012, 258, 3438–3443 CrossRef CAS.
- X. Li, H. Wang, J. T. Robinson, H. Sanchez, G. Diankov and H. Dai, J. Am. Chem. Soc., 2009, 131, 15939–15944 CrossRef CAS.
- A. Kumar, A. L. M. Reddy, A. Mukherjee, M. Dubey, X. Zhan, N. Singh, L. Ci, W. E. Billups, J. Nagurny, G. Mital and P. M. Ajayan, ACS Nano, 2011, 5, 4345–4349 CrossRef CAS.
- S. Wakeland, R. Martinez, J. K. Grey and C. C. Luhrs, Carbon, 2010, 48, 3463–3470 CrossRef CAS.
- Y. Wang, J. Liu, L. Liu and D. D. Sun, Nanoscale Res. Lett., 2011, 6, 241 CrossRef.
- E. C. Salas, Z. Sun, A. Luttge and J. M. Tour, ACS Nano, 2010, 4, 4852–4856 CrossRef CAS.
- G. Wang, F. Qian, C. W. Saltikov, Y. Jiao and Y. Li, Nano Res., 2011, 4, 563–570 CrossRef CAS.
- L. Q. Xu, W. J. Yang, K. G. Neoh, E. T. Kang and G. D. Fu, Macromolecules, 2010, 43, 8336–8339 CrossRef CAS.
- I. Kaminska, M. R. Das, Y. Coffinier, J. Niedziolka-Jonsson, J. Sobczak, P. Woisel, J. Lyskawa, M. Opallo, R. Boukherroub and S. Szunerits, ACS Appl. Mater. Interface, 2012, 4, 1016–1020 CrossRef CAS.
- Y. Lei, Z. Tang, R. Liao and B. Guo, Green Chem., 2011, 13, 1655–1658 RSC.
- P. Khanra, T. Kuila, N. H. Kim, S. H. Bae, D. S. Yu and J. H. Lee, Chem. Eng. J., 2012, 183, 526–533 CrossRef CAS.
- T. Wu, X. Wang, H. Qiu, J. Gao, W. Wang and Y. Liu, J. Mater. Chem., 2012, 22, 4772–4779 RSC.
- S. Zhang, Y. Shao, H. Liao, M. H. Engelhard, G. Yin and Y. Lin, ACS Nano, 2011, 5, 1785–1791 CrossRef CAS.
- D. Yang, A. Velamakanni, G. Bozoklu, S. Park, M. Stoller, R. D. Piner, S. Stankovich, I. Jung, D. A. Field, C. A. Ventrice and R. S. Ruoff, Carbon, 2009, 47, 145–152 CrossRef CAS.
- C. Go'mez-Navarro, R. T. Weitz, A. M. Bittner, M. Scolari, A. Mews, M. Burghard and K. Kern, Nano Lett., 2007, 7, 3499–3503 CrossRef CAS.
- Y. Zhu, M. D. Stoller, W. Cai, A. Velamakanni, R. D. Piner, D. Chen and R. S. Ruoff, ACS Nano, 2010, 4, 1227–1233 CrossRef CAS.
- M. J. McAllister, J. Li, D. H. Adamson, H. C. Schniepp, A. A. Abdala, J. Liu, M. Herrera-Alonso, D. L. Milius, R. Car, R. K. Prud'home and I. A. Aksay, Chem. Mater., 2009, 19, 4396–4404 CrossRef.
- C. Mattevi, G. Eda, S. Agnoli, S. Miller, K. A. Mkhoyan, O. Celik, D. Mastrogiovanni, G. Granozzi, E. Garfunkel and M. Chhowalla, Adv. Funct. Mater., 2009, 19, 2577–2583 CrossRef CAS.
- C. Valles, J. D. Nunez, A. M. Benito and W. K. Maser, Carbon, 2012, 50, 835–844 CrossRef CAS.
- Z. González, C. Botas, P. Álvarez, S. Roldán, C. Blanco and R. Santamaría, Carbon, 2012, 50, 828–834 CrossRef.
- M. Acik, G. Lee, C. Mattevi, A. Pirkle, R. M. Wallace, M. Chhowalla, K. Cho and Y. Chabal, J. Phys. Chem. C, 2011, 115, 19761–19781 CAS.
- H. L. Guo, X. F. Wang, Q. Y. Qian, F. B. Wang and X. H. Xia, ACS Nano, 2009, 3, 2653–2659 CrossRef CAS.
- Y. Shao, J. Wang, M. Engelhard, C. Wang and Y. Lin, J. Mater. Chem., 2010, 20, 743–748 RSC.
- Y. Harima, S. Setodoi, I. Imae, K. Komaguchi, Y. Ooyama, J. Ohshita, H. Mizota and J. Yano, Electrochim. Acta, 2011, 56, 5363–5368 CrossRef CAS.
- S. J. An, Y. Zhu, S. H. Lee, M. D. Stoller, T. Emilsson, S. Park, A. Velamakanni, J. An and R. S. Ruoff, J. Phys. Chem. Lett., 2010, 1, 1259–1263 CrossRef CAS.
- G. K. Ramesha and S. Sampath, J. Phys. Chem. C, 2009, 113, 7985–7989 CAS.
- Z. Wang, X. Zhou, J. Zhang, F. Boey and H. Zhang, J. Phys. Chem. C, 2009, 113, 14071–14075 CAS.
- L. J. Cote, R. Cruz-Silva and J. Huang, J. Am. Chem. Soc., 2000, 131, 11027–11032 CrossRef.
- Y. H. Ng, A. Iwase, A. Kudo and R. Amal, J. Phys. Chem. Lett., 2010, 1, 2607–2612 CrossRef CAS.
- W. Chen, L. Yan and P. R. Bangal, Carbon, 2010, 48, 1146–1152 CrossRef CAS.
- P. Yao, P. Chen, L. Jiang, H. Zhao, H. Zhu, D. Zhou, W. Hu, B. H. Han and M. Liu, Adv. Mater., 2010, 22, 5008–5012 CrossRef CAS.
- C. G. Navarro, J. C. Meyer, R. S. Sundaram, A. Chuvilin, S. Kurasch, M. Burghard, K. Kern and U. Kaiser, Nano Lett., 2010, 10, 1144–1148 CrossRef.
- O. Akhavan and E. Ghaderi, J. Phys. Chem. C, 2009, 113, 20214–20220 CAS.
- G. Williams, B. Seger and P. V. Kamat, ACS Nano, 2008, 2, 1487–1491 CrossRef CAS.
- L. Guardia, S. V. Rodil, J. I. Paredes, R. Rozada, A. Martinez-Alonso and J. M. D. Tascon, Carbon, 2012, 50, 1014–1024 CrossRef CAS.
- J. G. Radich and P. V. Kamat, ACS Catal., 2012, 2, 807–816 CrossRef CAS.
- L. Z. Fan, J. L. Liu, R. Ud-din, X. Yan and X. Qu, Carbon, 2012, 50, 3724–3730 CrossRef CAS.
- B. Zhao, P. Liu, Y. Jiang, D. Pan, H. Tao, J. Song, T. Fang and W. Xu, J. Power Sources, 2012, 198, 423–427 CrossRef CAS.
- J. Ye, H. Zhang, Y. Chen, Z. Cheng, L. Hu and Q. Ran, J. Power Sources, 2012, 212, 105–110 CrossRef CAS.
- Y. Chen, X. Zhang, D. Zhang and Y. Ma, Mater. Lett., 2012, 68, 475–477 CrossRef CAS.
- D. Zhang, X. Zhang, Y. Chen, C. Wang and Y. Ma, Electrochim. Acta, 2012, 69, 364–370 CrossRef CAS.
- D. Mhamane, S. M. Unni, A. Suryawanshi, O. Game, C. Rode, B. Hannoyer, S. Kurungot and S. Ogale, J. Mater. Chem., 2012, 22, 11140–11145 RSC.
- Z. Lei, L. Lu and X. S. Zhao, Energy Environ. Sci., 2012, 5, 6391–6399 CAS.
- Y. Zhu, S. Murali, M. D. Stoller, K. J. Ganesh, W. Cai, P. J. Ferreira, A. Pirkle, R. M. Wallace, K. A. Cychosz, M. Thommes, D. Su, E. A. Stach and R. S. Ruoff, Science, 2011, 332, 1537–1541 CrossRef CAS.
- Z. J. Li, B. C. Yang, S. R. Zhang and C. M. Zhao, Appl. Surf. Sci., 2012, 258, 3726–3731 CrossRef CAS.
- J. J. Yoo, K. Balakrishnan, J. Huang, V. Meunier, B. G. Sumpter, A. Srivastava, M. Conway, A. Leela, M. Reddy, J. Yu, R. Vajtai and P. M. Ajayan, Nano Lett., 2011, 11, 1423–1427 CrossRef CAS.
- M. D. Stoller, S. Park, Y. Zhu, J. An and R. S. Ruoff, Nano Lett., 2008, 8, 3498–3502 CrossRef CAS.
- Y. Wang, Z. Shi, Y. Huang, Y. Ma, C. Wang, M. Chen and Y. Chen, J. Phys. Chem. C, 2009, 113, 13103–13107 CAS.
- X. Du, P. Guo, H. Song and X. Chen, Electrochim. Acta, 2010, 55, 4812–4819 CrossRef CAS.
- Y. Zhu, S. Murali, M. D. Stoller, A. Velamakanni, R. D. Piner and R. S. Ruoff, Carbon, 2010, 48, 2118–2121 CrossRef CAS.
- W. Yang, J. H. Xu, Y. Xie, Y. Xu, G. Zhao and G. Q. Lin, Tetrahedron: Asymmetry, 2006, 17, 1769–1774 CrossRef CAS.
- Z. H. Yang, R. Zeng, G. Yang, Y. Wang, L. Z. Li, Z. S. Lv, M. Yao and B. Lai, J. Ind. Microbiol. Biotechnol., 2008, 35, 1047–1051 CrossRef CAS.
- A. M. Fonseca, F. J. Q. Monte, M. C. de Oliveira, M. C. Mattos, G. A. Cordell and R. B. Filho, J. Mol. Catal. B: Enzym., 2009, 57, 78–82 CrossRef CAS.
- M. Bohn, K. Leppchen, M. Katzberg, A. Lang, J. Steingroewer, J. Weber, T. Bley and M. Bertau, Org. Biomol. Chem., 2007, 5, 3456–3463 CAS.
- B. Xie, J. Yang, Q. Yang and W. Yuan, J. Mol. Catal. B: Enzym., 2009, 61, 284–288 CrossRef CAS.
- F. Li, J. Cui, X. Qian, R. Zhang and Y. Xiao, Chem. Commun., 2005, 1901–1903 RSC.
- T. Utsukihara, O. Misumi, N. Katao, T. Kuroiwa and C. A. Horiuchi, Tetrahedron: Asymmetry, 2006, 17, 1179–1185 CrossRef CAS.
- G. Kumaraswamy and S. Ramesh, Green Chem., 2003, 5, 306–308 RSC.
| This journal is © The Royal Society of Chemistry 2013 |