Cholesterol – a biological compound as a building block in bionanotechnology
Leticia
Hosta-Rigau
a,
Yan
Zhang
ab,
Boon M.
Teo
a,
Almar
Postma
c and
Brigitte
Städler
*a
aiNANO, Aarhus University, Aarhus, Denmark. E-mail: bstadler@inano.au.dk; Tel: +45 8715 6668
bState Key Laboratory for Modification of Chemical Fibers and Polymer Materials, College of Material Science and Engineering, Donghua University, Shanghai 201620, People's Republic of China
cCSIRO Materials Science and Engineering, Ian Wark Laboratory, Bayview Ave, Clayton, Victoria 3168, Australia
First published on 5th November 2012
Abstract
Cholesterol is a molecule with many tasks in nature but also a long history in science. This feature article highlights the contribution of this small compound to bionanotechnology. We discuss relevant chemical aspects in this context followed by an overview of its self-assembly capabilities both as a free molecule and when conjugated to a polymer. Further, cholesterol in the context of liposomes is reviewed and its impact ranging from biosensing to drug delivery is outlined. Cholesterol is and will be an indispensable player in bionanotechnology, contributing to the progress of this potent field of research.
 Leticia Hosta-Rigau | Leticia Hosta-Rigau received her PhD in Chemistry from the University of Barcelona, Spain, in 2009, under the supervision of Prof. F. Albericio. Her research was focused on the synthesis of peptides with antitumor properties by Solid Phase Peptide Synthesis. During 2010–2011 she worked as a post-doctoral researcher in the group of Prof. F. Caruso at the Department of Chemical and Biomolecular Engineering at the University of Melbourne, Australia. Currently she is a post-doctoral research fellow at iNANO, Aarhus University, Denmark, where she received a Marie Curie individual fellowship. Her main interest is the development of artificial cells towards a functional biomedical platform with potential in enzyme-therapy. |
 Yan Zhang | Yan Zhang is currently a PhD student in the College of Material Science and Engineering at Donghua University, China. Prior to this, she received her MS degree in Tianjin Polytechnic University, China. Currently, she works as an exchange PhD student in the group of Assistant Prof. Brigitte Städler at iNANO, Aarhus University, Denmark. Her research involves the assembly of advanced capsules towards their application in drug delivery. |
 Boon M. Teo | Boon M. Teo received her PhD from The University of Melbourne, Australia in 2010 working on ultrasonic polymer synthesis. Currently, she is working as a post-doc in Assistant Prof. Brigitte Städler's group at Aarhus University, Denmark. Her research interests include emulsion polymerization, anisotropic polymer colloids and drug delivery systems. |
 Almar Postma | Almar Postma received his PhD from The University of New South Wales, under the supervision of Prof. T. P. Davis and Drs G. Moad & M. O'Shea (2005) in the fields of controlled radical polymerisation and reactive extrusion. From 2005 to 2008 he held a postdoctoral fellowship position with Prof. F. Caruso at The University of Melbourne working on both polymer-based capsular drug delivery vehicles and with a startup company (iCeutica) in pharmaceutical nanoparticulate reformulations. He re-joined CSIRO as a Research Scientist in 2008. His current research interests lie at the interface of polymer design/synthesis and their applications in nanobiomedicine and electoactive materials. |
 Brigitte Städler | Brigitte Städler received her PhD from ETH Zürich, Switzerland, followed by over two years as a post-doc in the group of Prof. F. Caruso at the University of Melbourne, Australia. Since 2010, she is Assistant Professor at iNANO, Aarhus University, Denmark after she obtained a Sapere Aude Starting Grant from the Danish Council for Independent Research, Technology and Production Sciences. Her research group works on the development and characterization of smart nature-inspired drug delivery vehicles and on the establishment of microfluidic platforms to address questions in biomedicine. |
Introduction
Bionanotechnology is aiming to learn from nature's approach to assemble and run “nano-machines” and to mimic these concepts by engineering functional nano-devices e.g. making use of the self-assembly capability of biological building blocks or imitate nature's way of motility.1–3 In the latter case, attempts to copy the flagella of bacteria4,5 or the use of motor proteins6 have been considered. In the former case, the self-assembly of DNA,7,8 peptides,9,10 or lipids11,12 into a variety of nanostructures has been demonstrated and their complexity is permanently increasing and functionality is implemented. Conjugates with biomolecules e.g. cholesterol, have proven to be indispensable building blocks. Cholesterol, an essential component of mammalian cells, has been described by Michael S. Brown and Joseph L. Goldstein as “the most highly decorated small molecule in biology”,13 and this statement could probably be extended for its application in bionanotechnology as well.Looking back, the term “cholesterol” was introduced in the beginning of the 20th century from the term “cholesterine”, coined by Chevreul in 1816 “Je nommerai cholesterine, de χoλη, bile, et ςερεoς, solide, la substance cristallisée des calculs biliaires humains” (I will call cholesterin of χoλη, bile, and ςερεoς, solid, crystalline substance of human gallstones).14 However, cholesterol as a substance was already known in the late 1700's as an alcohol soluble fraction separated from human gallstones observed by Poulletier de la Salle. In 1789 De Fourcroy had prepared a large quantity of this substance, believing that it is the same as “blanc de baleine” or spermaceti.15 He labeled the crystalline fraction “adipocire” or fatty wax, a material similar to that obtained from acid treated grave wax. (Grave was a substance formed normally in damp areas of cemeteries from the decomposition of cadavers.) Chevreul later showed that it differed from these compounds by treatment with potassium hydroxide and melting point comparisons.16
Initial forays into elucidation of the structure of cholesterol showed it to be an alcohol17 in the course of the preparation of esters, and later it was shown to be a secondary alcohol18 through its conversion into the ketone cholesterone. The presence of the double bond was established in 1868. Further structural elucidation through parallel studies on derivatives of cholesterol and chemically related bile acids, with verification by X-ray studies,19 allowed the established formula to be arrived at as described in reviews by Windaus and Neukirchen.20,21
In nature, cholesterol is heterogeneously distributed in cellular membranes, with the highest content found in the plasma membrane.22 Most nucleated cells synthesise cholesterol in a regulated process consisting of multiple steps that mainly take place within the endoplasmic reticulum (ER).23–25 Alternatively, cholesterol can be taken up by the cells from lipoproteins.26 There are various pathways to traffic cholesterol to the plasma membrane in order to maintain the high concentration.27 When the cholesterol level reaches a threshold level which is regulated by the sphingomyelin content of the cell, cholesterol is transported to the ER for esterification and storage.28
The first link between cholesterol and human health was discovered in 1843, when Vogel showed that cholesterol was a major component of arterial plaques,29 and it has since then been intensively investigated.30,31 Cholesterol was consequently determined to be widely distributed in the animal kingdom and its isomeric forms, phytosterol, were shown to frequent the vegetable kingdom.32 Sterol research in the mid- to late 20th century progressed to biomimetic chemistry,33 tracer work,34 advanced structural determination of cholesterol using high-field NMR and X-ray diffraction methods,35 with a focus on the regulation of cholesterol synthesis and human physiology.36,37
Cholesterol's predominant characteristic is its ability to modulate the physicochemical properties (e.g. fluidity23 and permeability38) of the cell membrane. Due to this, cholesterol affects a number of cellular processes. For instance, cholesterol influences the behaviour of membrane proteins e.g. too much cholesterol and ordering will slow down the diffusion of membrane proteins and increase the bending modulus of the membrane.39 Cholesterol also acts as a precursor for the synthesis of bile acids40 (which are stored in the gallbladder and help to solubilise fats), and is also important for the metabolism of fat soluble vitamins, including vitamins A, D, E, and K.41 It is the major precursor for the synthesis of vitamin D and various steroid hormones.42 Recently, cholesterol has also been found to play an important role in membrane trafficking, sorting processes, and cell signalling processes, where researchers have suggested that it is involved in the formation of lipid rafts in the plasma membrane, as represented by G protein-coupled receptor signalling.39,43,44
The aim of this feature article is to outline the impact that the small biomolecule cholesterol has made in the field of bionanotechnology (Scheme 1). We are reviewing aspects and progresses in this area made possible and/or considerably improved and facilitated due to the presence of cholesterol. In the first part, we will outline the chemistry of cholesterol, starting with fundamental aspects, followed by cholesterol modifications and, finally, the synthesis of cholesterol-containing polymers will be discussed. The next section will address the state-of-the-art and challenges involving the self-assembly of cholesterol and cholesterol-modified polymers into nanostructures. The last part of this feature article will consider the effect of cholesterol on liposomes in bionanotechnology either as a stabiliser in drug delivery and in analytical science, or as an anchoring unit in engineering of biosensors, for the decoration of liposomes with polyethylene glycol (PEG), or in the assembly of capsosomes. The involvement of cholesterol in each of these aspects will be demonstrated by giving selected examples. Overall, we hope to demonstrate that the small molecule cholesterol contributes significantly to the field of bionanotechnology and its many applications.
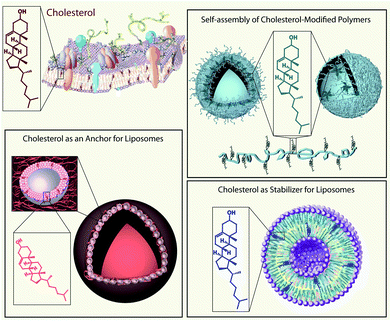 | ||
| Scheme 1 Schematic illustration of the applications of cholesterol (a small biomolecule found in biological cell membranes (top left)) in the field of bionanotechnology: self-assembly of cholesterol-containing polymers into nano-structures e.g. polymersomes (top right), cholesterol as a constituent of liposomes (bottom right), and cholesterol as an anchoring unit for liposomes e.g. for capsosome assembly (bottom left). | ||
The chemistry of cholesterol
Cholesterol is an amphiphatic molecule, a compound possessing both hydrophilic and lipophilic properties. It contains both a polar hydroxyl group on one side and a non-polar tetracylic steroidal ring structure with a branched hydrocarbon tail on the other (Fig. 1a). Further, cholesterol is comprised of a rigid planar subunit due to the trans-configuration of the rings and a flexible iso-octyl side chain ‘tail’. The combination of this rigidity and flexibility gives cholesterol the property of orientational ordering and display of liquid crystalline behaviour in combination with amphiphilic lipid membrane binding capabilities.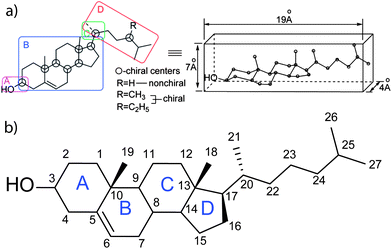 | ||
| Fig. 1 (a) A structural depiction of a cholesterol molecule highlighting the four domains of functional importance ((A) important for polarity and hydrogen bonding, (B) sterol rings, its conformation is important as it provides a rigid planar skeleton, (C) controls the conformation of the side chain, and (D) branched aliphatic side chain) (left) to function in membranes as a flat elongated compound. The structure presumed to form in cellular membranes (right). Reprinted with permission from ref. 37. Copyright (2011) American Chemical Society. (b) Cholesterol molecule showing the tetracyclic steroid frame (A–D) with numbering of carbon atoms of the sterol nucleus and side chain based on the conventional numbering system.17,254 | ||
Conjugation of cholesterol
The majority of conjugation strategies used to conjugate cholesterol onto molecules, biomolecules, or macromolecules of interest rely on the hydroxyl group in the 3′ position of the cholesterol ‘A’ ring (Fig. 1b). The most common are esterification, carbamate, and carbonate bond formation using chloroformate derivatives. These strategies allow common carboxylic acids, amines, or hydroxyl functional groups which are found on the most biologically important or other relevant molecules, to be coupled onto cholesterol. Other strategies applied to conjugate to cholesterol are illustrated in Scheme 2 and discussed in the next paragraphs.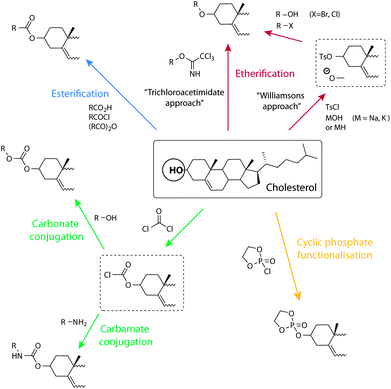 | ||
| Scheme 2 Schematic illustration of synthetic opportunities and conjugation strategies to the cholesterol alcohol. Clockwise from the top: etherification through the trichloroacetimidate approach and via Williamson's ether synthesis, dioxaphospholane cyclic phosphate functionalisation, carbamate and carbonate conjugation through a chloroformate, and esterification. | ||
Of further interest, Achalkumar et al. have recently reviewed the chemistry and approaches used to make cholesterol conjugate linkers for tethering onto phospholipid bilayers.45
A practical challenge with any conjugation strategy used for obtaining amphiphiles based on cholesterol is the choice of solubilising conditions. The challenge is to find a solvent that will dissolve both the hydrophobic cholesterol, and, at the same time, the hydrophilic material onto which it is conjugated. Typical solvent choices range from chloroform, dichloromethane, and dioxane, to tetrahydrofuran (THF). Furthermore, derivatives based on cholesterol often have a strong tendency to self-assemble. This often complicates chemistry isolation procedures such as water washes during solvent extraction steps. Nonetheless, chemical synthetic procedures for coupling cholesterol are well represented in the literature to indicate that these issues can be overcome.
Chloroformate
A common strategy for cholesterol conjugation is the use of the chloroformates. Cholesterol chloroformate, a commercially available compound, is simply combined with the material to be conjugated onto, which carries an amine, to give a carbamate cholesterol derivative. The addition of the components is usually performed under cooled anhydrous conditions and in the presence of a catalytic amount of a base like triethylamine or 4-dimethylaminopyridine (DMAP). This strategy has been used for the coupling of cholesterol onto, for example, the primary amines of 3′-O-(6-aminohexyl)-uridine46 or polyethylenimine (PEI).47 Also commonly applied, alcohols can be coupled to cholesterol chloroformate to give the corresponding carbonate. As an example, the alcohol of hydroxylethyl (meth)acrylate (HEMA) was coupled to cholesterol, as a way of placing a short spacer between the cholesterol and the vinyl group.48Esterification
The hydroxyl group lends itself well for esterification reactions and, as such, several approaches have been used. Acylation of the hydroxyl moiety of cholesterol with commercially available, functional acid chlorides, such as acryloyl chloride49,50 and methacryloyl chloride,51–53 allow appropriate vinyl esters to be synthesised in high yields. Further, the acid chloride can be obtained via reaction of the appropriate carboxylic acid with thionyl or oxalyl chloride prior to the reaction with cholesterol.54,55 Acid bromides, such as (2-bromoisobutyryl bromide) can also be efficiently coupled onto.56 Steglich esterification using N,N′-dicyclohexylcarbodiimide and DMAP, has been used successfully to react a carboxylic acid with the alcohol of cholesterol (Fig. 2a).57,58 Ring opening of succinic anhydride with cholesterol can also be used to form a cholesterol ester with a terminal carboxylic acid which can then be used for further chemical attachments.59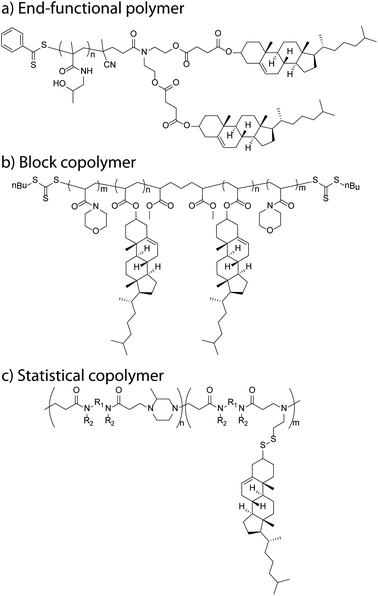 | ||
| Fig. 2 Schematic examples of cholesteryl-functional polymers: (a) bis-cholesteryl end-functional P(HPMA),57 (b) ABA triblock copolymer (PNAM-b-PChA-b-PNAM),94 and (c) poly(amidoamine)-co-(amidoamine disulfide cholesteryl) copolymer.102 | ||
Etherification
Ether formation from the hydroxyl group is a common strategy for the attachment of PEG and sugars to cholesterol. For example, cholesterol functionalised monomers containing a diethyleneglycol60 spacer and tetraethylene glycol10 spacer, attached via an ether bond, have been synthesised. Williamson ether synthesis is also a popular synthetic approach which can be carried out by reaction of the alkoxide of the alcohol with an alkylating agent carrying the ‘R’ group. Alternatively, to combine two alcohols together, one of them can be initially tosylated (using p-toluene sulfonyl chloride) and allowed to react with the other alcohol to form the corresponding ether. This was successfully carried out for the synthesis of a norbornene based monomer61 and a vinyl monomer containing an aliphatic spacer.62A glycolipid library based on acylated cholesteryl-b-galactosides was synthesised for the elucidation and construction of a vaccine against Lyme disease. Here, the glycosylation of cholesterol with galactose, essentially an ether bond formation, was achieved using a trichloroacetimidate approach.63 Several 3-cholesteryl 6-(glycosylthio)hexyl ether glycolipids were also prepared via thiol alkylation using 3-cholesteryl 6-iodohexyl ether. The latter was formed from cholesteryl p-toluenesulfonate via three steps.64
Miscellaneous chemistry
An example of the cyclic phosphate functionalisation of cholesterol was published by Iwasaki and Akiyoshi as a cholesteryl-functional monomer strategy for the synthesis of biodegradable and biocompatible polyphosphates.65 The synthesis of this monomer was achieved by the addition of cholesterol to a cooled solution of 2-chloro-2-oxo-1,3,2-dioxaphospholane in ether. However, only a 33% yield was achieved after recrystallisation.Monomers and polymers
The conjugation techniques previously mentioned are commonly applied to the synthesis of cholesterol-based (cholesteryl) monomers for the synthesis of polymers. Esterifications and carbamate formation are typically used to add the appropriate polymerisable or polymer initiating group to cholesterol. These can then be used to create a diversity of polymer architectures such as end-functional polymers, homopolymers, copolymers and block copolymers.Polymer end-functionalisation
Ring opening polymerisation (ROP)66 is one approach that has been used for adding efficiently cholesteryl end-functionality to polymers. The polymerisation is simply initiated by an alcohol, in this case cholesterol, which conveniently ring opens the monomers without the use of a catalyst. This approach has been used to synthesise polyesters, polylactide (PLA), polyglycolide and their copolymers, poly(lactide-co-glycolide) (PLGA)67 and, with the addition of a catalyst Sn(Oct)2, ε-caprolactone, which ring opens to give poly(ε-caprolactone).68 Polycarbonates based on trimethylene carbonate69 or 2,2-dimethyltrimethylene carbonate70 were also easily synthesised without the use of a catalyst.Klok et al. used ROP to great effect to synthesise L-lactic acid oligomer linkers initiated by cholesterol and terminated by cholesterol, or bioactive drugs, fluorescent markers,71 and generation 1, 2 and 3 L-lysine dendrons.72
End-functionalisation of a radical polymer with a cholesteryl moiety can be performed simply by the use of a cholesteryl-functional initiator. An example is the use of a cholesteryl radical initiator in conjunction with a cholesteryl-functional vinyl monomer. In this case 4,4-azobis(4-cyano-1-cholesteryl) pentanoate azo initiator was used to initiate the polymerisation of sodium 2-(acrylamido)-2-methylpropanesulfonate to give a cholesteryl end-capped and linear poly(sodium 2-(acrylamido)-2-methylpropanesulfonate).54 In another report, poly[N-(2-hydroxypropyl)methacrylamide]s (PHPMAs) with a terminal cholesteryl moiety either at one or both ends of the polymer chains have been synthesised (PHPMA-Chol and PHPMA-2-Chol). These were prepared by the radical polymerisation of N-(2-hydroxypropyl)methacrylamide, initiated with 4,4′-azobis-[(3-cholesteryl) 4-cyanopentanoate] in the presence of 2-mercaptoethanol and thiocholesterol chain-transfer reagents, respectively.73
A cholesteryl functional reversible addition fragmentation chain transfer (RAFT)74–78 agent with a single cholesteryl functionality placed in the RAFT re-initiating the ‘R’ group was used for the synthesis of ω-cholesteryl functional poly(polyethylene glycol) acrylate.58 A bis-cholesteryl functional ‘R’ group RAFT agent was used to place two cholesteryl ω-end groups onto poly(N,N-dimethyl acrylamide) (PDMA), PHPMA and copolymers thereof.57
Atom transfer radical polymerisation (ATRP)79–81 has also been used as a tool to initiate polymerisations with a cholesteryl-functional initiator.82 Xu et al. applied this technique to synthesise a biomimetic amphiphile from 2-(methacryloyloxy)ethyl phosphorylcholine from 10-cholesteryloxydecanyl-2-bromoisobutyrate.83,84 Similarly, cholesteryl-2-bromoisobutyrate was used to initiate various oligo(ethylene glycol) (meth)acrylates to produce copolymers with ω-bromine end-groups. These bromine end-groups were later transformed to reactive azides by nucleophilic substitution with sodium azide and, once assembled into micelles, allowed the azide–alkyne cycloaddition “click” reaction to occur in the corona of the micelle.56
Post-functionalisation of end-groups
The flexibility of RAFT agents allows for a symmetrical design of polymer end-group functionality via the RAFT agent. This can be achieved by using a RAFT agent that has a central bis-functional radical leaving/reinitiating the ‘R’ group and two thiocarbonylthio ‘Z’ groups on either side. Post-functionalisation can then be carried out through the aminolysis of the thiocarbonylthio end-groups which transform them into thiols. Such an approach has been used to react end group thiols with 3-cholesteryl 6-iodohexyl ether via nucleophilic reaction to make α,ω-di(cholest-5-en-3β-yl-6-oxyhexylthio) poly(N-isopropyl acrylamide) P(NiPAM).85 A pyridyldisulfide poly(oligo (ethyleneglycol) acrylate) RAFT polymer with a pre-attached bovine serum albumin (BSA), was functionalised with a cholesteryl group through disulfide exchange with thiocholesterol.86Homopolymers
One of the ways of introducing several cholesteryl functionalities along the length of a polymer chain is by homopolymerising a cholesteryl-functional monomer. Ruthenium-catalysed ring opening metathesis polymerisation (ROMP)87,88 of a monomer functionalised with both norbornyl and cholesteryl moieties has been utilised to make pendant cholesteryl-functional homopolymers with a variety of spacers present between the backbone and the cholesterol.55A technique that has been used more prolifically for introducing cholesteryl-functionality along a polymer chain is the radical polymerisation of cholesteryl-functionalised acrylate or methacrylate monomers to give pendant cholesteryl functionality to polymers. This was looked at as early as the late 60's and 70's by Hardy et al.89,90 and De Visser et al.49,50 More recently, cholesteryl polymerisation has seen resurgence when paired with controlled radical techniques like ATRP and RAFT. For example, a homopolymer based on cholesteryl acrylate (CA) using RAFT was synthesised and block extended with styrene.91
Block copolymers
Zhang et al. designed cholesterol monomers with a hydrophilic tetra(ethylene glycol) (TEG) spacer inserted between the mesogen and the acrylate (or methacrylate) polymerisable group in order to increase the water solubility of the monomer in their dispersion polymerisation system.10 From the poly(methacrylic acid (MAA)-co-PEG methacrylate (PEGMA)) copolymer macroRAFT agent, they synthesised the block copolymer P(MAA-co-PEGMA)-b-P(Chol-TEGMA) under dispersion polymerisation conditions, which allowed the formation of a block copolymer via self-assembly in minimal steps. In a different report, Liu et al. used a diethyleneglycol spacer between the cholesterol and the acrylate and made blocks with (benzyl protected) ascorbate sections.92 In our own research, we designed an amphiphilic block copolymer poly(N-vinyl pyrrolidone) P(NVP)-b-P(CA) which was obtained by polymerising a short CA oligomer section of, on average, 3 units from a P(NVP) macroRAFT agent.93 This macroRAFT agent was obtained via the controlled polymerisation of NVP with a xanthate RAFT agent. Alternatively, diblock copolymers have also been designed by growing the second block from a cholesteryl methacrylate (CMA) macroRAFT agent with a trimethylsilyl protected hydroxyethyl methacrylate (HEMA-TMS), giving P(CMA-b-HEMA-TMS). This block copolymer was further deprotected by the addition of concentrated HCl in a THF solution resulting in the clean product copolymer P(CMA-b-HEMA).51 Curiously, the authors reported problems with controlling of the polymerisation of the unprotected HEMA. Additionally, they initially observed difficulties polymerising CMA under ATRP conditions with methyl 2-bromopropionate as the initiator and copper(I) chloride as bipyridine as the catalyst–ligand system at 90 °C. Recently, we have looked at the synthesis of an ABA triblock copolymer in two steps from a bis ‘Z’ functional RAFT agent to form a CA macroRAFT agent, from which two N-acryloyl morpholine (NAM) arms can be grown (Fig. 2b).94 We designed the NAM![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :CA block ratio to be close to 2.5:1 to direct the spatial self-organisation of the polymer into polymersomes. A bis ‘R’ RAFT agent for the design of an ABA triblock with the cholesteryl as the ‘B’ block, for use in liquid crystalline block copolymers, has also been looked at by Shibaev et al.95
:CA block ratio to be close to 2.5:1 to direct the spatial self-organisation of the polymer into polymersomes. A bis ‘R’ RAFT agent for the design of an ABA triblock with the cholesteryl as the ‘B’ block, for use in liquid crystalline block copolymers, has also been looked at by Shibaev et al.95
ROMP can also be successfully used to synthesise well controlled block copolymers containing a cholesterol rich section. Here, a norborene functional cholesterol monomer is polymerised to form the second block from a poly(ethylene oxide) brush block. These block copolymers are assembled into nanoparticles for the delivery of doxorubicin and indomethacin, through acid labile carbamate linkers.61
Random or statistical copolymers
Copolymerisation is a convenient method for the introduction of cholesteryl pendants into the polymer chain at various mole percentages. Some examples include the free radical copolymerisation of acrylic acid (AA) with 5-acryloyloxypentyl cholesterate (Ch5A) to give P(Ch5A-co-AA),96 NiPAM with CA to give P(NiPAM-co-CA),97 HPMA terpolymerised with 6-methacrylamido hexanoyl hydrazine and cholest-5en-3β-yl 6-methacrylamido hexanoate,98 and our own work via RAFT mediated copolymerisation to give P(MAA-co-CMA) with a 7 mol% cholesterol content.52,99 Researchers at the Australian Centre for Nanomedicine have also recently looked at the copolymerisation of CMA with MAA.100 They varied the CMA content from 2 mol% to 8 mol% and together with dimethylamino ethyl methacrylates53 and RAFT mediated control, they were able to achieve a CMA content ranging from 2 mol% to 20 mol% whilst keeping their polymer molecular weights around 20–24 kDa with low molecular weight dispersities throughout. The amphiphilic copolymers showed pH-induced phase transitions in aqueous media and are envisioned to be useful for condensing and transporting siRNA due to their lipid membrane destabilizing properties.An example of a polyphosphate terpolymer containing pendant cholesteryl functionality, pendant ATRP initiating groups as well as 2-isopropyl-2-oxo-1,3,2-dioxaphospholane units were synthesised as a biodegradable graft terpolymer, a novel amphiphilic biomaterial which can function as a nanocarrier.65
Alternatively, post-functionalisation of polymers to introduce cholesterol has been performed on polymers containing amine or acid reactive groups, PEI,47 poly(L-lysine) (PLL)52 and sodium alginate,101 giving random copolymers. An interesting example of post-functionalisation is the Michael addition polymer poly(amidoamine) (PAA) (Fig. 2c).102 The nanoparticulate polymer network can be obtained using cystamine as a cross-linking agent which can be made to react with 2,2′-dithiodipyridine turning them into linear PAAs with dithiopyridyl side groups that are able to undergo a thiol exchange reaction with the commercially available thiocholesterol. The resultant products are examples of amphiphilic PAA–cholesterol conjugates in which lipophilic cholesterol moieties are linked to the hydrophilic PAA chain by disulfide bonds. These are relatively stable in blood but are cleavable under the reductive conditions within cells.
There are many more examples of cholesterol containing polymers, mostly with a focus on optical, liquid crystal and some general bioapplications; however, these are beyond the scope of this review. The reader is directed to reviews by Zhou et al. and Yusa for a more exhaustive list and description of these polymers.103,104
Self-assembly of cholesterol
As previously mentioned, cholesterol is structurally composed of different sections which play a role in its interesting self-assembly behaviour in a variety of multi-component solutions.105–108Helical ribbons were first discovered in human gallbladder bile, and it was found that these structures form as a precursor of gallstones upon dilution of bile.107 Konikoff et al. confirmed that cholesterol is the major constituent of these helical ribbons by various experimental techniques including density gradient centrifugation or X-ray diffraction. They also found that the helical ribbons themselves are precursors of stable cholesterol monohydrate crystals.106,107 Chung et al. used several model bile systems which consisted of a mixture of three types of chiral molecules in water: a bile salt, a phosphatidylcholine, and cholesterol. They demonstrated that helical ribbons formed with two distinctive pitch angles, 11.1 ± 0.5° and 53.7 ± 0.8°.105 Zastavker et al. then showed that the self-assembly of the helical ribbons with these two distinct pitch angles was not unique to model bile, but was a general phenomenon for a variety of four-component systems composed of a bile salt or surfactants, a phosphatidylcholine or fatty acids, a steroid analogue of cholesterol, and water.109 It was found that in undiluted solutions containing cholesterol, surfactants, and fatty acids or phospholipids, cholesterol is solubilised by micelles, i.e. cholesterol was contained in the core. These solutions contain both the free surfactants and the surfactants contained in micelles in equilibrium. Upon dilution, the equilibrium shifts from primarily micelles to primarily free surfactants. The micelles break up and release the cholesterol, causing the solution to become supersaturated with cholesterol. Subsequent dilution leads to the formation of large cholesterol monohydrate crystals. However, the exact structures that are formed and the sequence in which they form depend on the composition of the solution and the concentration of its components as shown in Fig. 3a.105
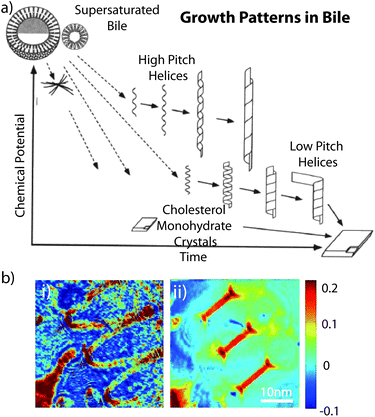 | ||
| Fig. 3 (a) Sequence and relative stability of metastable intermediates plotted as a functions of time after supersaturation of bile. Reproduced with permission from ref. 105. Copyright (1994) National Academy of Sciences, USA. (b) Images of a helical cholesterol ribbon. Phase images for angle of 0° illumination (i) and for aperture synthesis (ii). Reproduced with permission from ref. 113. | ||
In an attempt to assess the properties of helical ribbons, Starostin and coworkers studied the non-monotonic force-extension properties and predicted hysteresis behaviour for low-pitch ribbons of arbitrary material properties using a new model for inextensible elastic strips. It explained the first-order transition between two different helical states as experimentally observed. Furthermore, they revealed a new uncoiling scenario in which a ribbon of very low pitch shears under tension and successively releases a sequence of almost planar loops. They considered their results to be relevant for nanoscale devices such as force probes.110 Also, helical ribbons were thought to have potential serving as mesoscopic springs to measure or to exert forces on nanoscale biological objects. The spring constants of these helices depend on their submicroscopic thickness. Using quantitative phase microscopy, Khaykovich et al. discovered a quadratic relationship between the radius R and the thickness t of helical ribbons that form spontaneously in multicomponent cholesterol–surfactant mixtures. The quadratic relationship (R ∝ t2) between radius and thickness is a consequence of the crystal structure of the ribbons and enables the determination of the spring constant of any helices solely in terms of its observable geometrical dimensions.111 Choi et al. used high-speed synthetic aperture microscopy to image helical cholesterol ribbons in surfactant mixtures, as well as to measure the thickness of a nanoscale cholesterol helical ribbon (Fig. 3b) which was determined to be 68 nm.112,113 These fundamental findings provide the foundation towards applications of the helical ribbons.
Self-assembly of cholesterol-modified polymers into nanostructures
Due to its highly hydrophobic sterol skeleton, good biodegradability and biocompatibility, hydrophobic cholesterol is a good choice for the amphiphilic modification of many water-soluble polymers. Self-assembly of amphiphilic cholesterol-modified polymers has been a subject of great interest for the past few decades.71,73,101,114–116 Cholesterol units attached onto macromolecules as pendant(s) or as end-group(s) can induce formation of structures of well-defined morphology in water as a result of specific interactions among the cholesterol moieties.103,104 Though cholesterol has been used to modify many kinds of polymers,84,102,117–120 herein we will focus on cholesterol-modified polymers and their self-assembly behaviour with potential biomedical applications, e.g. drug delivery.Cholesterol-modified polysaccharides have attracted the interest of many researchers. Akiyoshi et al. prepared cholesteryl-bearing mannans (CHM) and cholesteryl pullulan (CHP). CHM and CHP can self-organise into nanoparticles and gels in semi-dilute solution due to the hydrophobic interaction of the cholesterol moieties.121 Cholesterol-modified chitosan (CS-CH) can form self-aggregated nanoparticles in physiological saline. The drug-loading and release behaviour of the CS-CH nanoparticles were investigated using Cyclosporine A (CyA) as a model drug. CyA was physically entrapped in the nanoparticles during the dialysis process, and the drug-loading was on the order of 6.2 wt% of the CS-CH self-aggregated nanoparticles.122 Yu et al. investigated cholesterol-modified glycol chitosan (CHGC), and used the CHGC self-aggregated nanoparticles for doxorubicin loading and delivery.123 They found that doxorubicin release from CHGC nanoparticles was dependent on the pH, with faster release at lower pH. This aspect was considered important, because the drug would be more efficiently released in the tumour tissue than in circulation. They reported that the pharmacokinetic parameters of doxorubicin changed in mice and more efficient tumor growth inhibition for doxorubicin-loaded CHGC compared to free doxorubicin was reported, making these nanoparticles promising carriers for insoluble anticancer drugs in cancer therapy. The Zhang group published a series of reports involving CS-CH124–128 and synthesised cholesterol-modified O-carboxymethyl chitosan (CCMC), cholesterol-modified chitosan conjugate with succinyl linkages (CHCS) and 6-O-cholesterol modified chitosan nanoparticles (O-CHCS NPs). The self-aggregation behaviour of the modified chitosan and chitosan nanoparticles due to the strong hydrophobic interaction of the cholesterol moieties was investigated. It was found that the degrees of substitution (DS) of the cholesterol moieties played an important role in the size and the surface property of the self-aggregated nanoparticles. Comparing the morphology and the stability of self-aggregated nanoparticles between CCMC and CHCS, the negatively charged carboxymethyl groups are advantageous for the formation of well-shaped and stable self-aggregated nanoparticles.126 They studied the drug loading and release property of CHCS and CCMC nanoparticles using Epirubicin (EPB) and paclitaxel, respectively.124,127 The results showed that the release rate decreased with increasing pH of the media. In PBS, the EPB release was very slow (∼24.9% in 48 h), while paclitaxel was continuously released over 84 h at 37 °C. The biodistribution of paclitaxel-loaded CCMC self-assembled nanoparticles showed that these nanoparticles seemed to significantly increase the uptake of paclitaxel in plasma, liver and spleen, but decreased the uptake in heart and kidney. Further, the interaction between BSA and self-aggregated CCMC and O-CHCS NPs with different DS of cholesterol moieties was assessed.125,129 The addition of O-CHCS NPs led to the decrease of the α-helical content of BSA and the increase of the β-strand content. The unfolding of BSA by a denaturant such as urea was largely suppressed upon interaction with CCMC self-aggregated nanoparticles.
Cholesterol is often used to modify PEG polymers leading to different self-assembly behaviour.130–137 Jia et al. synthesised amphiphilic liquid crystal block copolymers consisting of a cholesterol-based smectic liquid crystalline polymer block (PAChol) and a PEG block (Fig. 4i).135 They found that PEG45-b-PAChol and PEG114-b-PAChol (45 and 114 are the degrees of polymerisation of the PEG blocks) with different hydrophilic/hydrophobic weight ratios could self-assemble into polymer aggregates with different morphologies in THF/water and dioxane/water (Fig. 4ii). Adding water to a dilute solution of copolymers in dioxane yielded smectic polymer vesicles and/or nanofibers. By using THF instead, solid spherical aggregates were obtained upon water addition for the PEG45-b-PAChol copolymer. With proper block sizes and the use of appropriate organic co-solvents, these biocompatible cholesterol-based copolymers can self-assembly into aggregates with desirable shapes (e.g. vesicles or nanofibers) (Fig. 4iii), which have potential applications in drug delivery and material science.135 Nagahama et al. prepared a novel star-shaped copolymer derivative with cholesterol end groups, cholesterol-substituted 8-arm PEG-b-poly(L-lactide) (8-arm PEG-b-PLA-cholesterol). Based on the specific self-assembly between the cholesterol end groups, the 8-arm PEG-b-PLA-cholesterol aqueous solution exhibited instantaneous temperature-induced gelation at 34 °C.131 In addition, PEGylated poly(N-methyldietheneamine sebacate)-co-((cholesteryl oxocarbonylamido ethyl) methyl bis(ethylene) ammonium bromide) sebacate) (PEG-P(MDS-co-CES)) were synthesised, which could self-assemble into stable micelles of small size with a hydrophobic core and hydrophilic shell.136,137 Different cholesterol-grafting degrees of the PEG-P(MDS-co-CES) polymer were shown to have improved stability of micelle/DNA complexes in blood for systemic in vivo gene delivery. In another report, Manakker et al. investigated the rheological properties of a self-assembled hydrogel system composed of β-cyclodextrin (βCD)- and cholesterol-derived 8-arm star-shaped PEG.133 They found that the 8-arm polymer-based mixtures yielded tight viscoelastic networks, but their storage and loss moduli significantly deviated from those predicted by the Maxwell model. Rheological analysis also showed that the hydrogels were thermo-reversible. At low temperatures, the gels showed viscoelastic behaviour due to slow overall relaxation of the polymer chains. At higher temperatures, however, a reduced number of βCD–cholesterol complexes and concomitant faster chain relaxation processes eventually led to liquid-like behaviour.
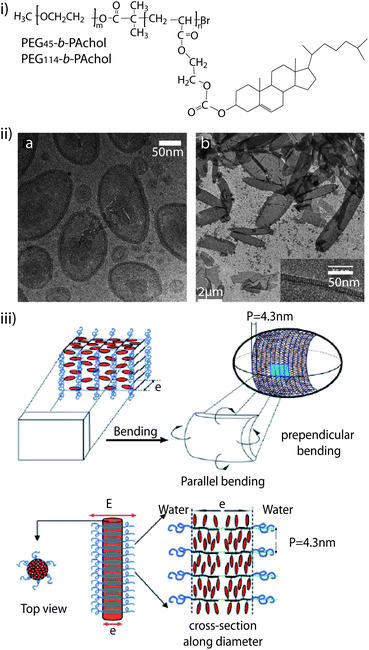 | ||
| Fig. 4 (i) Molecular structure of the diblock copolymer PEG-b-PAChol. (ii) Self-assembled structures of PEG45-b-PAChol in dioxane/water: cryo-transmission electron microscopy (TEM) image of vesicles of PEG45-b-PAChol10 (left) and cryo-TEM images of vesicles of PEG45-b-PAChol16 (right). (iii) Schematic presentation of the model for ellipsoidal smectic polymer vesicle (top) and smectic rod-like micelles (bottom). Reproduced with permission from ref. 135. | ||
Cholesterol-modified compounds can also be mixed with others, and self-assembled into different macroscopic vesicles. For example, the nucleoside 2′-deoxy-2′-aminouridine was modified with cholesterol, and the lipophilic nucleoside 2′-N-(2-(cholesteryl)-succinyl)-20-deoxy-20-aminouridine was obtained. When mixed with the unsaturated phospholipid dioleoylphosphatidylcholine, microtubes were formed. From confocal fluorescence microscopy images, the tubes appeared as open-ended cylindrical structures with outer diameters between 2 and 3 μm and lengths between 20 and 40 μm.138 In another report, the dialysis of a dimethyloxide solution of cholesterol-modified dextran (Chol-Dex) and PLA against water yielded hollow polymer nanocapsules with a highly stable structure and relatively narrow size distribution.139 The hollow capsules were thought to be formed by anchoring of most of the amphiphilic polysaccharide onto the surface of swelled aggregates as a result of phase separation of amphiphilic Chol-Dex from hydrophobic PLA. The capsules obtained have potential application as drug carriers or as biomembrane models.
We recently reported the self-assembly of an amphiphilic block copolymers containing cholesterol as the hydrophobic block into polymersomes (Fig. 5).94 We synthesised an ABA triblock copolymer with the ‘A’ segment consisting of poly(NAM), a non-ionic hydrophilic polymer with low-fouling properties similar to PEG, and the ‘B’ segment of poly(CA) as the hydrophobic part of the triblock copolymer. We demonstrated the stable, non-aggregated assembly of polymersomes with narrow polydispersity. We used these polymersomes as reservoirs in poly(vinyl alcohol) composite hydrogels and showed a significant decrease in viability of adhering myoblast cells when a cytotoxic compound was entrapped in the polymersomes, thus confirming the potential of this system in surface-mediated drug delivery, a biomedical approach which aims to deliver therapeutic compounds from the surface of an implantable device or a tissue culturing scaffold to adhering cells or to the surrounding media.
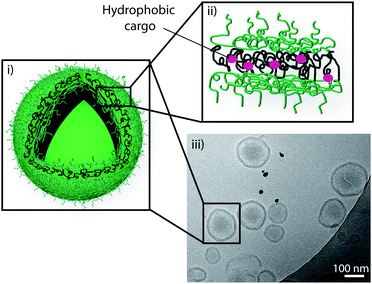 | ||
| Fig. 5 Schematic representation of a polymersome (i) assembled from an ABA triblock copolymer consisting of poly(N-acryloyl morpholine) and poly(cholesteryl acrylate) with entrapped cargo in the hydrophobic part of its polymer membrane (ii), and a cryo-TEM image confirming the assembly of polymersomes (iii). Reproduced with permission from ref. 94. | ||
In summary, based on the hydrophobic interaction, cholesterol-modified polymers can self-assemble into different micelles, nanoparticles, nanotubes, nanogels, or polymersomes. The size, shape and mechanical properties of these assemblies are designed for encapsulation, storage, release and delivery of therapeutics with potential application in biomaterials and biomedicine.
Cholesterol and liposomes
The final part of this feature article reviews the benefits of cholesterol incorporated into liposomes for biomedical applications. Two different aspects of cholesterol, namely its ability to act as stabiliser for lipid membranes and its ability to link lipid and polymeric structures together, are discussed. Examples of applications in drug delivery and biosensing will be outlined for both aspects.Cholesterol as a stabiliser
Liposomes are among the prominent candidates for various biomedical applications in bionanotechnology. Liposomes are spherical vesicles which consist of one or more lipid bilayer membrane(s), encapsulating a volume of aqueous medium (Fig. 6a).140 Glycerophospholipids, sphingolipids, and cholesterol are the predominant components used for their preparation. Their structural similarity to natural cell membranes,141 and their ability to mimic the behaviour of biological cell membranes make liposomes useful in various applications ranging from drug delivery,142,143 cosmetic,144 and food science145 to agriculture.145 However, control over the lipid bilayer permeability and stability towards solutes or biomolecules is often a challenge and can compromise their applications in the aforementioned fields. This aspect can be regulated by adjusting the cholesterol content of the lipid bilayer,146,147 since cholesterol affects the physicochemical properties of the lipid membrane. For an in-depth discussion, the reader is referred to the reviews by Ohvo-Rekilä et al. or Rog et al.148,149 The amphiphilic cholesterol is arranged in the lipid membrane by orienting the steroid ring parallel to the hydrocarbon chains of the lipids (phospholipids or sphingolipids) and the hydroxyl group encountering the aqueous phase. There are two physical states of a lipid bilayer in the absence of cholesterol: the gel state at lower temperatures and the liquid-crystalline state at higher temperatures. The individual lipid molecules of a lipid bilayer have temperature-dependent mobility (fluidity). All lipids have a characteristic temperature in which they undergo a transition from the gel to the liquid-crystalline phase, which is known as phase transition temperature (Tm). The phase behaviour of the lipid bilayer and thus Tm is determined by the van der Waals (VDW) interactions between adjacent lipid molecules. The interaction between the acyl chains of the lipids is mainly governed by two factors: the length of the chain and the packing of the lipids in the bilayer, i.e. longer tail lipids have more area to interact, which will, in turn, increase the strength of the interaction and, consequently, decrease the mobility of the lipid. The type of acyl chain (degree of saturation) plays an important role in how the lipids bind in the bilayer, since an unsaturated double bond can produce a kink in the alkane chain which will create extra free space within the bilayer, thus allowing for additional flexibility in the adjacent chains. Therefore, unsaturated lipids have a significantly lower Tm compared to saturated lipids. Incorporation of increasing amounts of cholesterol into the lipid bilayer induces an “intermediate” state by increasing and decreasing the fluidity of the hydrocarbon chain below and above Tm, respectively, and eventually eliminating Tm entirely. In the gel phase, the presence of cholesterol in the membrane weakens the VDW interactions between the hydrocarbon chains of the fatty acids and prevents lipid crystallisation, while in the liquid-crystalline phase, the hydrocarbon chains of the lipids which interact with the rigid, planar rings of the cholesterol, are partly immobilised. Specifically, cholesterol increases the degree of orientation and reduces the rate of motion in the liquid-crystalline phase of the lipid membrane. This leads to a laterally condensed membrane with increased packing density and, as a consequence, higher mechanical strength and lower permeability. Nature makes use of this aspect in many ways, but nanotechnological applications also rely strongly on this effect as outlined in the subsequent paragraphs.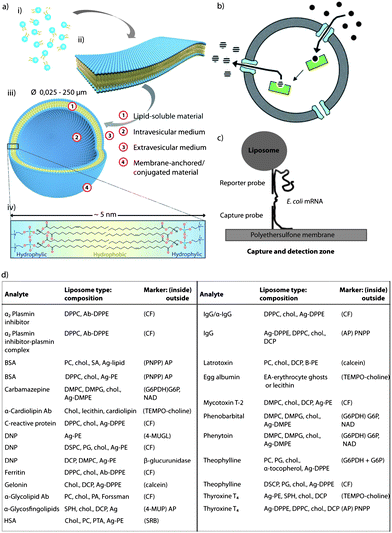 | ||
| Fig. 6 Cholesterol as stabiliser: (a) schematic illustration of the fundamental self-assembly process from individual phospholipid molecules (i) to bilayer membrane leaflets (ii), followed by transformation into liposomes (iii). A single bilayer consists of arranged individual lipid molecules with their hydrophobic tails facing each other and their hydrophilic head-groups facing toward the internal and external aqueous media (iv). Adapted with permission from ref. 140. (b) Schematic representation of liposomes with encapsulated enzymes. Substrates diffuse into the void of the liposome through porin channels, react with the enzymes, and the product is released back into the solution through diffusion. Reproduced with permission from ref. 141. (c) Schematic representation of a biosensor assay where a DNA capture probe is immobilised on a polyethersulfone membrane and a DNA reporter probe is coupled to the surface of a liposome. When a specific E. coli mRNA is present, a sandwich is formed between the capture probe, the E. coli mRNA and the reporter probe, capturing the liposome in the capture/detection zone. The number of liposomes is directly proportional to the amount of E. coli mRNA present. Reproduced with permission from ref. 203. (d) Table containing most common homogeneous complement mediated liposome immunoassays. Reprinted with permission from ref. 205. (ab, antibody; ag, antigen; BSA, bovine serum albumin; CF, carboxyfluorescein; chol., cholesterol; DCP, dicetylphosphate; DMPC, dimyristoylphosphatidylcholine; DMPG, 1,2-dimyristoyl-sn-glycero-3-phospho-(1′-rac-glycerol); DNP, dinitripheno(y)l(ated); DPPC, dipalmitoylphosphatidylcholine; DPPE, dipalmitoylphosphatidylethanolamine; DSPC, distearoylphosphatidylcholine; EA, egg albumin; HSA, human serum albumin; PA, palmitic acid; PC, phosphatidylcholine; PE, phosphatidylethanolamine; SA, stearylamine; and SPH, sphingomyeline). | ||
Drug delivery
Liposomes have been considered as drug delivery vehicles due to their colloidal size, ease of preparation, controllable surface and membrane properties, large carrying capacity, and biocompatibility for over 30 years.150 Since the liposomal shell can enclose or bind many different classes of substances, liposomes are successfully employed as therapeutic agents for the delivery of antibacterial,151 antiviral,152 and anticancer drugs,153 as well as hormones,154 enzymes,155 and nucleotides.153,156 However, two of the major drawbacks of liposomes as drug carriers are their often poor stability in blood circulation upon systemic application and the sometimes suboptimal control over cargo retention and release.157 In the latter case, drugs have to be transported to their site of action quantitatively, or the therapeutic cargo has to be gradually released.146,147Cholesterol has served to overcome both of the above-mentioned challenges. Since the addition of cholesterol reduces the fluidity of the lipid bilayer and increases the stability of the liposomal membrane,158 adjusting the cholesterol content of liposomes has been one of the prime ways to control the permeability to solutes both in vivo and in vitro. Typically, half-lives of passively encapsulated drugs range from minutes, in egg phosphocholine liposomes, to a dozen hours for cholesterol-containing liposomes.159 Owing to this increased cargo retention, the first liposomal formulation of the antitumor drug Doxorubicin (Doxil®) was approved in 1995 by the Food and Drug Administration (FDA) for medical use in oncology.160,161 Other cholesterol-containing liposomal formulations of drugs, i.e. Daunorubicin (DaunoXome®) used for the treatment of advanced HIV-associated Kaposi's sarcoma,162 Amphotericin B (AmBisome®, Amphotec®, and Ableet®) for the treatment of fungal infections in febrile neutropenic patients as well as for the treatment of aspergillosis, candidiasis, and cryptococcosis infections refractory to Amphotericin B,163,164 and cytarabine (DepoCyt®) for the treatment of lymphomatous meningitis,165 were FDA approved in 1996 (DaunoXome®), 1997 (AmBisome®), and 1999 (DepoCyt®), respectively, and are still on the market.164,166–168
Liposomal formulations containing cholesterol of different drugs, such as vincristine sulfate169 or cisplatin (Lipoplatin),170 have not been FDA approved yet but are on Phase II and Phase III clinical trials, respectively.
Analytical science
Interest in liposome technologies and applications for analytical purposes is constantly growing, and four major areas of activity can be identified, namely liquid chromatography (LC), capillary electrophoresis (CE), biosensors, and immunoassays.140 In all four analytical techniques, cholesterol plays an important role in liposome stabilisation.Here, just two examples to illustrate the importance of cholesterol are discussed. Wiedmer et al. employed 1-palmitoyl-2-oleyl-sn-glycero-3-phosphocholine (POPC) and POPC/PS liposomes with various amounts of cholesterol as carriers and steroidal sex hormones as model analytes. The study investigated the influence of cholesterol on the sizes and electrophoretic mobility of POPC and POPC/PS liposomes with special focus on the influence of cholesterol on possible selective interactions between the steroids and the liposomal membranes. Their results showed that the presence of cholesterol slightly increased the liposomal diameter, and significant differences in selectivity between the steroids and the liposomal membranes were achieved through the addition of cholesterol.186 Alternatively, liposomes have been used as coating materials in CE in order to mimic biological cell membranes, since cell membranes are mainly composed of phospholipids and cholesterol, and the interaction between the liposomes and the analytes has been studied.185,187 As an example, Hautala and collaborators showed the coating of silica capillaries with anionic liposomes comprising POPC, bovine brain PS, and cholesterol, and evaluated the coating effectiveness by examining the interactions of steroids with the coated capillaries. Their results show that the capillaries can be successfully coated with liposomes and those coated capillaries can be used to study the interactions between phospholipid bilayers and uncharged steroids. Since the steroids are neutral under the applied conditions, the separation is based on hydrophobic interactions with the liposome coating.187
An interesting example of cholesterol-containing liposomes in this context was reported by McNamara and co-workers, who developed a fluorescence-based oxygen nanosensor by encapsulating an oxygen-sensitive dye in the internal compartment of the liposomes.201 A major concern when developing liposomal fluorescence-based sensors is leaking of the dye molecules from the supportive matrix. To this end, McNamara and collaborators prepared liposomes with a lipid composition consisting of dimyristoylphosphatidylcholine, cholesterol, and dihexadecyl phosphate and showed high stability toward dye leaking at room temperature for 8 days, a fact which could be attributed to the addition of cholesterol to the lipid membrane.
Since safety of water and food supplies has to be ensured to avoid contamination of infectious agents, the detection of pathogenic organisms remains a crucial aspect. With the aim to address this issue, Baeumner et al. developed a nucleic acid based sensor, also known as a genosensor or gene probe, which is a highly sensitive and specific RNA biosensor for the rapid detection of viable Escherichia coli (E. coli) as an indicator of pathogenic organisms in water.203 Briefly, the mRNA from E. coli was extracted, purified, amplified, and then quantified with the biosensor. The biosensor is a membrane-based DNA/RNA hybridisation system and cholesterol-containing liposomes (a mixture composed of dipalmitoyl phosphatidyl choline (DPPC), cholesterol, dipalmitoyl phosphatidyl glycerol, and dipalmitoyl phosphatidyl ethanolamine) encapsulating sulphorhodamine for signal amplification. The method is based on the formation of a sandwich between a DNA-capture probe immobilised on a polyethersulfonate membrane, the target mRNA sequence, and a DNA-reported probe coupled to the surface of a liposome (Fig. 6c). When a specific E. coli mRNA is present, a sandwich is formed between the DNA capture probe attached onto the polyethersulfonate membrane, the E. coli mRNA, and the reporter probe attached to the liposomes, thus the liposomes being captured in the capture/detection zone. Since the number of liposomes is directly proportional to the amount of E. coli mRNA, when non-specific RNA is present instead of E. coli mRNA, the liposomes are not captured in the capture/detection zone, and no signal is reported in the detection zone.203
Fig. 6d depicts a table with the most commonly used liposomal compositions and the markers used to detect different analytes. As shown in the table, most liposomal compositions contain cholesterol to equip the lipid bilayer with additional stability.205 For instance, Kim and collaborators reported the creation of a liposome immunoassay for the detection of gentamicin using phospholipase C to release the fluorescent marker from the liposomes.206,207 Gentamicin, a drug frequently used for the treatment of serious Gram-negative microbial infections, was chosen as a model analyte.206 The liposomes were composed of a mixture of DPPC and cholesterol, since it was previously demonstrated that this composition allows the lysis of the liposomes by the lowest concentration of phospholipase C.206,207
Cholesterol as anchor
So far, the aspect of the cholesterol's ability to affect the membrane properties of the liposomes and the consequences for different biomedical applications have been discussed. This section considers the use of cholesterol for a different purpose, namely as an anchor either to immobilise the liposomes to a surface for biosensing applications, or to coat liposomes with specific molecules.Biosensors
Liposomes hold enormous potential for biosensing applications, not only as labels as discussed previously, but also for the establishment of a successful platform for high-throughput (membrane) protein sensing.208 Immobilised liposomes in an array format can act as supports for (membrane) proteins since the lipid bilayer provides a favorable environment that allows protein flexibility and movement while avoiding their denaturation and loss of function. These sensors are of paramount importance since proteins play a major role in disease developments and 60–70% of the drug targets are (membrane) proteins.209Approaches to immobilise intact liposomes include linkages such as avidin–biotin,210,211 interaction between antibody and antigen,212,213 disulfide bonds,214 or the chemical linkage between histidines,215 but also the chemisorption of vesicles onto gold films.214 However, when these types of linkages are used, the controlled immobilisation of different types of liposomes to predefined spots on the surface is impossible. This challenge has been addressed by tethering liposomes to solid supports via DNA-directed coupling, an approach that was first introduced by Niemeyer and co-workers.216
Several strategies have been developed for the DNA-labeling of intact vesicles by using functionalised lipids such as thiol-reactive maleimido-lipids217,218 and thiol derivatives of DNA.219 Alternately, several approaches which make use of cholesterol-modified DNA for spontaneous anchoring of liposomes to (patterned) substrates with immobilized complementary strands have been reported (Fig. 7ai).220–223 This non-covalent approach is advantageous compared to the previously mentioned concepts since it is largely independent of the lipid composition of the liposomes, does not require chemical modification of a lipid, has fast coupling rates, and there is no-need of introducing functionalised lipids. Therefore, by making use of a naturally occurring membrane constituent, unwanted side reactions between the functional groups and other membrane constituents are eradicated. However, cholesterol-based anchoring of DNA is relatively weak, i.e. it is reversible. In order to allow tagging of a type of liposomes with a specific DNA sequence, the cholesterol anchor has to be strengthened by using bivalent cholesterol DNA coupling.224 This approach allows sorting of red and green liposomes from a mixture (Fig. 7aii). In a continuation of this approach, ligand binding to liposome arrays equipped with G-protein coupled receptors has been demonstrated on the way to develop this platform into a heterogeneous, self-sorting biosensing platform (Fig. 7b).225
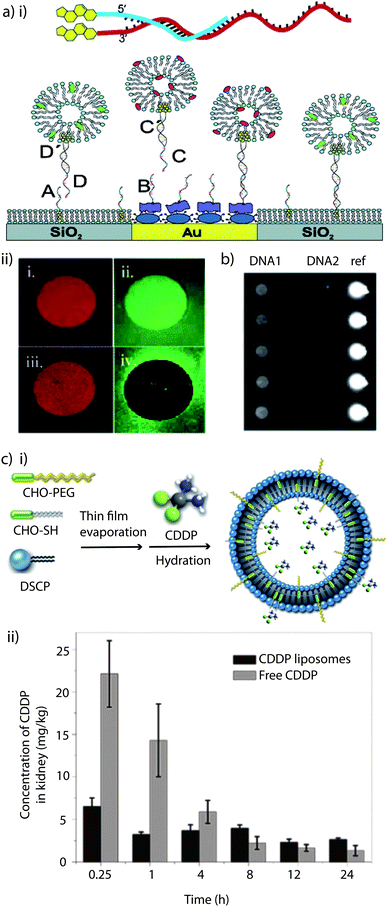 | ||
| Fig. 7 Cholesterol as anchor: (a) (i) schematic illustration of a tethered vesicle–DNA assembly, showing how the hybridisation of complementary cholesterol–DNA pairs forms the (mobile) tether. The DNA array was produced by preferential adsorption of biotin–BSA to Au spots, leaving the surrounding SiO2 substrate available for spontaneous supported phospholipid bilayer (SPB) formation, followed by the addition of NeutrAvidin, which binds to biotin–BSA-modified Au only. Biotin–DNAB and chol–DNAA specifically binds to NeutrAvidin/biotin–BSA/Au and SPB/SiO2, respectively, followed by the immobilization of liposomes tagged with the complementary DNA. (ii) Micrographs illustrating sorting of liposomes tagged with different DNA. Micrographs (1) and (2) were obtained by exposing the DNA-modified substrate to a mixture of red-labelled and green-labelled liposomes tagged with different monovalent coupled chol–DNA. (3) and (4) shows an identical experiment, but for a liposome mixture using bivalent coupled chol–DNA. Reprinted with permission from ref. 224. Copyright (2005) American Chemical Society. (b) Site-specific immobilisation of liposomes containing GPCRs is shown using ZeptoREADER imaging. Reprinted with permission from ref. 225. (c) (i) Schematic illustration of the preparation of cisplatin, (cis-diammine dichloroplatinum(II); CDDP) liposomes. Adapted with permission from ref. 158. (ii) In vivo CDDP concentration in kidney tissue of normal male Chinese Kun Ming (KM) mice, at predetermined time intervals after injection of CDDP loaded liposomes and free CDDP. Reprinted with permission from ref. 158. | ||
PEGylation
One of the main drawbacks of liposomes in drug delivery is their low stability in systemic circulation. Stealth liposomes were created when it was realised that neither mechanical nor electrostatic stabilisation could improve their performance significantly enough.157 It has been widely demonstrated that the incorporation of glycolipids such as monosialoganglioside GM1226 into the lipid bilayer or the coating of liposomes with hydrophilic polymer chains like polysaccharides226 or, preferentially, PEG226,227 becomes an efficient stabilisation tool by providing a steric barrier. The particular efficiency of surface-attached PEG chains has been explained by the combination of their water solubility and flexibility, which sterically stabilises the liposomes by preventing them from self-aggregation and fusion, as well as from opsonin adsorption, the first step towards clearance by the reticuloendothelial system.226–228 PEG coated liposomes remain in the blood for up to 100× longer than ordinary liposomes, thus increasing the pharmacological efficacy of the encapsulated agents,157 and passive targeting to tumors has been reported.227 The reader is referred to recent reviews229–232 for a more detailed discussion on the topic of PEGylated liposomes.There are different ways to coat liposomes with PEG such as by grafting it to the lipids by covalent bonding.226 Alternatively, cholesterol or phospholipids have been employed to anchor PEG to the liposome membrane, and it has been shown that cholesterol-PEG was easily incorporated, making cholesterol the more successful anchor moiety.104,158,226–228,233–235 They speculated that long anchor units are difficult to insert into the liposomal membranes or that there is a higher affinity between cholesterol and the constituent lipids.227 PEG-cholesterol has the added benefit that it confers cohesion on lipid bilayers by inducing tighter molecular packing and restricting the motions of the hydrocarbon chains, thus improving the cargo retention.226 Long-circulating liposomes containing cholesterol–PEG have already been successfully used in the cisplatin delivery in vitro and in vivo.158Fig. 7ci depicts a schematic illustration of liposomes containing a mixture of cholesterol-PEG and thiolated cholesterol for cisplatin encapsulation. The in vivo cisplatin concentrations in the kidney tissue of Chinese Kun Ming mice after injection of the liposome formulation and free cisplatin were compared (Fig. 7cii). The results showed that PEGylated liposomes could successfully avoid the peak-concentration in kidney aroused from free cisplatin, reducing the in vivo acute nephrotoxicity.158
Capsosomes
Sub-compartmentalised systems hold tremendous potential as cell mimics, which is an approach that aims to substitute for lost or missing cellular function.236 Artificial multi-compartment systems do not need to possess the entire complexity of biological cells, but a specific key feature, which will mostly depend on the application in mind (e.g. enzyme encapsulation for enzyme replacement therapy to provide a biological-inspired long term solution for patients with chronic diseases). Mimicking the hierarchical sub-compartmentalised structure of cells is essential to perform triggered, spatially separated, encapsulated (enzymatic cascade) reactions.52,237 Among the reported multi-compartment systems (e.g. vesosomes,238 liposomes within a liposome carrier, polymersomes within a polymersome,239 polymer capsule within a polymer capsule,240,241 polymer hydrogel capsules containing cubosomes242 or polymersomes243 and cellosomes,244 yeast cells associated with a polymer membrane)245,246 capsosomes, intact liposomes embedded into a polymer carrier capsule,52 are a novel, well-characterised system with potential in therapeutic cell mimicry. Capsosomes, as a dual composition system, can take advantage of the different properties of the sub-compartments and the carrier capsule. Liposomes, due to the inherently high impermeability of their lipid membranes, are ideal to encapsulate small or fragile cargo. The polymeric carriers, due to their semi-permeable nature, permit the interaction between the internal and the external environment while providing the structural integrity and help to overcome some of the limitations of liposomes such as instability under in vivo conditions and the lack of control over their degradation. Capsosomes are assembled via the LbL technique,247 by alternatingly depositing polymer and liposomes onto silica particles, followed by the assembly of the polymer carrier capsule and core dissolution (Fig. 8a).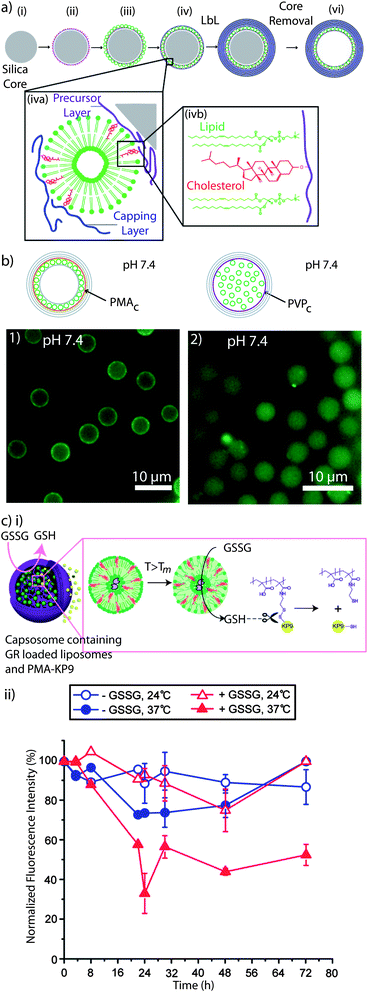 | ||
| Fig. 8 Capsosomes: (a) schematic illustration of the capsosome assembly. A silica particle (i) is coated with a cholesterol-functionalised polymer precursor layer (ii), liposomes (iii), and a cholesterol-functionalised polymer capping layer (iv), followed by subsequent polymer multilayer deposition (v), and removal of the silica template core (vi). Liposomes are adsorbed onto the polymer surface non-covalently with cholesterol-modified polymers (iva). Cholesterol is spontaneously incorporated into the lipid membrane (ivb). Reprinted with permission from ref. 99. (b) Fluorescence microscopy images of capsosomes containing fluorescently labelled liposomes using PMAc (1) or PVPc (2) as precursor and capping layers. While capsosomes assembled using PMAc contain the liposomal sub-units attached onto the carrier membrane, PVPc promotes the free-floating of the liposomes. Reprinted with permission from ref. 93. (c) (i) Release of encapsulated oligopeptide KP9 from capsosomes triggered by glutathione reductase (GR). Upon increasing the T above the Tm of the liposomal subunits, the entrapped GR interacts with the oxidised substrate glutathione (GSSG) and it is converted into its reduced product (GSH) which, in turn, induces the release of KP9 by reducing the disulfide bond. (ii) Normalised fluorescence intensity of capsosomes encapsulating GR within the liposomal subcompartments and fluorescently labelled KP9 upon incubation with GSSG and NADPH as a function of time. A decrease in the fluorescence intensity indicates a release of KP9. Reprinted with permission from ref. 253. | ||
With the aim of stably anchoring the liposomes into the polymer membrane, we employed cholesterol-modified polymers, i.e. cholesterol-containing PLL (PLLc),52,99 poly(methacrylic acid-co-cholesteryl methacrylate) (PMAc),52,99 and poly(N-vinyl pyrrolidone-block-cholesteryl acrylate) (P(NVPc)).93 We also showed that PMAc was the more successful polymer to anchor liposomes than PMA containing oleic acid (PMAoa).248 Interestingly, while PMAc, PMAoa, and PLLc led to the formation of capsosomes with the liposomal units associated with the polymer carrier capsule (Fig. 8b1), P(NVPc) promoted the “free-floating” of the liposomal sub-units in the void of the carrier capsule (Fig. 8b2).93 Since P(NVPc) is a cholesterol-containing uncharged block copolymer and contains the cholesterol moieties in a short block, while the others are copolymers, we speculated that in the former case the entanglement of the liposome-anchoring polymer with the carrier capsule is minimised and, therefore, allows for the detachment of the liposomes from the polymer membrane. The cholesterol-modified polymers allowed us to assemble capsosomes with up to 160000 liposomes within a 3 μm diameter carrier capsule.249 Further, P(NVPc), PMAc, or PMAoa, allowed for the subsequent assembly of the carrier capsule using the polymer pair P(NVP) and thiol-modified poly(methacrylic acid) PMASHvia hydrogen bonding, followed by crosslinking of the thiols in the film250 and P(NVP) release at physiological conditions upon disruption of the hydrogen bonds.251 Their functionality and their potential for biomedical applications were confirmed by encapsulating both hydrophilic52,93,99,249 and hydrophobic248,252 cargo within the liposomal sub-compartments and the demonstration of the payload activity in cell viability assays248,252 and for encapsulated enzymatic catalysis.52,93,253 A particularly interesting example is a two-step catalytic reaction in which the production of a substrate was employed to subsequently trigger cargo release by co-encapsulating glutathione reductase (GR) within cholesterol-containing liposomes and a disulfide-linked polymer–oligopeptide conjugate within the polymer carrier capsule (Fig. 8ci).253 By incubating these capsosomes with oxidised glutathione (GSSG) and by increasing the temperature above the Tm of the liposomal subunits, the entrapped enzyme was allowed to interact with the substrate GSSG and converted it into its reduced product (GSH) which subsequently induced the release of the encapsulated fluorescent oligopeptides from the capsosomes by reducing the disulfide bond, as shown by the decrease in fluorescence intensity over time (Fig. 8cii). This result brings capsosomes a step closer to potentially address a real medical problem by continuously generating a potent antioxidant while releasing a small molecule with therapeutic relevance.
Conclusions and future perspectives
This feature article gives an overview over the current use of cholesterol in bionanotechnology, including some fundamental aspects in cholesterol chemistry and polymer chemistry, the application of these polymeric materials into self-assembled structures, and the development of higher order nano-structures, along with their use in biosensing, analytical sciences, drug delivery, and cell mimicry.In terms of cholesterol chemistry, cholesterol chloroformate is commonly used to conjugate cholesterol. Further, esterification and etherification of the cholesterol's hydroxyl group are common ways to conjugate this molecule. A variety of cholesterol-containing end-functional polymers, homopolymers, and co- and block copolymers have been synthesised and used for the self-assembly of different micelles, nanoparticles, nanotubes, nanogels, or polymersomes. An interesting avenue that has not been investigated is conjugation onto cholesterol in other positions, besides the obvious hydroxyl, and how this effects its self-assembly and function. Also, the fine-tuning of the self-assembly process of cholesterol-modified polymers to yield structures with increased complexity would be another aspect to consider in the future. Although there are a few examples where the self-assembled nanostructures have been used in vivo, many of them were only examined in vitro using a model drug, if at all. A future challenge will be to consider specific medical conditions which require therapeutic compounds to be delivered and to assess the pharmacokinetics and pharmacodynamics of the cholesterol based delivery vehicle under biologically relevant conditions. An important question to address would be if the cholesterol-based delivery vehicles would inherently have an impact on cells or the health of animals in vivo.
Cholesterol has the ability to self-assemble from multi-component solutions. Many fundamental aspects are being understood and many applications are being thought of. The future challenge here will be the stabilization and post-modification of helical ribbons in order to integrate them in devices and use them as a tool in nanotechnology e.g. as nano-springs.
Cholesterol incorporated into liposomes also plays an important role in biomedical applications. Its ability to modulate the physicochemical properties of lipid bilayer has been employed to stabilise liposomes for their application in drug delivery or analytical science. In the former case, in order to make more liposome-based formulations commercially available for clinical applications, alternative concepts for stabilization and functionalization will have to be conceived. While cholesterol will remain an integral building block, cholesterol-modified polymers, instead of (cholesterol-)modified lipids, as a way to improve the stability and control over cargo retention and release, should be considered more. In analytical science, the use of cholesterol has a well-defined task, i.e. to stabilize the membrane. The future challenge in this context remains to successfully move from proof of concepts in research lab to commercially available products.
Further, cholesterol can be used to anchor other molecules such as PEG or DNA to the liposomes for their application in biosensing or as “stealth” drug carriers. In the latter case, the challenges are similar to the ones outlined above. One of the aims when using DNA-tagged liposomes in biosensing is to assemble a single-liposome array with the liposomes carrying different membrane proteins, and to find a means to analyse the protein–analyte interaction in a fast, quantitative and sensitive way. This would contribute considerably to the early detection of disease and identification of suitable drug candidates.
Cholesterol-modified polymers were found to be essential in the assembly of capsosomes as simple cell mimics. These have been well-characterized from a material point of view and diverse functionality has been demonstrated. In our research, we are convinced that capsosomes have the potential to address clinical issues, when the proper route of administration and the suitable cell type to mimic is chosen. However, we have yet to perform in vivo experiments and to consider actual medical conditions. These are major future challenges to be considered for further development of capsosomes in biomedicine.
Overall, cholesterol is a small molecule which has had a great impact in all the fields presented, and it will remain an indispensable player in various areas of bionanotechnology.
Acknowledgements
This work was supported by a Sapere Aude Starting Grant from the Danish Council for Independent Research, Technology and Production Sciences, Denmark.Notes and references
- D. S. Goodsell, Bionanotechnology: Lessons from Nature, Wiley, 2004 Search PubMed.
- J. Wang, ACS Nano, 2009, 3, 4–9 CrossRef CAS.
- S. J. Ebbens and J. R. Howse, Soft Matter, 2010, 6, 726–738 RSC.
- R. Dreyfus, J. Baudry, M. L. Roper, M. Fermigier, H. A. Stone and J. Bibette, Nature, 2005, 437, 862–865 CrossRef CAS.
- L. Zhang, K. E. Peyer and B. J. Nelson, Lab Chip, 2010, 10, 2203–2215 RSC.
- H. Hess, in Annual Review of Biomedical Engineering, ed. M. L. Yarmush, J. S. Duncan and M. L. Gray, 2011, vol. 13, pp. 429–450 Search PubMed.
- N. Michelotti, A. Johnson-Buck, A. J. Manzo and N. G. Walter, Wiley Interdiscip. Rev.: Nanomed. Nanobiotechnol., 2012, 4, 139–152 Search PubMed.
- R. M. Zadegan and M. L. Norton, Int. J. Mol. Sci., 2012, 13, 7149–7162 Search PubMed.
- A. Lakshmanan, S. G. Zhang and C. A. E. Hauser, Trends Biotechnol., 2012, 30, 155–165 CrossRef CAS.
- X. Zhang, S. Boisse, C. Bui, P.-A. Albouy, A. Brulet, M.-H. Li, J. Rieger and B. Charleux, Soft Matter, 2012, 8, 1130–1141 RSC.
- J. M. Schnur, Science, 1993, 262, 1669–1676 CrossRef CAS.
- D. Landesman-Milo and D. Peer, J. Controlled Release, 2012, 161, 600–608 Search PubMed.
- M. S. Brown and J. L. Goldstein, Science, 1986, 232, 34–47 CAS.
- M. E. Chevreul, Ann. Chim. Phys., 1816, 2, 339–372 Search PubMed.
- A. de Fourcroy, Ann. Chim., 1789, 3, 242–252 Search PubMed.
- M. E. Chevreul, Ann. Chim., 1815, 95, 5–50 Search PubMed.
- M. Berthelot, Ann. Chim. Phys., 1859, 56, 51–98 Search PubMed.
- O. Diels and E. Abderhalden, Ber. Dtsch. Chem. Ges., 1904, 37, 3092–3103 Search PubMed.
- J. D. Bernal, Nature, 1932, 129, 277–278 CAS.
- A. Windaus and K. Neukirchen, Ber. Dtsch. Chem. Ges., 1919, 52, 1915–1919 Search PubMed.
- A. Windaus, Hoppe-Seyler's Z. Physiol. Chem., 1932, 213, 147–187 Search PubMed.
- P. L. Yeagle, The Membranes of Cells, Academic Press, 1993 Search PubMed.
- P. L. Yeagle, Biochim. Biophys. Acta, 1985, 822, 267–287 CAS.
- P. L. Yeagle, The Structure of Biological Membranes, CRC Press, Boca Raton, 2005 Search PubMed.
- D. M. Medeiros and R. E. C. Wildman, Advanced Human Nutrition, Jones & Bartlett Publishers, 2011 Search PubMed.
- C. J. Fielding and P. E. Fielding, J. Lipid Res., 1995, 36, 211–228 Search PubMed.
- F. R. Maxfield and D. Wustner, J. Clin. Invest., 2002, 110, 891–898 CAS.
- F. J. Field, E. Born, S. Murthy and S. N. Mathur, J. Lipid Res., 1998, 39, 333–343 Search PubMed.
- J. Vogel, The Pathological Anatomy of the Human Body, H. Bailliere, 1847 Search PubMed.
- M. R. Hayden and S. C. Tyagi, Cardiovasc. Diabetol., 2004, 3, 1 Search PubMed.
- P. R. Moreno, J. Sanz and V. Fuster, J. Am. Coll. Cardiol., 2009, 53, 2315–2323 Search PubMed.
- C. Doree, Biochem. J., 1909, 4, 72–106 Search PubMed.
- W. S. Johnson, Bioorg. Chem., 1976, 5, 51–98 CrossRef CAS.
- R. E. Ostlund and D. E. Matthews, J. Lipid Res., 1993, 34, 1825–1831 Search PubMed.
- L. J. Goad and T. Akihisa, Analysis of Sterols, Blackie Academic & Professional, 1997 Search PubMed.
- J. L. Goldstein and M. S. Brown, Nature, 1990, 343, 425–430 CrossRef CAS.
- W. D. Nes, Chem. Rev., 2011, 111, 6423–6451 Search PubMed.
- K. Simons and W. L. C. Vaz, Annu. Rev. Biophys. Biomol. Struct., 2004, 33, 269–295 CrossRef CAS.
- E. Ikonen, Nat. Rev. Mol. Cell Biol., 2008, 9, 125–138 CrossRef CAS.
- N. B. Javitt, FASEB J., 1994, 8, 1308–1311 Search PubMed.
- D. R. Schmidt, S. R. Holmstrom, K. F. Tacer, A. L. Bookout, S. A. Kliewer and D. J. Mangelsdorf, J. Biol. Chem., 2010, 285, 14486–14494 Search PubMed.
- P. Insel, D. Ross, K. McMahon and M. Bernstein, Nutrition, Jones and Bartlett Publishers, 2004 Search PubMed.
- G. Vanmeer, Annu. Rev. Cell Biol., 1989, 5, 247–275 Search PubMed.
- V. Cherezov, D. M. Rosenbaum, M. A. Hanson, S. G. F. Rasmussen, F. S. Thian, T. S. Kobilka, H.-J. Choi, P. Kuhn, W. I. Weis, B. K. Kobilka and R. C. Stevens, Science, 2007, 318, 1258–1265 CrossRef CAS.
- A. S. Achalkumar, R. J. Bushby and S. D. Evans, Soft Matter, 2010, 6, 6036–6051 RSC.
- M. K. Bijsterbosch, E. T. Rump, R. L. A. D. Vrueh, R. Dorland, R. van Veghel, K. L. Tivel, E. A. L. Biessen, T. J. C. van Berkel and M. Manoharan, Nucleic Acids Res., 2000, 28, 2717–2725 CrossRef CAS.
- S.-O. Han, R. I. Mahato and S. W. Kim, Bioconjugate Chem., 2001, 12, 337–345 CrossRef CAS.
- S. Boisse, J. Rieger, A. Di-Cicco, P. A. Albouy, C. Bui, M. H. Li and B. Charleux, Macromolecules, 2009, 42, 8688–8696 CrossRef CAS.
- A. C. De Visser, K. De Groot, J. Feyen and A. Bantjes, J. Polym. Sci., Part A-1: Polym. Chem., 1971, 9, 1893–1899 Search PubMed.
- A. C. De Visser, J. Feyen, K. De Groot and A. Bantjes, J. Polym. Sci., Part B: Polym. Lett., 1970, 8, 805–808 Search PubMed.
- Y. Zhou and R. M. Kasi, J. Polym. Sci., Part A: Polym. Chem., 2008, 46, 6801–6809 CrossRef CAS.
- B. Städler, R. Chandrawati, A. D. Price, S.-F. Chong, K. Breheney, A. Postma, L. A. Connal, A. N. Zelikin and F. Caruso, Angew. Chem., Int. Ed., 2009, 48, 4359–4362 CrossRef CAS.
- S. Sevimli, S. Sagnella, M. Kavallaris, V. Bulmus and T. P. Davis, Polym. Chem., 2012, 3, 2057–2069 RSC.
- S. Yusa, M. Kamachi and Y. Morishima, Macromolecules, 2000, 33, 1224–1231 CrossRef CAS.
- S.-K. Ahn, L. T. Nguyen Le and R. M. Kasi, J. Polym. Sci., Part A: Polym. Chem., 2009, 47, 2690–2701 CrossRef CAS.
- J.-F. Lutz, S. Pfeifer and Z. Zarafshani, QSAR Comb. Sci., 2007, 26, 1151–1158 Search PubMed.
- J. T. Xu, L. Tao, C. Boyer, A. B. Lowe and T. P. Davis, Macromolecules, 2011, 44, 299–312 CrossRef CAS.
- J. Q. Liu, E. Setijadi, Y. K. Liu, M. R. Whittaker, C. Boyer and T. P. Davis, Aust. J. Chem., 2010, 63, 1245–1250 CrossRef CAS.
- Y. Zhu, Y. Zhou, Z. Chen, R. Lin and X. Wang, Polymer, 2012, 53, 3566–3576 Search PubMed.
- Y. Liu, Y. Wang, D. Zhuang, J. Yang and J. Yang, J. Colloid Interface Sci., 2012, 377, 197–206 Search PubMed.
- D. Smith, S. H. Clark, P. A. Bertin, B. L. Mirkin and S. T. Nguyen, J. Mater. Chem., 2009, 19, 2159–2165 RSC.
- Y. Zhou, S.-k. Ahn, R. K. Lakhman, M. Gopinadhan, C. O. Osuji and R. M. Kasi, Macromolecules, 2011, 44, 3924–3934 Search PubMed.
- G. Stübs, B. Rupp, R. R. Schumann, N. W. J. Schröder and J. Rademann, Chem.–Eur. J., 2010, 16, 3536–3544 Search PubMed.
- J. C. Chabala and T. Y. Shen, Carbohydr. Res., 1978, 67, 55–63 CrossRef CAS.
- Y. Iwasaki and K. Akiyoshi, Biomacromolecules, 2006, 7, 1433–1438 Search PubMed.
- A.-C. Albertsson and I. K. Varma, Biomacromolecules, 2003, 4, 1466–1486 CrossRef CAS.
- T. Zou, S.-X. Cheng and R.-X. Zhuo, Colloid Polym. Sci., 2005, 283, 1091–1099 Search PubMed.
- L. Zhang, Q.-R. Wang, X.-S. Jiang, S.-X. Cheng and R.-X. Zhuo, J. Biomater. Sci., Polym. Ed., 2005, 16, 1095–1108 Search PubMed.
- T. Zou, F. Li, S.-X. Cheng and R.-X. Zhuo, J. Biomater. Sci., Polym. Ed., 2006, 17, 1093–1106 Search PubMed.
- T. Wan, T. Zou, S.-X. Cheng and R.-X. Zhuo, Biomacromolecules, 2005, 6, 524–529 Search PubMed.
- H. A. Klok, J. J. Hwang, S. N. Iyer and S. I. Stupp, Macromolecules, 2002, 35, 746–759 CrossRef CAS.
- H. A. Klok, J. J. Hwang, J. D. Hartgerink and S. I. Stupp, Macromolecules, 2002, 35, 6101–6111 CrossRef CAS.
- K. Sugiyama, R. Hanamura and M. Sugiyama, J. Polym. Sci., Part A: Polym. Chem., 2000, 38, 3369–3377 Search PubMed.
- J. Chiefari, Y. K. Chong, F. Ercole, J. Krstina, J. Jeffery, T. P. T. Le, R. T. A. Mayadunne, G. F. Meijs, C. L. Moad, G. Moad, E. Rizzardo and S. H. Thang, Macromolecules, 1998, 31, 5559–5562 CrossRef CAS.
- G. Moad, E. Rizzardo and S. H. Thang, Acc. Chem. Res., 2008, 41, 1133–1142 CrossRef CAS.
- G. Moad, E. Rizzardo and S. H. Thang, Polymer, 2008, 49, 1079–1131 CrossRef CAS.
- G. Moad, E. Rizzardo and S. H. Thang, Aust. J. Chem., 2009, 62, 1402–1472 CrossRef CAS.
- G. Moad, E. Rizzardo and S. H. Thang, Aust. J. Chem., 2012, 65, 985–1076 Search PubMed.
- M. Kato, M. Kamigaito, M. Sawamoto and T. Higashimura, Macromolecules, 1995, 28, 1721–1723 CrossRef CAS.
- V. Percec and B. Barboiu, Macromolecules, 1995, 28, 7970–7972 CrossRef CAS.
- J.-S. Wang and K. Matyjaszewski, J. Am. Chem. Soc., 1995, 117, 5614–5615 CrossRef CAS.
- D. M. Haddleton, R. Edmonds, A. M. Heming, E. J. Kelly and D. Kukulj, New J. Chem., 1999, 23, 477–479 RSC.
- J. P. Xu, J. Ji, W. D. Chen and J. C. Shen, Macromol. Biosci., 2005, 5, 164–171 CrossRef CAS.
- J. P. Xu, J. Ji, W. D. Chen and J. C. Shen, J. Controlled Release, 2005, 107, 502–512 CrossRef CAS.
- F. Segui, X. P. Qiu and F. M. Winnik, J. Polym. Sci., Part A: Polym. Chem., 2008, 46, 314–326 CrossRef CAS.
- J. Liu, H. Liu, V. Bulmus, L. Tao, C. Boyer and T. P. Davis, J. Polym. Sci., Part A: Polym. Chem., 2010, 48, 1399–1405 CrossRef CAS.
- C. W. Bielawski and R. H. Grubbs, Prog. Polym. Sci., 2007, 32, 1–29 CrossRef CAS.
- S. Sutthasupa, M. Shiotsuki and F. Sanda, Polym. J., 2010, 42, 905–915 CrossRef CAS.
- G. Hardy and K. Nyitrai, Acta Chim. Hung., 1970, 65, 301–310 Search PubMed.
- G. Hardy, K. Nyitrai and F. Cser, Polymerization of Cholesteryl Acrylate in its Smectic Liquid Crystalline State, 1969 Search PubMed.
- S. J. He, Y. Zhang, Z. H. Cui, Y. Z. Tao and B. L. Zhang, Eur. Polym. J., 2009, 45, 2395–2401 Search PubMed.
- Y. J. Liu, Y. Z. Wang, D. Q. Zhuang, J. J. Yang and J. Yang, J. Colloid Interface Sci., 2012, 377, 197–206 Search PubMed.
- L. Hosta-Rigau, S. F. Chung, A. Postma, R. Chandrawati, B. Städler and F. Caruso, Adv. Mater., 2011, 23, 4082–4087 CrossRef CAS.
- L. Hosta-Rigau, B. E. B. Jensen, K. S. Fjeldsø, A. Postma, G. Li, K. N. Goldie, F. Albericio, A. N. Zelikin and B. Städler, Adv. Healthcare Mater., 2012, 1, 791–795 Search PubMed.
- V. Shibaev, M. Ivanov, N. Boiko and E. Chernikova, Dokl. Chem., 2009, 427, 183–185 Search PubMed.
- T. Kaneko, H. Nagasawa, J. P. Gong and Y. Osada, Macromolecules, 2003, 37, 187–191 Search PubMed.
- Z. Hongbo, L. Yaobang, Z. Haoyu and W. Xiaogong, Acta Polym. Sin., 2004, 1, 327–332 Search PubMed.
- S. K. Filippov, P. Chytil, P. V. Konarev, M. Dyakonova, C. Papadakis, A. Zhigunov, J. Plestil, P. Stepanek, T. Etrych, K. Ulbrich and D. I. Svergun, Biomacromolecules, 2012, 13, 2594–2604 Search PubMed.
- R. Chandrawati, B. Städler, A. Postma, L. A. Connal, S.-F. Chong, A. N. Zelikin and F. Caruso, Biomaterials, 2009, 30, 5988–5998 CrossRef CAS.
- S. Sevimli, F. Inci, H. M. Zareie and V. Bulmus, Biomacromolecules, 2012, 13, 3064–3075 Search PubMed.
- L. Yang, B. Zhang, L. Wen, Q. Liang and L.-M. Zhang, Carbohydr. Polym., 2007, 68, 218–225 Search PubMed.
- E. Ranucci, M. A. Suardi, R. Annunziata, P. Ferruti, F. Chiellini and C. Bartoli, Biomacromolecules, 2008, 9, 2693–2704 CrossRef CAS.
- Y. Zhou, V. A. Briand, N. Sharma, S.-k. Ahn and R. M. Kasi, Materials, 2009, 2, 636–660 Search PubMed.
- S.-I. Yusa, Int. J. Polym. Sci., 2012, 2012, 609767–609777 Search PubMed.
- D. S. Chung, G. B. Benedek, F. M. Konikoff and J. M. Donovan, Proc. Natl. Acad. Sci. U. S. A., 1993, 90, 11341–11345 CrossRef CAS.
- F. M. Konikoff, D. S. Chung, J. M. Donovan, D. M. Small and M. C. Carey, J. Clin. Invest., 1992, 90, 1155–1160 CrossRef CAS.
- F. M. Konikoff, D. E. Cohen and M. C. Carey, Mol. Cryst. Liq. Cryst. Sci. Technol., Sect. A, 1994, 248, 291–296 Search PubMed.
- B. Smith, Y. Zastavker and G. Benedek, Phys. Rev. Lett., 2001, 87, 2781011–2781014 Search PubMed.
- Y. V. Zastavker, N. Asherie, A. Lomakin, J. Pande, M. Donovan, J. M. Schnur and J. B. Benedek, Proc. Natl. Acad. Sci. U. S. A., 1999, 96, 7883–7887 CrossRef CAS.
- G. H. M. van der Heijden and E. L. Starostin, Phys. Rev. Lett., 2008, 101, 4.
- B. Khaykovich, N. Kozlova, W. Choi, A. Lomakin, C. Hossain, Y. Sung, R. R. Dasari, M. S. Feld and G. B. Benedek, Proc. Natl. Acad. Sci. U. S. A., 2009, 106, 15663–15666 Search PubMed.
- W. Choi, C. Fang-Yen, K. Badizadegan, S. Oh, N. Lue, R. R. Dasari and M. S. Feld, Nat. Methods, 2007, 717–719 Search PubMed.
- M. Kim, Y. Choi, C. Fang-Yen, Y. Sung, R. R. Dasari, M. S. Feld and W. Choi, Opt. Lett., 2011, 36, 148–150 Search PubMed.
- S.-I. Yusa, M. Kamachi and Y. Morishima, Langmuir, 1998, 14, 6059–6067 Search PubMed.
- Y. Jin, R. Xin, P. Ai and D. Chen, Int. J. Pharm., 2008, 350, 330–337 CrossRef CAS.
- Q. V. Bach, J. R. Moon, Y. S. Jeon, W.-S. Choe and J.-H. Kim, J. Appl. Polym. Sci., 2011, 120, 1685–1693 Search PubMed.
- X. Zhu and M. Nichifor, Acc. Chem. Res., 2002, 35, 539–546 CrossRef CAS.
- C.-S. Chaw, K.-W. Chooi, X.-M. Liu, C.-w. Tan, L. Wang and Y.-Y. Yang, Biomaterials, 2004, 25, 4297–4308 CrossRef CAS.
- X. Liu, K. P. Pramoda, Y. Yang, S. Y. Chow and C. He, Biomaterials, 2004, 25, 2619–2628 CrossRef CAS.
- H. Zeng, Y. Li, H. Zhang and X. Wang, Acta Polym. Sin., 2004, 1, 327–332 Search PubMed.
- E. Akiyama, N. Morimoto, P. Kujawa, Y. Ozawa, F. M. Winnik and K. Akiyoshi, Biomacromolecules, 2007, 8, 2366–2373 CrossRef CAS.
- X. Yuan, H. Li and Y. Yuan, Carbohydr. Polym., 2006, 65, 337–345 CrossRef CAS.
- J. Yu, Y. Li, L. Qiu and Y. Jin, J. Pharm. Pharmacol., 2009, 61, 713–719 CrossRef CAS.
- Y.-S. Wang, Q. Jiang, R.-S. Li, L.-L. Liu, Q.-Q. Zhang, Y.-M. Wang and J. Zhao, Nanotechnology, 2008, 19, 145101–145109 Search PubMed.
- Y. Wang, Q. Jiang, L. R. Liu and Q. Zhang, Polymer, 2007, 48, 4135–4142 Search PubMed.
- Y. Wang, L. Liu, J. Weng and Q. Zhang, Carbohydr. Polym., 2007, 69, 597–606 CrossRef CAS.
- Y.-S. Wang, L.-R. Liu, Q. Jiang and Q.-Q. Zhang, Eur. Polym. J., 2007, 43, 43–51 Search PubMed.
- X. Li, M. Chen, z. Zhou, L. Liu and Q. Zhang, Colloids Surf., B, 2012, 1, 136–141 Search PubMed.
- X. Li, M. Chen, W. Yang, Z. Zhou, L. Liu and Q. Zhang, Colloids Surf., B, 2012, 92, 136–141 Search PubMed.
- C.-S. Chern, H.-C. Chiu and Y.-C. Chuang, Polym. Int., 2004, 53, 420–429 Search PubMed.
- K. Nagahama, T. Ouchi and Y. Ohya, Adv. Funct. Mater., 2008, 18, 1220–1231 CrossRef CAS.
- C. J. Thompson, C. Ding, X. Qu, Z. Yang, I. F. Uchegbu, L. Tetley and W. P. Cheng, Colloid Polym. Sci., 2008, 286, 1511–1526 Search PubMed.
- F. van de Manakker, T. Vermonden, N. el Morabit, C. F. van Nostrum and W. E. Hennink, Langmuir, 2008, 24, 12559–12567 CrossRef CAS.
- D. Yang, J. Zhu, Z. Huang, H. Ren and Z. Zheng, Colloids Surf., B, 2008, 63, 192–199 Search PubMed.
- L. Jia, P.-A. Albouy, A. Di Cicco, A. Cao and M. Li, Polymer, 2011, 52, 2565–2575 CrossRef CAS.
- Y. Wang, C.-Y. Ke, C. W. Beh, S.-Q. Liu, S.-H. Goh and Y.-Y. Yang, Biomaterials, 2007, 28, 5358–5368 Search PubMed.
- J. P. Xu, J. Ji, W. D. Chen and J. C. Shen, in Asbm6: Advanced Biomaterials Vi, ed. X. Zhang, J. Tanaka, Y. Yu and Y. Tabata, 2005, vol. 288–289, pp. 465–468 Search PubMed.
- P. Pescador, N. Brodersen, H. A. Scheidt, M. Loew, G. Holland, N. Bannert, J. Liebscher, A. Herrmann, D. Huster and A. Arbuzova, Chem. Commun., 2010, 46, 5358 RSC.
- L. Long, X. Yuan, J. Chang, Z. Zhang, M. Gu, T. Song, Y. Xing, X. Yuan, S. Jiang and J. Sheng, Carbohydr. Polym., 2012, 87, 2630–2637 Search PubMed.
- A. Jesorka and O. Orwar, Annu. Rev. Anal. Chem., 2008, 1, 801–832 Search PubMed.
- B.-W. Park, D.-Y. Yoon and D.-S. Kim, Biosens. Bioelectron., 2010, 26, 1–10 CrossRef CAS.
- J. Thongborisute, A. Tsuruta, Y. Kawabata and H. Takeuchi, J. Drug Targeting, 2006, 14, 147–154 Search PubMed.
- Y. Gerelli, S. Barbieri, M. T. Di Bari, A. Deriu, L. Cantu, P. Brocca, F. Sonvico, P. Colombo, R. May and S. Motta, Langmuir, 2008, 24, 11378–11384 CrossRef CAS.
- P. M. B. Goncalves Maia Campos, F. B. de Camargo Junior, J. P. de Andrade and L. R. Gaspar, Photochem. Photobiol., 2012, 88, 748–752 Search PubMed.
- T. M. Taylor, P. M. Davidson, B. D. Bruce and J. Weiss, Crit. Rev. Food Sci. Nutr., 2005, 45, 587–605 CrossRef CAS.
- C. Kirby, J. Clarke and G. Gregoriadis, Biochem. J., 1980, 186, 591–598 CAS.
- C. Kirby, J. Clarke and G. Gregoriadis, FEBS Lett., 1980, 111, 324–328 Search PubMed.
- H. Ohvo-Rekilä, B. Ramstedt, P. Leppimaki and J. P. Slotte, Prog. Lipid Res., 2002, 41, 66–97 CrossRef CAS.
- T. Rog, M. Pasenkiewicz-Gierula, I. Vattulainen and M. Karttunen, Biochim. Biophys. Acta, Biomembr., 2009, 1788, 97–121 CrossRef CAS.
- B. J. Boyd, Expert Opin. Drug Delivery, 2008, 5, 69–85 Search PubMed.
- E. A. Trafny, M. Antos-Bielska and J. Grzybowski, J. Microencapsulation, 1999, 16, 419–429 Search PubMed.
- B. Ludewig, F. Barchiesi, M. Pericin, R. M. Zinkernagel, H. Hengartner and R. A. Schwendener, Vaccine, 2000, 19, 23–32 Search PubMed.
- D. D. Lasic, J. J. Vallner and P. K. Working, Curr. Opin. Mol. Ther., 1999, 1, 177–185 Search PubMed.
- M. A. Kisel, L. N. Kulik, I. S. Tsybovsky, A. P. Vlasov, M. S. Vorob'yov, E. A. Kholodova and Z. V. Zabarovskaya, Int. J. Pharm., 2001, 216, 105–114 CrossRef CAS.
- M. M. Gaspar, R. PerezSoler and M. E. M. Cruz, Cancer Chemother. Pharmacol., 1996, 38, 373–377 Search PubMed.
- F. Eckstein, Expert Opin. Biol. Ther., 2007, 7, 1021–1034 Search PubMed.
- D. D. Lasic and D. Papahadjopoulos, Science, 1995, 267, 1275–1276 CrossRef CAS.
- Y. Kuang, J. Liu, Z. Liu and R. Zhuo, Biomaterials, 2012, 33, 1596–1606 Search PubMed.
- B. Ceh, M. Winterhalter, P. M. Frederik, J. J. Vallner and D. D. Lasic, Adv. Drug Delivery Rev., 1997, 24, 165–177 CrossRef CAS.
- Y. C. Barenholz, J. Controlled Release, 2012, 160, 117–134 Search PubMed.
- D. C. Drummond, O. Meyer, K. L. Hong, D. B. Kirpotin and D. Papahadjopoulos, Pharmacol. Rev., 1999, 51, 691–743 CAS.
- Anonymous, J. Int. Assoc. Phys. AIDS Care, 1996, 2, 50–51 Search PubMed.
- G. W. Boswell, D. Buell and I. Bekersky, J. Clin. Pharmacol., 1998, 38, 583–592 Search PubMed.
- K. V. Clemons and D. A. Stevens, Antimicrob. Agents Chemother., 2004, 48, 1047–1050 Search PubMed.
- Anonymous, Biotechnol. Law Rep., 1999, 18, 257 Search PubMed.
- Relevant homepages, http://www.daunoxome.com/, http://www.multivu.com/mnr/54292-galen-chemotherapy-agent-daunoxome-liposomal-daunorubicin-kaposi-s-sarcoma.
- J. R. Azanza Perea and J. Barberan, Rev. Esp. Quimioter., 2012, 25, 17–24 Search PubMed.
- M. C. Chamberlain, J. Neuro-Oncol., 2012, 109, 143–148 Search PubMed.
- D. A. Thomas, H. M. Kantarjian, W. Stock, L. T. Heffner, S. Faderl, G. Garcia-Manero, A. Ferrajoli, W. Wierda, S. Pierce, B. Lu, S. R. Deitcher and S. O'Brien, Cancer, 2009, 115, 5490–5498 Search PubMed.
- G. P. Stathopoulos and T. Boulikas, J. Drug Delivery, 2012, 2012, 581363–581373 Search PubMed.
- S. K. Wiedmer, M. S. Jussila and M. L. Riekkola, TrAC, Trends Anal. Chem., 2004, 23, 562–582 CrossRef CAS.
- A. Gomez-Hens and J. M. Fernandez-Romero, TrAC, Trends Anal. Chem., 2005, 24, 9–19 CrossRef CAS.
- P. Lundahl and Q. Yang, J. Chromatogr., A, 1991, 544, 283–304 CrossRef CAS.
- P. Lundahl and F. Beigi, Adv. Drug Delivery Rev., 1997, 23, 221–227 Search PubMed.
- X. Y. Liu, C. Nakamura, Q. Yang, N. Kamo and J. Miyake, J. Chromatogr., A, 2002, 961, 113–118 CrossRef CAS.
- M. Sandberg, P. Lundahl, E. Greijer and M. Belew, Biochim. Biophys. Acta, Gen. Subj., 1987, 924, 185–192 Search PubMed.
- E. Krause, M. Dathe, T. Wieprecht and M. Bienert, J. Chromatogr., A, 1999, 849, 125–133 CrossRef CAS.
- Q. Yang, X. Y. Liu, M. Yoshimoto, R. Kuboi and J. Miyake, Anal. Biochem., 1999, 268, 354–362 CrossRef CAS.
- Q. Yang, X. Y. Liu, S. Ajiki, M. Hara, P. Lundahl and J. Miyake, J. Chromatogr., Biomed. Appl., 1998, 707, 131–141 CrossRef CAS.
- X. Y. Liu, Q. Yang, C. Nakamura and J. Miyake, J. Chromatogr., Biomed. Appl., 2001, 750, 51–60 CrossRef CAS.
- X. Liu, B. Testa and A. Fahr, Pharm. Res., 2011, 28, 962–977 Search PubMed.
- S. Chapelle and M. Gillesbaillien, Biochim. Biophys. Acta, Lipids Lipid Metab., 1983, 753, 269–271 Search PubMed.
- X. Liu, P. Fan, M. Chen, H. Hefesha, G. K. E. Scriba, D. Gabel and A. Fahr, Helv. Chim. Acta, 2010, 93, 203–211 Search PubMed.
- Y. X. Zhang, R. Zhang, S. Hjerten and P. Lundahl, Electrophoresis, 1995, 16, 1519–1523 CrossRef CAS.
- J. M. Cunliffe, N. E. Baryla and C. A. Lucy, Anal. Chem., 2002, 74, 776–783 CrossRef CAS.
- S. K. Wiedmer, M. S. Jussila, J. M. Holopainen, J. M. Alakoskela, P. K. J. Kinnunen and M. L. Riekkola, J. Sep. Sci., 2002, 25, 427–437 Search PubMed.
- J. T. Hautala, M. V. Linden, S. K. Wiedmer, S. J. Ryhanen, M. J. Saily, P. K. J. Kinnunen and M. L. Riekkola, J. Chromatogr., A, 2003, 1004, 81–90 CrossRef CAS.
- N. Dale, S. Hatz, F. M. Tian and E. Llaudet, Trends Biotechnol., 2005, 23, 420–428 CrossRef CAS.
- A. Mueller, Mini-Rev. Med. Chem., 2005, 5, 231–239 CAS.
- S. K. Arya, M. Datta and B. D. Malhotra, Biosens. Bioelectron., 2008, 23, 1083–1100 CrossRef CAS.
- M. Badihi-Mossberg, V. Buchner and J. Rishpon, Electroanalysis, 2007, 19, 2015–2028 CrossRef CAS.
- H. Nakamura and I. Karube, Anal. Bioanal. Chem., 2003, 377, 446–468 CrossRef CAS.
- S. Andreescu and J. L. Marty, Biomol. Eng., 2006, 23, 1–15 CrossRef CAS.
- G. B. Sukhorukov, A. L. Rogach, B. Zebli, T. Liedl, A. G. Skirtach, K. Kohler, A. A. Antipov, N. Gaponik, A. S. Susha, M. Winterhalter and W. J. Parak, Small, 2005, 1, 194–200 CrossRef CAS.
- I. Willner, R. Baron and B. Willner, Biosens. Bioelectron., 2007, 22, 1841–1852 CrossRef CAS.
- D. Trau and R. Renneberg, Biosens. Bioelectron., 2003, 18, 1491–1499 CrossRef CAS.
- B. M. Wohl and J. F. J. Engbersen, J. Controlled Release, 2012, 158, 2–14 CrossRef CAS.
- J. Kim, J. W. Grate and P. Wang, Chem. Eng. Sci., 2006, 61, 1017–1026 CrossRef CAS.
- J. Kim, J. W. Grate and P. Wang, Trends Biotechnol., 2008, 26, 639–646 CrossRef CAS.
- T. Nguyen, K. P. McNamara and Z. Rosenzweig, Anal. Chim. Acta, 1999, 400, 45–54 CrossRef CAS.
- K. P. McNamara and Z. Rosenzweig, Anal. Chem., 1998, 70, 4853–4859 CrossRef CAS.
- R. Jelinek and S. Kolusheva, Biotechnol. Adv., 2001, 19, 109–118 CrossRef CAS.
- A. J. Baeumner, R. N. Cohen, V. Miksic and J. H. Min, Biosens. Bioelectron., 2003, 18, 405–413 CrossRef CAS.
- J. R. Chen, Y. Q. Miao, N. Y. He, X. H. Wu and S. J. Li, Biotechnol. Adv., 2004, 22, 505–518 CrossRef CAS.
- H. A. H. Rongen, A. Bult and W. P. vanBennekom, J. Immunol. Methods, 1997, 204, 105–133 CrossRef CAS.
- C. K. Kim and K. M. Park, J. Immunol. Methods, 1994, 170, 225–231 Search PubMed.
- S. J. Lim and C. K. Kim, Anal. Biochem., 1997, 247, 89–95 CrossRef CAS.
- M. Bally, K. Bailey, K. Sugihara, D. Grieshaber, J. Voeroes and B. Stadler, Small, 2010, 6, 2481–2497 CrossRef CAS.
- K. Lundstrom, Cell. Mol. Life Sci., 2006, 63, 2597–2607 CrossRef CAS.
- Y. Barenholz, Curr. Opin. Colloid Interface Sci., 2001, 6, 66–77 CrossRef CAS.
- L. S. Jung, J. S. Shumaker-Parry, C. T. Campbell, S. S. Yee and M. H. Gelb, J. Am. Chem. Soc., 2000, 122, 4177–4184 CrossRef CAS.
- T. ShibataSeki, J. Masai, T. Tagawa, T. Sorin and S. Kondo, Thin Solid Films, 1996, 273, 297–303 Search PubMed.
- C. R. MacKenzie, T. Hirama, K. K. Lee, E. Altman and N. M. Young, J. Biol. Chem., 1997, 272, 5533–5538 CrossRef CAS.
- I. Stanish, J. P. Santos and A. Singh, J. Am. Chem. Soc., 2001, 123, 1008–1009 CrossRef CAS.
- T. Stora, Z. Dienes, H. Vogel and C. Duschl, Langmuir, 2000, 16, 5471–5478 CrossRef CAS.
- C. M. Niemeyer, T. Sano, C. L. Smith and C. R. Cantor, Nucleic Acids Res., 1994, 22, 5530–5539 CrossRef CAS.
- B. Chaize, M. Nguyen, T. Ruysschaert, V. le Berre, E. Trevisiol, A. M. Caminade, J. P. Majoral, G. Pratviel, B. Meunier, M. Winterhalter and D. Fournier, Bioconjugate Chem., 2006, 17, 245–247 CrossRef CAS.
- B. Städler, M. Bally, D. Grieshaber, J. Voeroes, A. Brisson and H. M. Grandin, Biointerphases, 2006, 1, 142–145 Search PubMed.
- C. Yoshina-Ishii and S. G. Boxer, J. Am. Chem. Soc., 2003, 125, 3696–3697 CrossRef CAS.
- S. Svedhem, I. Pfeiffer, C. Larsson, C. Wingren, C. Borrebaeck and F. Hook, ChemBioChem, 2003, 4, 339–343 CrossRef CAS.
- B. Städler, D. Falconnet, I. Pfeiffer, F. Hook and J. Voros, Langmuir, 2004, 20, 11348–11354 CrossRef.
- J. J. Benkoski and F. Hook, J. Phys. Chem. B, 2005, 109, 9773–9779 CrossRef CAS.
- M. R. Dusseiller, B. Niederberger, B. Stadler, D. Falconnet, M. Textor and J. Voros, Lab Chip, 2005, 5, 1387–1392 RSC.
- I. Pfeiffer and F. Hook, J. Am. Chem. Soc., 2004, 126, 10224–10225 CrossRef CAS.
- K. Bailey, M. Bally, W. Leifert, J. Voeroes and T. McMurchie, Proteomics, 2009, 9, 2052–2063 CrossRef CAS.
- S. Beugin, K. Edwards, G. Karlsson, M. Ollivon and S. Lesieur, Biophys. J., 1998, 74, 3198–3210 CrossRef CAS.
- I. Sugiyama and Y. Sadzuka, Biol. Pharm. Bull., 2007, 30, 208–211 Search PubMed.
- S. Beugin-Deroo, M. Ollivon and S. Lesieur, J. Colloid Interface Sci., 1998, 202, 324–333 Search PubMed.
- V. P. Torchilin, Nat. Rev. Drug Discovery, 2005, 4, 145–160 CrossRef CAS.
- Y. Ikeda and Y. Nagasaki, in Polymers in Nanomedicine, ed. S. Y. T. Kunugi, 2012, vol. 247, pp. 115–140 Search PubMed.
- S. Qian, C. Li and Z. Zuo, Curr. Drug Metab., 2012, 13, 372–395 Search PubMed.
- R. R. Sawant and V. P. Torchilin, AAPS J., 2012, 14, 303–315 Search PubMed.
- C. Carrion, J. C. Domingo and M. A. de Madariaga, Chem. Phys. Lipids, 2001, 113, 97–110 Search PubMed.
- X. B. Zhao, N. Muthusamy, J. C. Byrd and R. J. Lee, J. Pharm. Sci., 2007, 96, 2424–2435 Search PubMed.
- C. Li, J. Cui, C. Wang, L. Zhang, X. Xiu, Y. Li, N. Wei, Y. Li and L. Zhang, J. Pharm. Pharmacol., 2011, 63, 376–384 Search PubMed.
- Y. Zhang, W. C. Ruder and P. R. LeDuc, Trends Biotechnol., 2008, 26, 14–20 CrossRef CAS.
- B. Städler, A. D. Price and A. N. Zelikin, Adv. Funct. Mater., 2011, 21, 14–28 CrossRef.
- E. T. Kisak, B. Coldren, C. A. Evans, C. Boyer and J. A. Zasadzinski, Curr. Med. Chem., 2004, 11, 199–219 CrossRef CAS.
- H. C. Chiu, Y. W. Lin, Y. F. Huang, C. K. Chuang and C. S. Chern, Angew. Chem., Int. Ed., 2008, 47, 1875–1878 CrossRef CAS.
- B. G. De Geest, S. De Koker, K. Immesoete, J. Demeester, S. C. De Smedt and W. E. Hennink, Adv. Mater., 2008, 20, 3687–3691 CrossRef CAS.
- O. Kulygin, A. D. Price, S. F. Chong, B. Stadler, A. N. Zelikin and F. Caruso, Small, 2010, 6, 1558–1564 CrossRef CAS.
- C. D. Driever, X. Mulet, A. P. R. Johnston, L. J. Waddington, H. Thissen, F. Caruso and C. J. Drummond, Soft Matter, 2011, 7, 4257–4266 RSC.
- H. Lomas, A. P. R. Johnston, G. K. Such, Z. Zhu, K. Liang, M. P. Van Koeverden, S. Alongkornchotikul and F. Caruso, Small, 2011, 7, 2109–2119 CrossRef CAS.
- R. F. Fakhrullin and V. N. Paunov, Chem. Commun., 2009, 2511–2513 RSC.
- R. Chandrawati, M. P. van Koeverden, H. Lomas and F. Caruso, J. Phys. Chem. Lett., 2011, 2, 2639–2649 Search PubMed.
- Y. Wang, L. Hosta-Rigau, H. Lomas and F. Caruso, Phys. Chem. Chem. Phys., 2011, 13, 4782–4801 RSC.
- M.-L. De Temmerman, J. Demeester, S. C. De Smedt and J. Rejman, Nanomedicine, 2012, 7, 771–788 Search PubMed.
- L. Hosta-Rigau, R. Chandrawati, E. Saveriades, P. D. Odermatt, A. Postma, F. Ercole, K. Breheney, K. L. Wark, B. Städler and F. Caruso, Biomacromolecules, 2010, 11, 3548–3555 CrossRef CAS.
- R. Chandrawati, L. Hosta-Rigau, D. Vanderstraaten, S. A. Lokuliyana, B. Städler, F. Albericio and F. Caruso, ACS Nano, 2010, 4, 1351–1361 CrossRef CAS.
- S.-F. Chong, R. Chandrawati, B. Staedler, J. Park, J. Cho, Y. Wang, Z. Jia, V. Bulmus, T. P. Davis, A. N. Zelikin and F. Caruso, Small, 2009, 5, 2601–2610 CrossRef CAS.
- A. N. Zelikin, J. F. Quinn and F. Caruso, Biomacromolecules, 2006, 7, 27–30 CrossRef CAS.
- L. Hosta-Rigau, B. Städler, Y. Yan, E. C. Nice, J. K. Heath, F. Albericio and F. Caruso, Adv. Funct. Mater., 2010, 20, 59–66 CrossRef CAS.
- R. Chandrawati, P. D. Odermatt, S.-F. Chong, A. D. Price, B. Städler and F. Caruso, Nano Lett., 2011, 11, 4958–4963 CrossRef CAS.
- P. Blandon, in Cholesterol: Chemistry, Biochemistry and Pathology, ed. R. P. Cook, Academic Press. Inc., New York, 1958, ch. 2 Search PubMed.
| This journal is © The Royal Society of Chemistry 2013 |