 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Tracking stem cells in tissue-engineered organs using magnetic nanoparticles
Roxanne
Hachani
a,
Mark
Lowdell
b,
Martin
Birchall
c and
Nguyễn Thi Kim
Thanh
*a
aDepartment of Physics & Astronomy, University College London, Gower Street, London WC1E 6BT, UK. E-mail: roxanne.hachani.12@ucl.ac.uk; ntk.thanh@ucl.ac.uk
bDepartment of Haematology, University College London, Medical School, Rowland Hill St., London NW3 2PF, UK. E-mail: m.lowdell@ucl.ac.uk
cUCL Ear Institute, The Royal National Throat Nose and Ear Hospital, 330 Grays Inn Road, London WC1X 8DA, UK. E-mail: m.birchall@ucl.ac.uk
First published on 23rd September 2013
Abstract
The use of human stem cells (SCs) in tissue engineering holds promise in revolutionising the treatment of numerous diseases. There is a pressing need to comprehend the distribution, movement and role of SCs once implanted onto scaffolds. Nanotechnology has provided a platform to investigate this through the development of inorganic magnetic nanoparticles (MNPs). MNPs can be used to label and track SCs by magnetic resonance imaging (MRI) since this clinically available imaging modality has high spatial resolution. In this review, we highlight recent applications of iron oxide and gadolinium based MNPs in SC labelling and MRI; and offer novel considerations for their future development.
 Roxanne Hachani | Roxanne Hachani is currently a PhD student at University College London, under the supervision of Prof. Nguyễn TK Thanh, Dr Mark Lowdell and Prof. Martin Birchall. She received her Bachelors of Science in Biochemistry from the University Claude Bernard Lyon 1 in 2010, and her Master of Science in Nanoscale engineering in 2012 from the Ecole Centrale de Lyon in collaboration with the National Institute of Applied Sciences Lyon and University Claude Bernard Lyon 1. Her current research projects focus on developing novel magnetic nanoparticles for labelling and imaging stem cells within tissue-engineered scaffolds. |
 Mark Lowdell | Dr Mark Lowdell is an assistant professor at University College London and Director of the Laboratory of Cellular Therapeutics at Royal Free Hospital London. His GMP facility produces somatic cell medicines and tissue engineered products and he holds MHRA qualified person status for their release for clinical trials. Dr Lowdell's has held and holds numerous appointments to academic and government advisory boards and is a member of the scientific advisory boards of Cell Medica UK and Coronado Bioscience in the USA. His team includes 9 GMP production scientists and 9 pre-clinical research scientists. |
 Martin Birchall | Martin Birchall is Professor of Laryngology at UCL, London and a practising airway surgeon. He co-led teams which delivered the worlds first stem cell based organ (trachea) transplants in an adult (Lancet, 2008) and a child (Lancet 2012), as well as the world's second documented laryngeal transplant. He has over 150 peer-reviewed publications, including Lancet, Nature, BMJ, and presently holds £8m in peer-reviewed research grants. |
 Nguyễn Thi Kim Thanh | Nguyễn Thi Kim Thanh FRSC CChem CSci MInstP is a Professor of Nanomaterials and a Royal Society University Research Fellow (http://www.ntk-thanh.co.uk). She has over 10 years of research experience in synthesis and biofunctionalisation of nanoparticles. She has been an invited speaker at over 90 institutes and scientific meetings. She has served on organisation and scientific committees for many major international conferences on Nanoparticles research (e.g., Scientific chair of Faraday Discussion 175:“Physical Chemistry of Functionalised Biomedical Nanoparticle and organiser of UK Colloids 2014”. Recently she edited a seminal book: “Magnetic nanoparticles: from fabrication to clinical applications” published by CRC Press/Taylor and Francis. |
Mesenchymal stem cells
The twenty first century has seen numerous scientific discoveries in the field of stem cell research which have led us to broaden our understanding of cell biology. Stem cell-based therapies are expected to have an important impact on the treatment and cure of wide spread diseases such as diabetes, neurodegenerative or cardiovascular diseases. While the first in vivo transplantation of stem cell trachea implant was successful,1 being able to track the stem cells in vivo would allow us to monitor their biodistribution, viability in tissue engineered organs and to understand their interactions with the recipient's cells and the mechanical pathways following transplantation.Mesenchymal stem cells (MSCs) constitute a valuable population of SCs; they differentiate into cells of various tissue types: cartilage, bone, muscle, ligament, tendon and adipose (Fig. 1).2–5 Hence, understanding their molecular regulation and their migration within scaffolds and in vivo is essential.6,7 The use of stem cells in tissue engineering faces the challenge of understanding the kinetics, distribution and function of the cells after transplantation.
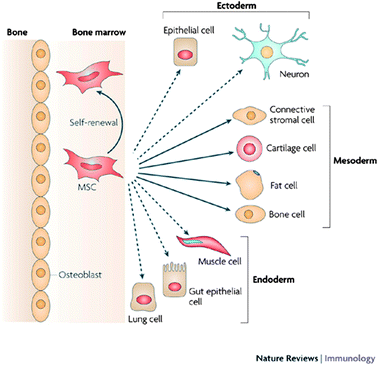 | ||
| Fig. 1 Lineage potential of adult human MSCs. This figure shows the ability of MSCs in the bone marrow cavity to self-renew (curved arrow) and to differentiate (straight, solid arrows) towards the mesodermal lineage. The reported ability to transdifferentiate into cells of other lineages (ectoderm and endoderm) shown by dashed arrows remains controversial in vivo. Reproduced with permission from ref. 5. | ||
This review aims to investigate the use of MNPs as a tool to label and track MSCs in vivo, as well as exploring the possibility of active targeting to increase the specificity of nanoparticle-labeled cells.
Tracking stem cells by various imaging modalities
Similar to molecular-based contrast agents, nanoparticles can be used to enhance the resolution of imaging while offering better biocompatibility, toxicity and biodistribution. There are various imaging modalities clinically available: optical imaging, magnetic resonance imaging (MRI), computed tomography (CT), ultrasound (US), positron emission tomography (PET) or single photon emission computed tomography (SPECT). Each technique has its own advantages and drawbacks but combining techniques can compensate for drawbacks in any individual technique (Table 1).8–10 For example, a multimodal nanoparticle can combine a fluorescent probe, for high sensitivity, and an MRI probe for high spatial resolution. Cellular imaging with NPs can be a complementary tool to standard molecular-based methods for the early detection of diseases, understanding their processes, and is now developed more and more to follow the efficacy of therapy11 or labelled cells.| Source of imaging | Spatial resolution | Tissue penetrating depth | Sensitivity | Types of probe | |
|---|---|---|---|---|---|
| Magnetic resonance imaging (MRI) | Radiowave | 25–100 μm | No limit | mM to μM (low) | Para- (Gd3+) or superparamagnetic (Fe3O4) materials |
| Positron emission tomography (PET) | γ-ray | 1–2 mm | No limit | pM (high) | Radionuclides (18F, 11C, 13N, 15O, 124I, 64Cu) |
| Computed tomography (CT) | X-ray | 50–200 μm | No limit | Not well characterized | High atomic number atoms (iodine, barium sulphate) |
| Optical fluorescence imaging | Visible or near-infrared light | in vivo, 2–3 mm; in vitro, sub-μm | <1 cm | nM to pM (medium) | Fluorescent dyes, quantum dots |
Nanoparticles (NPs) have potential as contrast enhancers in medical imaging to track the implantation, retention and migration of MSCs, which are necessary before cellular therapy can be validated. NPs have a high surface area to volume ratio, a property which can help control their biodistribution allowing longer lifetimes for them to reach their targets, and the surface chemistry can be adjusted to direct nanoparticle trafficking.
Recognition of targeted cells by specific receptors can be achieved through optimising the surface properties of NPs and its functionalisation by coupling targeting moieties which recognise specific receptors of targeted cells.
Nanoparticle-based cellular imaging
While there is a wide variety of imaging techniques used clinically, different factors must be considered when developing NPs for SC tracking such as: sensitivity, resolution, specificity and exposure of individual patients to secondary effects (Table 2).| Advantages | Disadvantages | References | |
|---|---|---|---|
| Multimodality | Integrating several imaging functions on a single platform. Recently, tri-modal imaging probes for MRI/PET/optical imaging have been designed by combining 64Cu-DOTA chelates to MRI/optical imaging probes. | Non specific imaging of cells; reproducibility of synthesis; interference of imaging modalities | 12 and 13 |
| Contrast | Generating enhanced contrast (lipid-based NPs, Gd-based NPs, iron oxide NPs) | There must still be enough nanoparticles to generate sufficient contrast within a cell, while avoiding toxic effects. | 14–17 |
| Large surface area to volume ratio | Integration of larger amounts of contrast agents and ability to control the circulation time of NPs |
Interaction with plasma proteins in vivo may alter the nanoparticle's properties. Larger amounts of contrast agents may lead to secondary effects. Gadolinium chelating ligands commercially available may be used to functionalise the surface of NPs and prevent against toxic side-effects of free gadolinium ions such as:
– The disruption of any calcium-dependant reaction (blockage of calcium channels, neurotransmission etc.) since Gd3+ has a similar size to Ca2+ – Gd3+ precipitates at physiological pH so this may block the reticuloendothelial system (RES) by impairing the phagocytic function of Kupffer cells. |
18–20 |
All these, when taken into account, lead to the selection of techniques which are of clinical interest.8,21 In this review, we will focus on MRI as this is most commonly studied for NP-based stem cell tracking.
Magnetic resonance imaging (MRI)
MRI is amongst today's most studied imaging modalities for cellular tracking and labelling as it presents numerous advantages such as a high spatial15,16 and temporal resolution, and the use of non-ionizing radiation.In medical applications, MRI detects the signal of the protons present in water, which will respond to a magnetic field in an environment-dependant manner (i.e. different tissues). Contrast agents are used to enhance the sensitivity by influencing the relaxation time of the protons after application of a radiofrequency pulse, and this can be done in two ways. In an isotropic system without any chemical exchange: longitudinal or spin-lattice relaxation (T1) and transverse or spin–spin relaxation (T2).
MRI positive contrast agents (T1)
Contrast agents for longitudinal relaxation of protons mainly comprise of paramagnets as these possess a large number of free electrons (lanthanides for example) and shorten T1, thus forming bright contrast. This is the case of gadolinium-based chelates which are clinically used as contrast agents (Fig. 2).17,22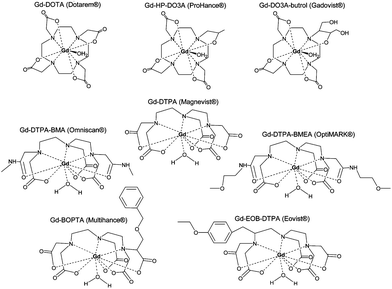 | ||
| Fig. 2 Commercially available Gd3+ chelate MRI contrast agents: (a) Gd DOTA, (b) Gd DTPA, (c) Gd sulfonamide, (d) Gd β-galactosidase, (e) Gd esterase, (f) Gd bpatcn-QD, (g) Gd glutamate. Reprinted with permission from ref. 85. Copyright 2010 American Chemical Society. | ||
However, precaution must be taken when using contrast agents containing gadolinium because of recent cases of nephrogenic systemic fibrosis (NSF) in patients with renal failure who were administered Omniscan®.13
This is why gadolinium cations Gd3+ are chelated when administered to prevent toxicity. Complexes of Gd3+ are now widely used in routine MRIs and can be conjugated with NPs to render these applicable to T1 MRI imaging.23 These chelating ligands are commercially available and they aim to prevent toxic side-effects of free gadolinium ions (Table 2).
The complexes which chelate Gd3+: DOTA (1,4,7,10-tetraazacyclododecane-1,4,7,10-tetraacetic acid) and DTPA (diethylenetriaminepentaacetic dianhydride) are stable and soluble in physiological medium. They have a low osmotic potential and are rapidly eliminated from the body by renal excretion, so this confers the minimal biological impact desired.
Although DTPA was the first chelate to be commercially available, DOTA is preferred to DTPA. Indeed, it is more advantageous in terms of high relaxivity, blood clearance rate in vivo, higher binding coefficient to Gd3+ (more stable in vivo in terms of thermodynamics and kinetics) and for the same image quality a smaller dose is required than DTPA.24
The main drawback of using this contrast agent is that it must be administered in high concentrations to produce an effective contrast. This is because of diffusion, low relaxivity (the change in relaxation rate) per gadolinium ion and dilution phenomena in vivo after injection. This is where the nanoparticles are beneficial because they increase the quantity of contrast agents per NP allowing a higher concentration of contrast agent. They can also potentially lead to an easier targeting as it is the case in tumours with passive targeting by the enhanced permeability and retention (EPR) effect. A recent study also confirmed the use of gadolinium not only to label MSCs but also to determine their viability after transplantation depending on the signal obtained.25
MRI negative contrast agents (T2)
Several types of NPs including superparamagnetic iron oxide nanoparticles (SPIONs) and ultra small SPIONs (USPIOs) were developed in the 1980s and have now been clinically approved by the EMA (European Medicines Agency) and FDA (Food and Drug Administration). Negative contrast can be provided by superparamagnetic iron oxide NPs which shorten the T2 relaxation time and produce a darker image. Two principal factors play a role in the contrast enhancement: the iron oxide core and its coating. The shortening of T2 is determined by transverse relaxation of hydrogen protons of water molecules surrounding the superparamagnetic iron oxide NPs.26Iron oxide NPs have proven particularly useful by their biocompatibility.27–30 The proton relaxation per atom due to iron is greater than the one which can be attributed to paramagnetic elements such as gadolinium, hence smaller amounts of contrast agent can be administered. For in vivo applications, superparamagnetic nanoparticles also present the advantage of being magnetic only in the presence of an external magnetic field, and it has been proven that iron oxide is safely degraded and metabolised by living organisms.31,32
Imaging properties of MNPs depend on their physicochemical characteristics
The MRI contrast enhancement by magnetic nanoparticles is dependent on their composition, size, surface properties and degree of aggregation (Table 3). It has been demonstrated for example that size is a critical parameter influencing the transverse relaxivity r2,33,34 while the surface contact influences the longitudinal relaxivity r1.35 This is confirmed by the quantum-mechanical outer sphere theory which expresses the T2 relaxivity constant of iron oxide NPs26,36 such as:| 1/T2 = (256π2γ2/405)V* Ms2α2/D(1 + L/α) |
| Nanoparticle | Core size (nm) | Hydrodynamic diameter (nm) | r 1 (mM−1 s−1) | r 2 (mM−1 s−1) | Ref. |
|---|---|---|---|---|---|
| Ferumoxide (feridex) γFe2O3, Fe3O4 – dextran | 4.96 | 160 | 10.1 | 120 | 27 |
| (0.47 T) | (0.47 T) | ||||
| Ferucarbotran (resovist) Fe3O4 – carboxydextran | 4 | 60 | 186 | 27 | |
| Fe3O4–PEG–(NH2)2 | 12 | 49 | 22 (1.5 T) | 191 (1.5 T) | 74 |
| Fe3O4 | 9 | 265 (1.5 T) | 56 | ||
| γFe2O3 – alendronate | 10 | 32.5 | — | 287 (4.7 T) | 75–77 |
| γFe2O3 – neridronate | 10 | 38.1 | — | 277 (4.7 T) | |
| γFe2O3 – zoledronate | 10 | 37.5 | — | 254 (4.7 T) | |
| γFe2O3 – risedronate | 10 | 28.8 | — | 267 (4.7 T) | |
| Fe3O4-casein | 15 | 30 | — | 273 (3 T) | 78 |
| Fe3O4-dimercaptosuccinic acid | 9.2 | 70 | 18.5 (1.5 T) | 317 (1.5 T) | 79 |
| Fe3O4-dextran | 20–25 | 91 | 320 | 80 | |
| Fe3O4-triethylene glycol | 11 | 9.5 (1.5 T) | 337 (1.5 T) | 81 | |
| Fe3O4-(1,2-distearoyl-sn-glycero-3-phosphoethanolamine-N-[methoxy(polyethylene glycol)] copolymer)-mPEG1000 | 14 | 28.6 | 385 | 36 | |
| (0.47 T) |
On the other hand, the shortening of T1 depends solely on the direct exchange of energy between hydrogen protons in water molecules and the paramagnetic element's outer shell electrons, hence an indirect correlation with the surface of NPs.
Clinical MRI (1.5 T scanners with radiofrequency coils) is not ideal for imaging of single cells after systemic administration. The spatial resolution needed would require long scanning times, which is not feasible in clinical settings. The latter are still adequate for magnetically labeled cells administered locally (for example, in the case of an implanted scaffold). This requires a compromise between the minimum number of cells detectable and the concentration of NPs to ensure there is no effect on cell viability or proliferation. Schäfer et al. recently determined that at least 1000 SPIO-labelled MSCs could be visualised by MRI in vitro after exposition to a magnetic field.37 A recent study showed that a relatively high incubation concentration (10 μg USPIOs per 105 cells) and long incubation times (21 h) were needed to detect a minimum of 250![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 MSCs in vivo.38 Most notably, in comparison, the in vitro detection threshold of cells after incubation with gadolinium oxide nanoparticles at a Gd concentration of 0.2 g per 5 × 105 cells for 2 h was estimated to be about 10000 cells.39 Yang et al. were able to compare the efficiency of MSC labeling with SPIONs and paramagnetic chelates. Iron oxide nanoparticle labeling had a detection threshold of 12500 cells in vitro, while gadolinium chelates labeling could be detected for at least 50000 cells.40
000 MSCs in vivo.38 Most notably, in comparison, the in vitro detection threshold of cells after incubation with gadolinium oxide nanoparticles at a Gd concentration of 0.2 g per 5 × 105 cells for 2 h was estimated to be about 10000 cells.39 Yang et al. were able to compare the efficiency of MSC labeling with SPIONs and paramagnetic chelates. Iron oxide nanoparticle labeling had a detection threshold of 12500 cells in vitro, while gadolinium chelates labeling could be detected for at least 50000 cells.40
New strategies have been explored to overcome the limits of clinical MRI when studying the biodistribution of a single cell. These include the use of high field MRI scanners (for example 9.4 T, 17.6 T),41 cryogenic probes,15 or high temperature superconducting coils16 that can be mounted on clinical 1.5 T devices, which allow the resolution at a single cell level even after systemic administration.
MRI is a high resolution-low sensitivity technique leading to a near cellular resolution (25–50 μm). Current research focuses on its combination with other higher sensitivity-low resolution techniques such as Positron Emission Tomography (PET), Single Photon Emission Computed Tomography (SPECT) or NIRF imaging (Near Infrared Fluorescence).
Indeed, multimodal nanoparticles incorporating several imaging probes on a single platform are being developed. This is to overcome the limitations of a single modality by complementing it with another modality (Fig. 3).42 The main disadvantages of MRI currently are the loss of signal with time and the lack of specificity. Nanoparticles can be taken up by non specific cells in vivo after implanted stem cells undergo apoptosis or degradation, or their exocytosis can occur under stress conditions.43–46 To overcome these limits, chitosan coated NPs have been developed as they allow an enhanced uptake and long term tracking of cells through limited endo-lysosomal trafficking and exocytosis.47,48 Recently, hydrophilic SPIONs coated with glucosaminic acid (GA) were shown to enhance cellular uptake of NPs.49 Furthermore, once the MSCs divide or differentiate the concentration of nanoparticles decreases rapidly hence the MRI signal is lost with time. Encapsulation of iron oxide nanoparticles in polymeric microparticles has been a new strategy explored to help maintain a strong MRI signal and reduce exocytosis in order to enable the longitudinal tracking of SCs.
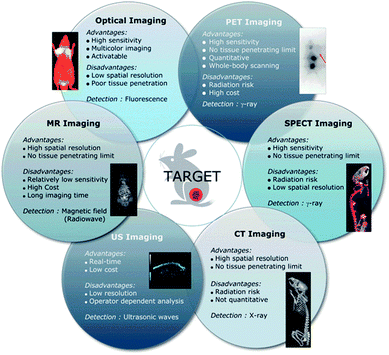 | ||
| Fig. 3 Advantages and limitations of current imaging modalities available clinically. Reproduced from ref. 40 with permission from the Royal Society of Chemistry. | ||
Nanoparticles for in vivo tracking of transplanted MSCs
NPs are fabricated from different building blocks which can be organic (e.g., polymeric micelles, dendrimers) or inorganic (e.g., iron, gold, silver, silica). It is now relevant to present some of the most common NPs currently studied for the tracking of MSCs.Although several contrast agents that have been clinically approved and tested, nanomaterials are gaining more attention since their properties (size, shape, surface chemistry…) can be finely tuned for specific applications.50,51
Indeed, quantum dots (QDs) which are semiconductor nanocrystals, are frequently used for cellular optical imaging. QDs exhibit a size-dependent fluorescence emission wavelength, enhanced photo-stability properties making them the ideal tools for long term cell labelling and tracking.52 PEG coated CdSe/ZnS QDs were used to label MSCs transplanted in nude mice in 2008 by Lei et al.53 24 h after transplantation, the QDs- hMSCs were visualised under fluorescence imaging showing significant accumulation in the liver, lung and spleen. More recently, they were used to label and image rat pancreatic stem cells and were once again proven to have no effects on cell viability and proliferation.54 However, all studies have been done in animal models as there is great concern in regards to the toxicity of QDs through their degradation and the release of cadmium in vivo.
Magnetic ferrite NPs have also been studied for cellular labelling, but to a lesser extent. Indeed, rat neural stem cells labelled with the CoPt hollow nanoparticles were injected into cultured spinal cord slices and observed over time.55 This study indicated that MRl can effectively detect low numbers of labelled cells due to the enhanced contrast provided by the cobalt-based nanoparticles which provide a stronger response to magnetic fields than iron oxide for example. While the NPs are efficient tools for MRI and are biologically stable for long periods of time, the problem of introducing platinum in vivo needs to be addressed before clinical applications may be pursued.
Iron oxide NPs
Amongst magnetic nanomaterials, manganese ferrite NPs possess the highest magnetic moment per unit, making them the nanosystems with the highest transverse relaxivity amongst ferrites.56However, iron oxide NPs have been coveted as multifunctional imaging and therapy tools because of their biocompatibility and their well-known chemical synthesis57 which leads to a highly uniform and crystalline structure. As mentioned previously, SPIONs and USPIOs are available commercially for clinical applications, hence increased research focus on these particles.
Some iron oxide-based NPs commercially available or recently developed in research laboratories are listed for efficient T2 contrast comparison (Table 3). Iron oxide NPs such as magnetite (Fe3O4) or its oxidized form maghemite (γ-Fe2O3) are currently the most studied systems for cellular labelling and biomedical imaging. Indeed, their non-toxicity and non-immunogenicity is essential for pre-clinical and clinical use.
Due to their clinical approval, commercial iron oxide NPs have been used to label and track various types of stem cells after their transplantation such as bone marrow stromal stem cells,58 embryonic stem cells,59 neural stem cells,60,61 hematopoietic progenitor cells.62–64 Syková and Jendelová have shown that the use of Endorem® can be used effectively to track the migration of transplanted mouse embryonic stem cells (ESC) to sites of injury by MRI over extended periods of time.65
A study in 2012 showed that in comparison to Gd chelates, iron oxide NPs did not allow the differentiation of viable or dead cells, thus leading to a misleading MRI signal.25 Also, Bos et al. in 2004 showed that while SPIO labelled MSCs injected in rat kidney and liver were able to differentiate properly, above 100 μg Fe ml−1, their proliferation was decreased.66 This is something to consider for long term imaging of cells and their migratory capacity after transplantation.
These FDA-approved NPs have now all been discontinued and a variety of SPIONs are developed with physico-chemical properties which may be finely tuned.
The hydrophobic nature of SPIONs implies that their surface must be rendered stable and biocompatible by coating with biomolecules or polymers such as dextran, polyethylene glycol (PEG), citrate and chitosan (Fig. 4).67 The coating will influence the imaging properties of the NP as it may hinder the diffusion or interaction with water molecules. In addition, the coating minimizes non-specific interactions and prevents aggregation of the iron oxide cores.
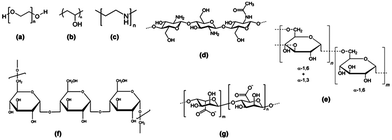 | ||
| Fig. 4 Structure of polymeric stabilizers of magnetic nanoparticles (a) PEG, (b) PVA, (c) PEI, (d) chitosan, (e) dextran, (f) pullulan, and (g) alginate. Reprinted with permission from ref. 65. Copyright 2012 American Chemical Society. | ||
Dextran is a polysaccharide commonly used to stabilise iron oxide nanoparticles as it has been shown to have a particular affinity for their surface through hydrogen bonding.68,69 However, several years ago, it was demonstrated that after incubating hMSCs with dextran coated nanoparticles during 2 hours at 25 mg Fe ml−1, no labeling of hMSCs was detectable. More recently, Babič and colleagues also confirmed the low affinity of dextran coated iron oxide nanoparticles and rat MSCs, even after long incubation times of up to 72 h with rat MSCs.70
The coating of NPs is essential as proven by K. Andreas et al. who recently confirmed an increased cellular uptake and more intense MRI hypointensity of citrate SPIONS compared to commercial SPIONs at the same iron concentration (Fig. 5).71 Recent research has studied the increased cellular uptake of negatively charged NPs with an increased density of carboxyl groups, which probably occurs through electrostatic adsorption on negatively charged cellular membranes.72
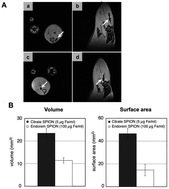 | ||
| Fig. 5 In vivo and ex vivo visualization of SPION-labeled MSCs. In vivo MR imaging showed areas of hypointensity in rat muscle tissue in the axial (A; a and c) and sagittal (A; b and d) view albeit with a larger area of hypointensity seen for citrate SPION-labeled MSCs (A; a and b) than for Endorem SPION-labeled cells (A; c and d). Consistently, transplanted citrate SPION-labeled MSCs showed considerably higher hypointense volume and surface area compared to Endorem SPION-labeled cells (B). Reprinted from ref. 69, Copyright (2012) with permission from Elsevier. | ||
Labelling of hMSCs has been successfully achieved with magnetic iron oxide silica core–shell nanoparticles.73 These nanostructures allow bimodal imaging with the incorporation of the organic dye fluorescein isothiocyanate (FITC) in the silica shell. The high labelling efficiency was demonstrated with the need for a short incubation time of 1 h and a low concentration of iron (0.5 μg ml−1).
No cytotoxicity was present after incubation, thus cells preserved their differentiation abilities and imaging of low number of cells (105) was possible with a clinical grade 1.5 T MRI imager.
More recently, ultra magnetic liposomes have been constructed with a highly efficient encapsulation and concentration of ultra- small iron oxide nanoparticles inside a liposome.82 This structure possesses a high blocking temperature, enhanced heating properties for hyperthermia and a high r2 value leading to better contrast in MRI. Another recent study was conducted with ‘nanoflowers’ constituted by multi-core NPs made of maghemite cores.83,84 The three-dimensional organization of the cores with their close spherical contact, identical crystal orientation and the absence of a matrix lead to cooperative behaviour. This in turn leads to an enhanced magnetic moment and r2 relaxivity while maintaining the hydrodynamic diameter below 40 nm which is ideal for biological applications.
In regards to MRI, these nanostructures are potentially efficient as it was demonstrated single tumour cells could still be distinguished 6 days after they had been labelled.
Gadolinium based NPs
As positive MRI contrast agents, Gd-based NPs have been less studied than iron oxide NPs for labelling of stem cells. Recently, Shi and colleagues have used 6 nm anionic europium (Eu3+) doped Gd2O3 nanoparticles for bimodal labelling and imaging of hMSCs.39 Europium provides a fluorescence signal whereas the oxide core constitutes a positive CA for MRI with a relaxivity value r1 = 13.2 mM−1 s−1, which is greater than that of commercially available Gd-chelates. While the labelling had no negative effect on cell viability and differentiation, no studies were conducted to determine the fate of these NPs within the cells and over time.The coating also plays an important role in the MR tracking of cells by influencing the relaxation properties of the NPs. Indeed, it has been shown that the transfer of gadolinium oxide NPs from an organic solvent such as diethylene glycol (DEG) to capping with a PEG chain allows a significant increase of the longitudinal relaxivity constant.85,86
Tracking stem cells using magnetic nanoparticles (MNPs)
Tracking SCs in vivo has been hindered by the inability to find a suitable imaging technique for obtaining dynamic information and cellular resolution during several days or weeks in a non-invasive manner. Recently, MRI has become a well-studied technique for imaging SCs at a quasi-cellular level by labelling with iron oxide NPs. While this has been studied on human MSCs in vitro or on animal SCs in vivo,66,87 few studies have yet to report reliable results on labelling hMSCs in vivo within tissue-engineered scaffolds.Pre-clinical studies have largely been restricted to small rodents. For example in 2002, Modo et al. studied the transplantation of stem cells in ischemia-damaged rat hippocampus by MRI.88 However, these cells successfully migrated and integrated the host's parenchyma and it was challenging to differentiate these from the host cells by MRI hence the need for labelling with an efficient contrast agent allowing identification of small clusters at high resolution. While gadolinium rhodamine dextran was an efficient T1 and T2 contrast agent (r1 = 17 mM−1 s−1), the fluorescence signal was lost after 7 days due to dilution of the signal following division of the cells. The dual imaging allowed detection of the cells by overlaying histology and MRI without need for another technique.
Lee and colleagues labelled hMSCs with commercial fluorescent NPs (NEO-LIVE Magnoxide 675) and monitored their movement from inside the bone marrow cavity of rats to an osteochondral defect for up to 21 days.42,89 This allowed them to conclude on the efficacy of platelet-derived growth factor (PDGF-AA) as a chemotactant for hMSCs.
Most notably, the novel approach of labelling stem cells in vivo was successfully achieved by Heike Daldrup-Link's team by injecting ferumoxytol, an FDA-approved anemia treatment composed of iron oxide nanoparticles within the bone marrow of rodents. The mesenchymal stem cells were then injected into knee injury sites of rats, and were visualised over several weeks.90 This new strategy has yet to be tested with humans but holds great promise for the development of stem cell therapies. Indeed, this would decrease substantially risks of contamination and biologic alteration of MSCs associated with ex vivo labelling procedures.
Challenges and future considerations
The main challenge to overcome is the dilution of signal due to cell division and exocytosis of the NPs. In cell populations which divide rapidly, such as hMSCs, NPs will be distributed evenly or unevenly amongst daughter cells. After few cycles of cell division, only a fraction of the cell population is labelled and the signal becomes undetectable. A way to overcome this is to genetically transfect the cells to express certain reporter genes such as the Green Fluorescent Protein (GFP) for fluorescence imaging or luciferase for bioluminescence. This has been widely employed in pre-clinical animal studies. However, the effect of genetically modified cells in humans has to be established before thinking about potential clinical applications.Research regarding the impact of magnetically labelled human MSCs on chondrogenesis remains insufficient. This must be carefully studied as SCs are long-lived cells that will persist and divide in vivo. While many groups have shown that there is no negative impact on the differentiation of human MSCs,91,92 some have shown impaired chondrogenesis.93–96 Whilst not yet been fully understood, it could be due to a dose-dependent disruption of iron metabolism involved in chondrogenesis and interference with the organisation of the cytoskeleton. This has been demonstrated with the inhibition of osteogenesis and the promotion of cell migration after labelling hMSCs with Ferucarbotran, an ionic SPION clinically approved as a contrast agent for MRI of the liver.97 The inhibition occurred in a dose-dependent manner and could be correlated to the presence of free irons following the lysosomal degradation of the nanoparticles.
To overcome the problem of dilution of the contrast agent signal due to cell division and exocytosis of the NPs, a recent report illustrated the efficiency of encapsulating 10 nm iron oxide NPs within poly(lactide-co-glycolide) microparticles of 0.8 μm in diameter.98 Indeed, this system allowed a 5-fold increased r2 relaxivity (from 61.16 mM−1 s−1 to 316.6 mM−1 s−1) and 2-fold increased retention within MSCs compared to the iron oxide NPs alone. After 18 days, only microparticle-labelled cells showed fluorescence in more than 15% of cells whereas nanoparticle-labelled cells no longer emitted a fluorescence signal. This was also the case of the MRI signal which allowed visualization of the microparticles 12 days after labelling, and of nanoparticles only up to 6 days. These enhanced properties could be to a size-dependant exocytosis which is slower for larger particles and a clustering of the NPs within the microparticles.
As discussed, imaging is useful as a non-invasive method of tracking MSCs in vivo to comprehend the underlying processes essential to the development of tissue-engineering based therapies. The nanosystems must be sensitive, non-toxic, hydrophilic, non-immunogenic and must be cleared through the renal system. These MNPs which can be used for multimodal imaging may be further functionalized towards imaging specific targets such as a defined cell type for example.
Strategies for active targeting of transplanted MSCs
Composition, size, morphology and surface chemistry can now be tailored to improve magnetic properties but also affect the behaviour of MNPs in vivo.99,100 Biomedical applications require stability in physiological conditions. Modifying the surface's chemistry is essential for their colloidal stability, biocompatibility, and to avoid aggregation in vivo.101 Cell surface labelling is not ideal for in vivo applications due to potential recognition by the RES and their subsequent clearance, hence the interest in functionalizing the surface of NPs to increase their cellular uptake, to yield specific recognition by MSCs and to minimize their recognition by the RES. Active targeting strategies will vary according to the applications desired. For example for cancer, it is a common strategy to graft a vector on the NP that will bind to a receptor or biomarker strongly overexpressed by cancer cells. The aim is to increase selectivity by tumour cells, minimize secondary effects on healthy cells therefore decreasing the dose and impact of treatments administered.Targeting of NPs depends on their physicochemical properties
Fundamental properties such as the size and shape of NPs will influence their ability to target specific cells. In a recent study by Jiang et al., multivalent grafting of herceptin on NPs of 2 different sizes was engineered to allow the recognition of the ErbB2 receptor overexpressed on human breast cancer cells.102 While very small NPs (≤25 nm) had a good biodistribution they did not allow the presentation of multiple ligands to the cell receptors. This could be achieved with larger NPs (≥50 nm) and with a higher binding affinity. A compromise between the biodistribution and binding ability can be achieved with NPs of average size 25–50 nm.In addition, the shape of the NPs will influence their targeting ability. Several studies have demonstrated that relative to targeted spherical NPs, rod-shaped NPs will have a longer blood circulation time and enhanced cell binding properties.103–106
The surface plays an essential role in the biocompatibility and stability of NPs in vivo. In the case of iron oxide NPs, dextran is a polysaccharide often used for coating because of its biocompatibility and high affinity for the surface of iron oxide NPs by polar interaction through hydrogen bonding for example.69 However, labelling of hMSCs with dextran coated NPs has proven difficult without the use of transfection agents.70,107 A frequently used polymer is polyethylene glycol (PEG) which renders the NPs hydrophilic and extends their blood circulation times by steric repulsion of opsonins.108,109 This polymer is non toxic, non immunogenic and has been approved for clinical applications.
Transfection agents to increase cellular uptake of MNPs
Due to the limited phagocytic capacity of MSCs a poor cellular uptake of commercially available MNPs leads to limited MRI sensitivity. MNPs are highly phagocytized by Kupffer cells of the RES, thus different surface modification strategies have been explored to increase their cellular uptake.110 The cellular uptake of NPs will depend on the size, shape and surface chemistry of NPs, but also on the cell line. A strategy widely studied is the use of transfection agents which often are positively charged peptides to facilitate the internalization of NPs by cells. However, this requires a purification step of the cells to minimize non-specific uptake by other cell populations in vivo. Also, they may pose the problem of toxicity if their ratio is not controlled and if used on their own.111,112 The HIV Tat peptide has been conjugated to dextran-coated iron oxide NPs to increase cellular uptake.113,114 More recently, Ferucarbotran was labelled with protamine sulphate during a 4 h incubation period thus increasing the internalization of NPs by hMSCs.95 Ferumoxide and protamine sulphate have also been used to label hMSCs for imaging of the cells over 12 weeks.115 This study has led to a better understanding of the homing of hMSCs after their local intra-articular injection.Active targeting of MNPs to MSCs
To gain in specificity, reduce side effects and toxicity, active targeting is done by linking biomolecules on therapeutic vectors, such as MNPs, to recognise receptors on specific cells. The receptor must be accessible (at the surface and close to blood stream), specific to the stem cell, and recognised by a ligand of strong affinity and specificity. In the case of an antibody, the availability of internalized monoclonal antibodies and the limited number of cell membrane receptors must be addressed.MSCs have been identified through their expression of specific cell surface markers such as the following clusters of differentiation, CD73, CD90, CD105 (endoglin, SH2), CD106 and CD309.116–118 The latter is also known as the vascular endothelial growth factor receptor or KDR. In contrast to hematopoietic stem cells, they do not express CD45, CD34, and rapidly down regulate CD133 upon culture. However, the expression of cell surface markers is complex and not yet fully understood as it may be influenced by factors secreted by accessory cells in the initial passages, and the in vitro expression of MSC markers may differ with their expression in vivo119 as well as their differentiation stages.
Being able to target specific markers would allow us to gain a deeper understanding on the evolution and becoming of hMSCs once transplanted. Furthermore, this would allow targeted labelling of MSCs in vivo. As mentioned previously, the novel approach of actively targeting SCs in vivo has yet to be investigated although it would help minimize alteration of MSCs. This approach has been demonstrated successfully with endothelial progenitor cells labelled with CdSe quantum dots and their differentiation into mature endothelial cells within a vascular bypass graft being followed by optical imaging.120 Also, specifically targeting surface markers of MSCs may be useful for tissue-engineered scaffolds which are seeded with selected and isolated cell populations, thus minimising non specific interactions with other cell populations which may occur in vivo.
It has been shown that hMSCs play an essential role by mobilizing into the peripheral blood when needed or migrating across endothelial cells from the blood into tissues. Angiogenesis requires migration and adhesion of endothelial cells on the extra cellular matrix (ECM) to form new blood vessels. This phenomenon requires adhesion molecules such as integrins which are heterodimeric transmembrane cell surface receptors which participate in cell–cell and cell–ECM interactions. They link cells to their surroundings and to proteins of the ECM (collagen, laminin, fibronectin…). Various integrin molecules, such as α1, α2, α3, α4, α5, αv, β1, β3, and β4, are known to be expressed on hMSCs. Adhesion molecules such as VCAM-1, ICAM-1, ICAM-3, ALCAM, and endoglin are expressed on this cell line.121
The strategy of using nanoprobes to target specific cells has yet to be explored for mesenchymal stem cells, but has been thoroughly investigated for tumour cells. Indeed, active targeting of cancer cells has been developed with multimodal nanoprobes functionalized by the cRGD peptide. The tripeptide RGD (Arginin – Glycin – Aspartate) is present in numerous proteins of the ECM and is a ligand of the integrin αvβ3 which is specifically upregulated by tumour endothelial cells for angiogenesis. The affinity is related to the protein's conformation. The integrin αvβ3 has an extracellular V-type structure in which each subunit has a “closed headpiece bent” conformation. Physiologically, it corresponds to a weak affinity state. The fixation of a ligand, such as the RGD peptide, will lead to a conformational change in which the affinity is increased. Then we obtain the activated receptor in which the cytoplasmic domains are far. Cyclic peptides are more often used than the linear equivalent since the latter was shown, in clinical trials, to accumulate strongly in the liver and not in the desired tumour cells.122 Also, the linear form of the peptide can have conformations with different affinities for the integrin αvβ3 and are more sensitive to proteolysis.
The team of J. L. Coll and P. Dumy was able to target the integrin αvβ3 with the target peptide RAFT(c-[RGDfk])4.123 The interest of polymers like the RAFT peptide is the possibility to present multiple copies of the RGD peptide. It has a better affinity towards the integrin αvβ3 (known to prefer multivalent interactions), improved target internalization in cells, and better signal to noise ratio in vivo.
Recently, active targeting of tumour cells using RGD functionalized nanoparticles has been developed for targeted drug delivery as specific targeting was shown to be efficient in numerous studies.124–127
However, the idea of targeting specific cells by surface markers remains a challenge. While surface markers of specific cells can be determined, their specificity remains the bottleneck of this issue. This may not raise concerns when dealing with an isolated cell population however in vivo applications require further study into identifying specific surface markers to target.
Conclusions
Iron oxide-based NPs have been extensively studied for the labelling and MR imaging of hMSCs. However, positive contrast agents such as gadolinium-based NPs hold great promise as a non-invasive and efficient imaging tool. Both nanoparticles remain interesting by the ability to tune their magnetic properties and surface chemistry. Novel multimodal NPs also allow the use of MRI in complement to other imaging modalities such as PET, SPECT or optical imaging to overcome the limits of MRI. The latter includes the need for high concentrations of contrast agent administered to obtain sufficient contrast,55 which is a key point explored for the development of future nanoprobes in cell labelling and imaging. However, further studies need to be conducted to address questions which remain before clinical applications of stem cell therapy can be considered.Outstanding questions
While this review focuses on targeted imaging of hMSCS, some general questions remain regarding nanoparticle-based cell tracking:• How to maximize the labeling efficiency of hMSCs with various NPs and minimize non-specific uptake of NPs by other cell populations?
• How to gather functional information about the MSCs once administered?
• What happens to the nanoparticles inside the cells?
• How do we monitor the change of the nanoparticles within the cells?
• Which characteristics of the nanoparticles can we modify so they can remain in the cells long enough and be eliminated safely afterwards?
• How can we obtain quantitative information from the imaging output?
• What do we need to prove in terms of toxicology to establish nanoparticles as a safe means by which cells can be labelled and tracked in vivo?
Acknowledgements
Nguyễn T. K. Thanh thanks the Royal Society for her University Research Fellowship. Martin Birchall is supported by Medical Research Council TSCRC GRANT G0902411. Roxanne Hachani thanks UCL School of Life and Medical Sciences for her Grand Challenge PhD studentship.Notes and references
- P. Macchiarini, P. Jungebluth, T. Go, M. A. Asnaghi, L. E. Rees, T. A. Cogan, A. Dodson, J. Martorell, S. Bellini, P. P. Parnigotto, S. C. Dickinson, A. P. Hollander, S. Mantero, M. T. Conconi and M. A. Birchall, Lancet, 2008, 372, 2023–2030 CrossRef.
- R. J. Deans and A. B. Moseley, Exp. Hematol., 2000, 28, 875–884 CrossRef CAS.
- R. S. Tuan, G. Boland and R. Tuli, Arthritis Res. Ther., 2003, 5, 32–45 CAS.
- J. J. Minguell, A. Erices and P. Conget, Exp. Biol. Med., 2001, 226, 507–520 CAS.
- A. Uccelli, L. Moretta and V. Pistoia, Nat. Rev. Immunol., 2008, 8, 726–736 CrossRef CAS PubMed.
- G. Chamberlain, J. Fox, B. Ashton and J. Middleton, Stem Cells, 2007, 25, 2739–2749 CrossRef CAS PubMed.
- M. F. Pittenger, A. M. Mackay, S. C. Beck, R. K. Jaiswal, R. Douglas, J. D. Mosca, M. A. Moorman, D. W. Simonetti, S. Craig and D. R. Marshak, Science, 1999, 284, 143–147 CrossRef CAS.
- M. Rudin and R. Weissleder, Nat. Rev. Drug Discovery, 2003, 2, 123–131 CrossRef CAS PubMed.
- T. F. Massoud and S. S. Gambhir, Genes Dev., 2003, 17, 545–580 CrossRef CAS PubMed.
- J. Cheon and J.-H. Lee, Acc. Chem. Res., 2008, 41, 1630–1640 CrossRef CAS PubMed.
- D. P. Cormode, T. Skajaa, Z. A. Fayad and W. J. M. Mulder, Arterioscler., Thromb., Vasc. Biol., 2009, 29, 992–1000 CrossRef CAS PubMed.
- S. R. Cherry, in Annual Review of Biomedical Engineering, Annual Reviews, Palo Alto, 2006, vol. 8, pp. 35–62 Search PubMed.
- J. Zwerner, Y. Kim, D. Fiorentino and H. Richmond, Arch. Dermatol., 2007, 143, 1025–1030 CrossRef PubMed.
- R. Alwi, S. Telenkov, A. Mandelis, T. Leshuk, F. Gu, S. Oladepo and K. Michaelian, Biomed. Opt. Express, 2012, 3, 2500–2509 CrossRef CAS PubMed.
- A. A. Faraj, N. Luciani, J. Kolosnjaj-Tabi, E. Mattar, O. Clement, C. Wilhelm and F. Gazeau, Contrast Media Mol. Imaging, 2013, 8, 193–203 CrossRef PubMed.
- P. Smirnov, M. Poirier-Quinot, C. Wilhelm, E. Lavergne, J.-C. Ginefri, B. Combadière, O. Clément, L. Darrasse and F. Gazeau, Magn. Reson. Med., 2008, 60, 1292–1297 CrossRef PubMed.
- J. P. Goullé, A. Cattanéo, E. Saussereau, L. Mahieu, M. Guerbet and C. Lacroix, Ann. Pharm. Fr., 2009, 67, 335–339 CrossRef PubMed.
- D. E. Owens Iii and N. A. Peppas, Int. J. Pharm., 2006, 307, 93–102 CrossRef PubMed.
- X. Duan and Y. Li, Small, 2013, 9, 1521–1532 CrossRef CAS PubMed.
- A. Mahapatro and D. K. Singh, J. Nanobiotechnol., 2011, 9, 55 CrossRef CAS PubMed.
- M. Edmundson, N. T. Thanh and B. Song, Theranostics, 2013, 3, 573–582 CrossRef CAS PubMed.
- P. Caravan, J. J. Ellison, T. J. McMurry and R. B. Lauffer, Chem. Rev., 1999, 99, 2293–2352 CrossRef CAS PubMed.
- K. W. Y. Chan and W. T. Wong, Coord. Chem. Rev., 2007, 251, 2428–2451 CrossRef CAS PubMed.
- K. Nwe, M. Bernardo, C. A. S. Regino, M. Williams and M. W. Brechbiel, Bioorg. Med. Chem., 2010, 18, 5925–5931 CrossRef CAS PubMed.
- J. Guenoun, A. Ruggiero, G. Doeswijk, R. C. Janssens, G. A. Koning, G. Kotek, G. P. Krestin and M. R. Bernsen, Contrast Media Mol. Imaging, 2013, 8, 165–174 CrossRef CAS PubMed.
- P. Gillis and S. H. Koenig, Magn. Reson. Med., 1987, 5, 323–345 CrossRef CAS.
- C. W. Jung and P. Jacobs, Magn. Reson. Imaging, 1995, 13, 661–674 CrossRef CAS.
- L. Josephson, J. Lewis, P. Jacobs, P. F. Hahn and D. D. Stark, Magn. Reson. Imaging, 1988, 6, 647–653 CrossRef CAS.
- D. D. Stark, R. Weissleder, G. Elizondo, P. F. Hahn, S. Saini, L. E. Todd, J. Wittenberg and J. T. Ferrucci, Radiology, 1988, 168, 297–301 CAS.
- P. Reimer and T. Balzer, Eur. J. Radiol., 2003, 13, 1266–1276 Search PubMed.
- M. Levy, N. Luciani, D. Alloyeau, D. Elgrabli, V. Deveaux, C. Pechoux, S. Chat, G. Wang, N. Vats, F. Gendron, C. Factor, S. Lotersztajn, A. Luciani, C. Wilhelm and F. Gazeau, Biomaterials, 2011, 32, 3988–3999 CrossRef CAS PubMed.
- A. S. Arbab, L. B. Wilson, P. Ashari, E. K. Jordan, B. K. Lewis and J. A. Frank, NMR Biomed., 2005, 18, 383–389 CrossRef CAS PubMed.
- H. W. Duan, M. Kuang, X. X. Wang, Y. A. Wang, H. Mao and S. M. Nie, J. Phys. Chem. C, 2008, 112, 8127–8131 CAS.
- U. I. Tromsdorf, N. C. Bigall, M. G. Kaul, O. T. Bruns, M. S. Nikolic, B. Mollwitz, R. A. Sperling, R. Reimer, H. Hohenberg, W. J. Parak, S. Forster, U. Beisiegel, G. Adam and H. Weller, Nano Lett., 2007, 7, 2422–2427 CrossRef CAS PubMed.
- J. M. Shin, R. M. Anisur, M. K. Ko, G. H. Im, J. H. Lee and I. S. Lee, Angew. Chem., Int. Ed., 2009, 48, 321–324 CrossRef CAS PubMed.
- S. Tong, S. J. Hou, Z. L. Zheng, J. Zhou and G. Bao, Nano Lett., 2010, 10, 4607–4613 CrossRef CAS PubMed.
- R. Schäfer, R. Bantleon, R. Kehlbach, G. Siegel, J. Wiskirchen, H. Wolburg, T. Kluba, F. Eibofner, H. Northoff, C. D. Claussen and H. P. Schlemmer, BMC Cell Biol., 2010, 11 CrossRef PubMed.
- A. B. Mathiasen, L. Hansen, T. Friis, C. Thomsen, K. Bhakoo and J. Kastrup, Stem Cells Int., 2013, 2013, 353105 Search PubMed.
- Z. L. Shi, K. G. Neoh, E. T. Kang, B. Shuter and S. C. Wang, Contrast Media Mol. Imaging, 2010, 5, 105–111 CAS.
- C.-Y. Yang, M.-F. Tai, S.-T. Chen, Y.-T. Wang, Y.-F. Chen, J.-K. Hsiao, J.-L. Wang and H.-M. Liu, J. Appl. Phys., 2009, 105, 07B314 CrossRef.
- A. Stroh, C. Faber, T. Neuberger, P. Lorenz, K. Sieland, P. M. Jakob, A. Webb, H. Pilgrimm, R. Schober, E. E. Pohl and C. Zimmer, NeuroImage, 2005, 24, 635–645 CrossRef PubMed.
- D.-E. Lee, H. Koo, I.-C. Sun, J. H. Ryu, K. Kim and I. C. Kwon, Chem. Soc. Rev., 2012, 41, 2656–2672 RSC.
- J. Terrovitis, M. Stuber, A. Youssef, S. Preece, M. Leppo, E. Kizana, M. Schar, G. Gerstenblith, R. G. Weiss, E. Marban and M. R. Abraham, Circulation, 2008, 117, 1555–1562 CrossRef PubMed.
- A. K. A. Silva, C. Wilhelm, J. Kolosnjaj-Tabi, N. Luciani and F. Gazeau, Pharm. Res., 2012, 29, 1392–1403 CrossRef CAS PubMed.
- S. C. Berman, C. Galpoththawela, A. A. Gilad, J. W. M. Bulte and P. Waczak, Magn. Reson. Med., 2011, 65, 564–574 CrossRef PubMed.
- E. Pawelczyk, A. S. Arbab, A. Chaudhry, A. Balakumaran, P. G. Robey and J. A. Frank, Stem Cells, 2008, 26, 1366–1375 CrossRef CAS PubMed.
- Z. Shi, K. G. Neoh, E. T. Kang, B. Shuter, S.-C. Wang, C. Poh and W. Wang, ACS Appl. Mater. Interfaces, 2008, 1, 328–335 CrossRef PubMed.
- S. H. Bakhru, E. Altiok, C. Highley, D. Delubac, J. Suhan, T. K. Hitchens, C. Ho and S. Zappe, Int. J. Nanomed., 2012, 7, 4613–4623 CrossRef CAS PubMed.
- C. Yu, J. Zhao, Y. Guo, C. Lu, X. Ma and Z. Gu, J. Biomed. Mater. Res., Part A, 2008, 87, 364–372 CrossRef PubMed.
- P. Guo, L. Cui, Y. Wang, M. Lv, B. Wang and X. S. Zhao, Langmuir, 2013, 29, 8997–9003 CrossRef CAS PubMed.
- A. Akbarzadeh, M. Samiei and S. Davaran, Nanoscale Res. Lett., 2012, 7, 1–13 CrossRef PubMed.
- B. S. Shah and J. J. Mao, in Moleuclar Imaging: Methods and Protocols, ed. K. Shah, 2011, vol. 680, pp. 61–75 Search PubMed.
- Y. Lei, H. Tang, L. Yao, R. Yu, M. Feng and B. Zou, Bioconjugate Chem., 2007, 19, 421–427 CrossRef PubMed.
- S. Danner, H. Benzin, T. Vollbrandt, J. Oder, A. Richter and C. Kruse, Int. J. Cell Biol., 2013, 2013, 12 Search PubMed.
- X. Meng, H. C. Seton, L. T. Lu, I. A. Prior, N. T. K. Thanh and B. Song, Nanoscale, 2011, 3, 977–984 RSC.
- J. H. Lee, Y. M. Huh, Y. Jun, J. Seo, J. Jang, H. T. Song, S. Kim, E. J. Cho, H. G. Yoon, J. S. Suh and J. Cheon, Nat. Med., 2007, 13, 95–99 CrossRef CAS PubMed.
- Y. W. Jun, J. H. Lee and J. Cheon, Angew. Chem., Int. Ed., 2008, 47, 5122–5135 CrossRef CAS PubMed.
- J. Guo, J.-K. Shen, L. Wang, L. Xiao, R.-J. Zhang, W.-F. Luo, Z.-G. Gong, J. Sun, H. Xu, P. Sirois and K. Li, Appl. Biochem. Biotechnol., 2011, 163, 636–648 CrossRef CAS PubMed.
- R. T. Castaneda, A. Khurana, R. Khan and H. E. Daldrup-Link, J. Visualized Exp., 2011, e3482 Search PubMed.
- S. Wang, Y. Zhao, X. Yan, K. Fu and L. Wang, Neural Regener. Res., 2010, 5, 970–974 Search PubMed.
- M. Song, W. K. Moon, Y. Kim, D. Lim, I.-C. Song and B.-W. Yoon, Korean J. Radiol., 2007, 8, 365–371 CrossRef.
- T. J. England, P. M. W. Bath, M. Abaei, D. Auer and D. R. E. Jones, Cytotherapy, 2013, 15, 384–390 CrossRef CAS PubMed.
- H. E. Daldrup-Link, M. Rudelius, G. Piontek, S. Metz, R. Bräuer, G. Debus, C. Corot, J. Schlegel, T. M. Link, C. Peschel, E. J. Rummeny and R. A. J. Oostendorp, Radiology, 2005, 234, 197–205 CrossRef PubMed.
- A. S. Arbab, G. T. Yocum, A. M. Rad, A. Y. Khakoo, V. Fellowes, E. J. Read and J. A. Frank, NMR Biomed., 2005, 18, 553–559 CrossRef CAS PubMed.
- E. V. A. SykovÁ and P. JendelovÁ, Ann. N. Y. Acad. Sci., 2005, 1049, 146–160 CrossRef PubMed.
- C. Bos, Y. Delmas, A. Desmouliere, A. Solanilla, O. Hauger, C. Grosset, I. Dubus, Z. Ivanovic, J. Rosenbaum, P. Charbord, C. Combe, J. W. M. Bulte, C. T. W. Moonen, J. Ripoche and N. Grenier, Radiology, 2004, 233, 781–789 CrossRef PubMed.
- L. H. Reddy, J. L. Arias, J. Nicolas and P. Couvreur, Chem. Rev., 2012, 112, 5818–5878 CrossRef CAS PubMed.
- S. Laurent, D. Forge, M. Port, A. Roch, C. Robic, L. Vander Elst and R. N. Muller, Chem. Rev., 2008, 108, 2064–2110 CrossRef CAS PubMed.
- O. Veiseh, J. W. Gunn and M. Q. Zhang, Adv. Drug Delivery Rev., 2010, 62, 284–304 CrossRef CAS PubMed.
- M. Babič, D. Horak, M. Trchova, P. Jendelova, K. Glogarova, P. Lesny, V. Herynek, M. Hajek and E. Sykova, Bioconjugate Chem., 2008, 19, 740–750 CrossRef PubMed.
- K. Andreas, R. Georgieva, M. Ladwig, S. Mueller, M. Notter, M. Sittinger and J. Ringe, Biomaterials, 2012, 33, 4515–4525 CrossRef CAS PubMed.
- V. Mailander, M. R. Lorenz, V. Holzapfel, A. Musyanovych, K. Fuchs, M. Wiesneth, P. Walther, K. Landfester and H. Schrezenmeier, Mol. Imag. Biol., 2008, 10, 138–146 CrossRef PubMed.
- C. W. Lu, Y. Hung, J. K. Hsiao, M. Yao, T. H. Chung, Y. S. Lin, S. H. Wu, S. C. Hsu, H. M. Liu, C. Y. Mou, C. S. Yang, D. M. Huang and Y. C. Chen, Nano Lett., 2007, 7, 149–154 CrossRef CAS PubMed.
- A. Ruiz, Y. Hernandez, C. Cabal, E. Gonzalez, S. Veintemillas-Verdaguer, E. Martinez and M. P. Morales, Nanoscale, 2013 10.1039/C3NR01412F.
- F. Benyettou, Y. Lalatonne, I. Chebbi, M. Di Benedetto, J. M. Serfaty, M. Lecouvey and L. Motte, Phys. Chem. Chem. Phys., 2011, 13, 10020–10027 RSC.
- Y. Lalatonne, M. Monteil, H. Jouni, J. M. Serfaty, O. Sainte-Catherine, N. Lièvre, S. Kusmia, P. Weinmann, M. Lecouvey and L. Motte, J. Osteoporosis, 2010, 747852 CAS.
- L. Motte, F. Benyettou, C. de Beaucorps, M. Lecouvey, I. Milesovic and Y. Lalatonne, Faraday Discuss., 2011, 149, 211–225 RSC.
- J. Huang, L. Wang, R. Lin, A. Y. Wang, L. Yang, M. Kuang, W. Qian and H. Mao, ACS Appl. Mater. Interfaces, 2013, 5, 4632–4639 CAS.
- A. G. Roca, S. Veintemillas-Verdaguer, M. Port, C. Robic, C. J. Serna and M. P. Morales, J. Phys. Chem. B, 2009, 113, 7033–7039 CrossRef CAS PubMed.
- C. Chouly, D. Pouliquen, I. Lucet, J. J. Jeune and P. Jallet, J. Microencapsulation, 1996, 13, 245–255 CrossRef CAS PubMed.
- D. Maity, P. Chandrasekharan, C.-T. Yang, K.-H. Chuang, B. Shuter, J.-M. Xue, J. Ding and S.-S. Feng, Nanomedicine, 2010, 5, 1571–1584 CrossRef CAS PubMed.
- G. Bealle, R. Di Corato, J. Kolosnjaj-Tabi, V. Dupuis, O. Clement, F. Gazeau, C. Wilhelm and C. Menager, Langmuir, 2012, 28, 11843–11851 CrossRef PubMed.
- L. Lartigue, P. Hugounenq, D. Alloyeau, S. P. Clarke, M. Lévy, J.-C. Bacri, R. Bazzi, D. F. Brougham, C. Wilhelm and F. Gazeau, ACS Nano, 2012, 6, 10935–10949 CrossRef CAS PubMed.
- L. Lartigue, P. Hugounenq, D. Alloyeau, S. P. Clarke, M. Levy, J. C. Bacri, R. Bazzi, D. F. Brougham, C. Wilhelm and F. Gazeau, ACS Nano, 2012, 6, 10935–10949 CrossRef CAS PubMed.
- A. A. Guay-Begin, P. Chevallier, L. Faucher, S. Turgeon and M. A. Fortin, Langmuir, 2012, 28, 774–782 CrossRef CAS PubMed.
- M. A. Fortin, R. M. Petoral, F. Soderlind, A. Klasson, M. Engstrom, T. Veres, P. O. Kall and K. Uvdal, Nanotechnology, 2007, 18, 395501 CrossRef.
- R. M. Guo, N. Cao, F. Zhang, Y. R. Wang, X. H. Wen, J. Shen and X. T. Shuai, Eur. J. Radiol., 2012, 22, 2328–2337 CrossRef PubMed.
- M. Modo, D. Cash, K. Mellodew, S. C. R. Williams, S. E. Fraser, T. J. Meade, J. Price and H. Hodges, NeuroImage, 2002, 17, 803–811 CrossRef.
- J. M. Lee, B. S. Kim, H. Lee and G. I. Im, Mol. Ther., 2012, 20, 1434–1442 CrossRef CAS PubMed.
- A. Khurana, F. Chapelin, G. Beck, O. D. Lenkov, J. Donig, H. Nejadnik, S. Messing, N. Derugin, R. C.-F. Chan, A. Gaur, B. Sennino, D. M. McDonald, P. J. Kempen, G. A. Tikhomirov, J. Rao and H. E. Daldrup-Link, Radiology, 2013, 269, 186–197 CrossRef PubMed.
- E. S. M. Lee, J. Chan, B. Shuter, L. G. Tan, M. S. K. Chong, D. L. Ramachandra, G. S. Dawe, J. Ding, S. H. Teoh, O. Beuf, A. Briguet, K. C. Tam, M. Choolani and S. C. Wang, Stem Cells, 2009, 27, 1921–1931 CrossRef CAS PubMed.
- A. Balakumaran, E. Pawelczyk, J. Q. Ren, B. Sworder, A. Chaudhry, M. Sabatino, D. Stroncek, J. A. Frank and P. G. Robey, Plos One, 2010, 5, e11462 Search PubMed.
- L. Kostura, D. L. Kraitchman, A. M. Mackay, M. F. Pittenger and J. W. M. Bulte, NMR Biomed., 2004, 17, 513–517 CrossRef PubMed.
- J. W. M. Bulte, D. L. Kraitchman, A. M. Mackay and M. F. Pittenger, Blood, 2004, 104, 3410–3412 CrossRef CAS PubMed.
- T. D. Henning, E. J. Sutton, A. Kim, D. Golovko, A. Horvai, L. Ackerman, B. Sennino, D. McDonald, J. Lotz and H. E. Daldrup-Link, Contrast Media Mol. Imaging, 2009, 4, 165–173 CrossRef CAS PubMed.
- S. J. H. Soenen, U. Himmelreich, N. Nuytten and M. De Cuyper, Biomaterials, 2011, 32, 195–205 CrossRef CAS PubMed.
- Y. C. Chen, J. K. Hsiao, H. M. Liu, I. Y. Lai, M. Yao, S. C. Hsu, B. S. Ko, Y. C. Chen, C. S. Yang and D. M. Huang, Toxicol. Appl. Pharmacol., 2010, 245, 272–279 CrossRef CAS PubMed.
- C. Xu, D. Miranda-Nieves, J. A. Ankrum, M. E. Matthiesen, J. A. Phillips, I. Roes, G. R. Wojtkiewicz, V. Juneja, J. R. Kultima, W. Zhao, P. K. Vemula, C. P. Lin, M. Nahrendorf and J. M. Karp, Nano Lett., 2012, 12, 4131–4139 CrossRef CAS PubMed.
- A. K. Gupta and M. Gupta, Biomaterials, 2005, 26, 3995–4021 CrossRef CAS PubMed.
- P. Tartaj, M. D. Morales, S. Veintemillas-Verdaguer, T. Gonzalez-Carreno and C. J. Serna, J. Phys. D: Appl. Phys., 2003, 36, R182–R197 CrossRef CAS.
- Q. A. Pankhurst, J. Connolly, S. K. Jones and J. Dobson, J. Phys. D: Appl. Phys., 2003, 36, R167–R181 CrossRef CAS.
- W. Jiang, B. Y. S. Kim, J. T. Rutka and W. C. W. Chan, Nat. Nanotechnol., 2008, 3, 145–150 CrossRef CAS PubMed.
- J. H. Park, G. von Maltzahn, L. L. Zhang, A. M. Derfus, D. Simberg, T. J. Harris, E. Ruoslahti, S. N. Bhatia and M. J. Sailor, Small, 2009, 5, 694–700 CrossRef CAS PubMed.
- J. H. Park, G. von Maltzahn, L. L. Zhang, M. P. Schwartz, E. Ruoslahti, S. N. Bhatia and M. J. Sailor, Adv. Mater., 2008, 20, 1630–1635 CrossRef CAS PubMed.
- M. M. Arnida, A. Janat-Amsbury, C. M. Ray, Peterson and H. Ghandehari, Eur. J. Pharm. Biopharm., 2011, 77, 417–423 CrossRef CAS PubMed.
- B. D. Chithrani, A. A. Ghazani and W. C. W. Chan, Nano Lett., 2006, 6, 662–668 CrossRef CAS PubMed.
- J. A. Frank, B. R. Miller, A. S. Arbab, H. A. Zywicke, E. K. Jordan, B. K. Lewis, L. H. Bryant and J. W. M. Bulte, Radiology, 2003, 229, 610 CrossRef.
- J. M. Harris and R. B. Chess, Nat. Rev. Drug Discovery, 2003, 2, 214–221 CrossRef CAS PubMed.
- F. Alexis, E. Pridgen, L. K. Molnar and O. C. Farokhzad, Mol. Pharmaceutics, 2008, 5, 505–515 CrossRef CAS PubMed.
- P. Wunderbaldinger, L. Josephson and R. Weissleder, Bioconjugate Chem., 2002, 13, 264–268 CrossRef CAS PubMed.
- A. S. Arbab, G. T. Yocum, L. B. Wilson, A. Parwana, E. K. Jordan, H. Kalish and J. A. Frank, Mol. Imaging, 2004, 3, 24–32 CrossRef CAS PubMed.
- B. L. Strand, L. Ryan, P. I. Veld, B. Kulseng, A. M. Rokstad, G. Skjak-Braek and T. Espevik, Cell Transplant., 2001, 10, 263–275 CAS.
- M. Lewin, N. Carlesso, C. H. Tung, X. W. Tang, D. Cory, D. T. Scadden and R. Weissleder, Nat. Biotechnol., 2000, 18, 410–414 CrossRef CAS PubMed.
- L. Josephson, C. H. Tung, A. Moore and R. Weissleder, Bioconjugate Chem., 1999, 10, 186–191 CrossRef CAS PubMed.
- X. H. Jing, L. Yang, X. J. Duan, B. Xie, W. Chen, Z. Li and H. B. Tan, Jt., Bone, Spine, 2008, 75, 432–438 CrossRef PubMed.
- R. Calloni, E. A. Aparicio Cordero, J. A. Pegas Henriques and D. Bonatto, Stem Cells Dev., 2013, 22, 1455–1476 CrossRef CAS PubMed.
- H. J. Lee, B. H. Choi, B.-H. Min and S. R. Park, Arthritis Rheum., 2009, 60, 2325–2332 CrossRef CAS PubMed.
- D. Campbell and M. Pei, Cells, 2012, 1, 1107–1120 CrossRef.
- S. Gronthos, P. J. Simmons, S. E. Graves and P. G. Robey, Bone, 2001, 28, 174–181 CrossRef CAS.
- A. de Mel, J. T. Oh, B. Ramesh and A. M. Seifalian, Regener. Med., 2012, 7, 335–347 CrossRef CAS PubMed.
- M. K. Majumdar, M. Keane-Moore, D. Buyaner, W. B. Hardy, M. A. Moorman, K. R. McIntosh and J. D. Mosca, J. Biomed. Sci., 2003, 10, 228–241 CrossRef CAS.
- S. J. DeNardo, R. Liu, H. Albrecht, A. Natarajan, J. L. Sutcliffe, C. Anderson, L. Peng, R. Ferdani, S. R. Cherry and K. S. Lam, J. Nucl. Med., 2009, 50, 625–634 CrossRef CAS PubMed.
- E. Garanger, D. Boturyn, J.-L. Coll, M.-C. Favrot and P. Dumy, Org. Biomol. Chem., 2006, 4, 1958–1965 CAS.
- H. Hong, Y. Zhang, J. Sun and W. Cai, Nano Today, 2009, 4, 399–413 CrossRef CAS PubMed.
- W. Xu, T. Luo, P. Li, C. Zhou, D. Cui, B. Pang, Q. Ren and S. Fu, Int. J. Nanomed., 2012, 7, 915–924 CAS.
- F. Danhier, A. L. Breton and V. r. Préat, Mol. Pharmaceutics, 2012, 9, 2961–2973 CrossRef CAS PubMed.
- F. Danhier, B. Vroman, N. Lecouturier, N. Crokart, V. Pourcelle, H. Freichels, C. Jérôme, J. Marchand-Brynaert, O. Feron and V. Préat, J. Controlled Release, 2009, 140, 166–173 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2013 |