Oligonucleotide-templated chemical reactions: pushing the boundaries of a nature-inspired process
Claudia
Percivalle
ab,
Jean-François
Bartolo
a and
Sylvain
Ladame
*a
aDepartment of Bioengineering, Imperial College London, South Kensington campus, London SW7 2AZ, UK. E-mail: sladame@imperial.ac.uk; Fax: (+44) 02075949817; Tel: (+44) 02075945308
bDepartment of Chemistry, University of Pavia, 27100 Pavia, Italy
First published on 21st September 2012
Abstract
Widespread in nature, oligonucleotide-templated reactions of phosphodiester bond formation have inspired chemists who are now applying this elegant strategy to the catalysis of a broad range of otherwise inefficient reactions. This review highlights the increasing diversity of chemical reactions that can be efficiently catalysed by an oligonucleotide template, using Watson–Crick base-pairing to bring both reagents in close enough proximity to react, thus increasing significantly their effective molarity. The applications of this elegant concept for nucleic acid sensing and controlled organic synthesis will also be discussed.
Introduction
The concept of the oligonucleotide-templated reaction (OTR) is widely present in nature. Before it divides, a cell must duplicate its DNA. This natural process called DNA replication is probably one of the oldest examples of an oligonucleotide templated reaction where each strand of a DNA double helix serves as a template for the production of a complementary strand. The chemistry involved is the creation of a phosphodiester bond and is catalysed by an enzyme DNA polymerase. But there also exist enzymes that synthesise RNA from a DNA template (transcription by RNA polymerases) or DNA from a RNA template (reverse transcriptases). High fidelity transfer of genetic information during replication relies on the specificity of Watson–Crick hydrogen bonding in each new base pair (A always facing a T and G always facing a C) but also very importantly on the recognition of the overall shape of the DNA nucleobases by the polymerase. While all these biological processes proceed via formation of a phosphodiester bond, naturally occurring oligonucleotide-templated reactions also include the creation of peptide/amide bonds from an RNA template catalysed by ribonucleoproteins called ribosomes. In 1983, Kary Mullis developed the concept of a Polymerase Chain Reaction (PCR) where billions of copies of a DNA template can be produced by a repetitive cycle of (i) denaturation into DNA single strands, (ii) annealing to a combination of two carefully designed DNA primers, and (iii) elongation of the DNA primers by a DNA polymerase.1 This discovery represented a biotechnological breakthrough, allowing scientists to produce large amounts of DNA from only a very small fragment.OTRs commonly use sequence-specific hybridization to bring together a pair of small molecule substrates, thus leading to chemical bond formation. The first successful example of a non-enzymatic ligation of two thymidine hexanucleotides catalysed only by a polyadenosine template was reported in 1966 by Naylor and co-workers.2 The dodecathymidine was however obtained with a very poor yield (ca. 5%). Significantly more efficient systems were developed two decades later, notably by von Kiedrowski who reported the first auto-catalysed DNA-templated ligation of two trinucleotides using a self-complementary hexanucleotide template.3 Despite a low yield (ca. 12%) this system introduces the concept of catalytic turnover of the template (the ligation product being identical to the template, it participates in the subsequent steps of the catalytic cycle) in DNA-templated-reactions. Although conceptually very interesting, these various studies aiming at templating the formation of phosphodiester bonds between (poly)nucleotides all suffered from a number of severe drawbacks (low yields, applicability to the synthesis of short DNA strands only…) and had therefore very limited practical use. They however prepared the way for the modern developments of OTRs with valuable applications as sensing or synthetic tools. In the present article, we will review the increasing but yet limited diversity of chemistries that have been successfully catalysed by an oligonucleotide template. We will then highlight the most successful applications in this ever-growing field, focusing mainly on sequence and/or structure-specific nucleic acid sensing and DNA-templated organic synthesis. The main advantages and drawbacks of these conceptually elegant systems compared to well-established methodologies (e.g. molecular beacons for nucleic acid sensing; metallo- or organo-catalysis for controlled organic synthesis) will also be discussed.
Oligonucleotide-templated reactions (OTRs): a conceptually elegant strategy
The general concept of OTR relies on the use of sequence-specific Watson–Crick base pairing to bring together and promote the reaction between two molecules each covalently attached to a short strand of DNA. By increasing the effective molarity of two (or more) monomers, otherwise present in solution at concentrations too low to be favourable (typically in the nanomolar or micromolar range), the DNA templating effect dramatically increases the reaction rate, thus enabling controlled reaction between the monomers.Depending on the nature of the application, two main strategies have been reported (Fig. 1).
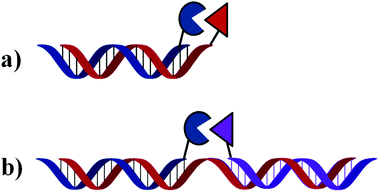 | ||
| Fig. 1 General mechanism of OTRs. Reaction occurs either at the end (a) or in the middle (b) of the homo- or hetero-duplex formed. | ||
1. The two DNA strands holding each monomer are complementary to each other. In this case, hybridisation between both strands will bring both reagents in close proximity and will promote the desired reaction occurring at the blunt end of the newly formed duplex.
2. The two DNA strands holding each monomer are complementary to neighbouring sequences of a third (and longer) oligonucleotide which serves as a template. Only upon simultaneous hybridisation of both functionalised oligonucleotides to the same template strand will both monomers be found in close enough proximity to react with each other. In this case, reaction will occur at the middle of the newly formed duplex (or three way junction).
The backbone of the template and of the complementary strands holding the reactive moieties can also be of different chemical nature. Although they are typically ribo- or deoxyribo-oligonucleotides, uncharged analogues capable of forming highly stable heteroduplexes via sequence-specific hybridisation to the DNA (or RNA) template of interest are also commonly used. Peptide Nucleic Acid4 (PNA) backbones have received particular attention due to their high chemical and enzymatic stability and their ability to form PNA![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :DNA and PNA:RNA heteroduplexes more stable that the corresponding DNA or RNA homoduplexes.
:DNA and PNA:RNA heteroduplexes more stable that the corresponding DNA or RNA homoduplexes.
Oligonucleotide-templated chemistries: from phosphodiester bond formation to organometallic catalysis, an increasing but yet limited diversity
The production of nucleic acids (or nucleic acid analogues) via RNA- or DNA-catalysed phosphodiester bond formation represents the gold example of oligonucleotide-templated chemistry (OTC).2,3 In the last decade, thanks to a better understanding of its basic principles, OTC has found increasing applications in the fields of synthetic methodology and chemical biology. Before discussing the most important and innovative applications, with particular emphasis on multi-step organic synthesis and nucleic acids sensing, we aim to provide an overview of the increasing (although limited) range of chemical reactions compatible with oligonucleotide templating.Nucleic acids and nucleic acid analogues
Following Orgel's pioneering work,5 many research groups used OTC strategies to generate linear and circular nucleic acid analogues via phosphodiester,6,7 thioester8 and selenoester9 bond formation.Early examples from the Kool and Letsinger groups include OT auto-ligations that consisted in an SN2 reaction between two oligonucleotides brought in close proximity as a result of their simultaneous hybridisation to a single complementary strand serving as a template: (1) an oligonucleotide functionalised at its 5′-end with various electrophiles (e.g., bromoacetyl, iodo, tosyl) and (2) a second oligonucleotide functionalised at its 3′-end with a phosphorothioate moiety.6,8
In 2002, Lynn and co-workers reported the first synthesis of an oligonucleotide analogue via a DNA-templated reductive amination,10 shortly followed by Rosenbaum and Liu.11 For instance, OTR between aldehyde-functionalised templates (e.g., benzaldehyde, or glyoxal linked reagents) and amine-modified oligonucleotides were successfully carried out at room temperature, in aqueous solution, in the presence of NaBH3CN at millimolar concentrations.12 Peptide bond formation (a and b, Fig. 2) is another example of OTR which was first applied to the synthesis of amide-linked DNA analogues.13 Oligonucleotide mimics such as PNA–oligomers14 and peptide–DNA conjugates15 were also synthesized using a similar approach that exploits OT amide formation.
Other analogues were synthesised that retained the integrity of the phospho-ribose backbone but involved chemical reactions between nucleobases. An early example of end-to-end DNA strand ligation exploited the well known ability of thymidines to undergo photo-dimerization (λexc > 290 nm).16 More recent strategies relied on the psoralene-thymidine photoreaction17 and stilbene-dimerisation.18 Later, Saito and co-workers exploited a [2 + 2] photo-cycloaddition between 5-vinyldeoxyuridine modified oligonucleotides to develop a reversible DNA-templated photoligation–photocleavage system.19 The reversibility of the system was guaranteed by the need of different excitation wavelengths to induce the photo-cycloaddition and photo-reversion processes (λexc/direct = 366 nm, λexc/invers = 302 nm).
However, OTRs are not limited to reactions that promote the formation of oligonucleotides or structural analogues of oligonucleotides. From the late 90s an increasing number of reactions have been designed that aimed to exploit DNA as a catalytic template. However, they still remain only a small fraction of the large pool of reactions used by synthetic chemists or found in nature. Whilst most of these new OTRs could be depicted as “coupling reactions”, a smaller number of reactions involving the transformation of functional groups have also been reported.20
Coupling reactions exploit DNA hybridization to mediate “ligation” of DNA-linked reactive groups. These reactions can be carried out either (i) with a non DNA-linked activator or catalyst or (ii) under conditions that do not require any other reagents aside from the oligonucleotide conjugates (“reagent-free”). A list of the most significant coupling reactions based on an oligonucleotide-templated strategy are summarised in Fig. 2.
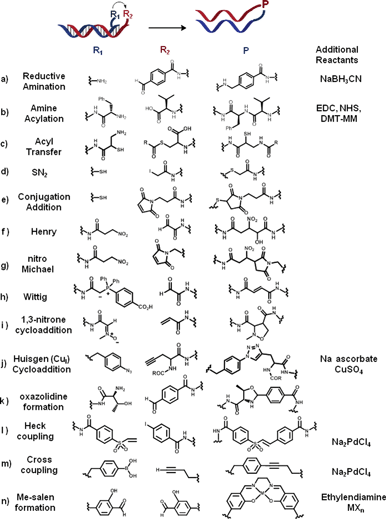 | ||
| Fig. 2 List of coupling reactions (a–n) successfully applied to the concept of OTC. Two complementary oligonucleotides are functionalised with two reactive moieties (R1 and R2). Upon hybridisation of the two complementary strands, R1 reacts with R2 to form a product P (in the absence or in the presence of additional reagents). | ||
Peptides and peptidomimetics
In 2002, Liu and co-workers applied OTC to the efficient synthesis of amides in water. They successfully coupled carboxylic acid-bearing reagents (e.g., phenylalanine, D-leucine) to an amine-functionalised template at pH 6.0 using 1-(3-dimethylaminopropyl)-3-ethylcarbodiimide (EDC), N-hydroxy-sulfo-succinimide (sulfo-NHS), and 4-(4,6-dimethoxy-1,3,5-triazin-2-yl)-4-methylmorpholinium chloride (DMT-MM) as carboxylic acid activators.12 OT–peptide bond formation was subsequently exploited for the development of macrocyclic small molecule libraries,21 but also for sequence-programmed multistep OT synthesis.22,23 An efficient multistep OT-synthesis of monocyclic and bicyclic N-acyl-oxazolidines was also recently achieved from aldehydes, amino-alcohols and acyl donor (k, Fig. 2).24 Considering the large amount of commercially available carboxylic acids and chiral amines, OTRs of amide formation have great potential for the efficient and controlled synthesis of products with increased structural and chemical diversity.Another OT strategy to generate peptidomimetics is the acyl transfer reaction between an activated thioester and an N-nucleophile. The DNA-catalysed transfer of a reporter group by native chemical ligation25 was first reported by Grossmann and Seitz.26 They used an iso-cystein (iCys)–PNA mediator for the acyl transfer of a thioester-modified PNA. The hybridization of both PNA probes triggered the trans-thioesterification, followed by an irreversible S → N acyl migration (c, Fig. 2). In 2011, Mc Kee et al. published a systematic study in which they have compared the reactivity of different N-nucleophiles (i.e. amines, hydrazines, benzylamines, aminooxy, hydrazides and hydrazone-nicotinates) using the same template as for OT acyl transfer.27 Unsurprisingly, most efficient acylation reactions (at neutral pH) were observed with N-nucleophiles characterised by a low pKa value and/or benefiting from the presence of α-effects.
Sequence-specific DNA-templated SN2 substitutions6–9 and additions to α,β-unsaturated systems (e.g., maleimide and vinyl sulfones) have also been shown to proceed in good yields with excellent sequence selectivity (d and e, Fig. 2).28 Different nucleophiles have been employed ranging from thiols to amines. Under optimised experimental conditions (pH 7.5, 25 °C, 250 mM NaCl, 60 nM concentration of template and reagent), only “matched” oligonucleotide sequences (i.e. complementary to the template) afforded the desired product, regardless of key parameters such as transition-state geometry, steric hindrance, and conformational flexibility.
Organometallic catalysis and carbon–carbon bond formation
Interestingly, few organometallic-catalysed reactions and reactions of carbon–carbon bond formation have also been recently added to the list of reactions suitable for oligonucleotide templating. This discovery could be considered one of the main breakthroughs in OTC considering the great importance these two broad classes of reactions have in synthetic organic chemistry.The first examples of nitro-aldol (Henry) and nitro-Michael additions were reported by Liu and co-workers.12 In this study, nitroalkene-linked oligonucleotides were shown to react with either aldehydes (Henry, Fig. 2f) or maleimides (Michael, Fig. 2g) with high efficiency and sequence specificity at pH 7.5–8.5 and 25 °C.
In the same article, Liu and co-workers expanded the scope of OTC reactions to the Wittig olefination. The first example was carried out using a stabilized phosphorus ylide reagent, as depicted in Fig. 2h. This molecule can react with aldehydes (e.g. benzaldehyde or glyoxal), in the presence of an oligonucleotide template, providing the corresponding E-olefin with yields as high as 90%, at pH 6.0–8.0 and 25 °C.12 Because it proceeds in good yields in aqueous solution (pH 8.5) without any further additives, this OT version of the famous Wittig reaction was subsequently further exploited in sequential multistep synthesis of triolefins.29 Recently a parallel multistep methodology has been developed, opening the opportunity to create oligomeric libraries of increased diversity one-pot.30
A DNA-templated version of the 1,3-dipolar cycloaddition has also been reported by the Liu group. One of the first attempts showed how a nitrone-linked reagent (i, Fig. 2) can react efficiently with activated alkenes as maleimides, vinyl sulfones and acrylamide in a sequence specific manner at pH 7.5.12 One year later, the same group reported that the Cu(I)-mediated Huisgen cycloaddition, best example of click-reaction,31 could be catalysed by introduction of a DNA-template: a 1,4-disubstituted triazoylalanine adduct was synthesised in 32% overall yield by reaction between propargyl-glycine and phenylazide-functionalised oligonucleotides in the presence of 500 μM copper(II) sulphate and sodium ascorbate (j, Fig. 2).32 Template-directed click reaction has then been used for oligonucleotide strand ligation,28 producing an unnatural extended DNA backbone linkage, and for sequence-specific conjugation of PNA–DNA, PNA–PNA with single nucleotide discrimination.33 Recently, the OT-click cycloaddition has been exploited as a novel biosensing strategy to probe Cu(II) ions with excellent selectivity and sensitivity.34
Another successful example of OT-organometallic reaction is the Palladium-mediated Heck coupling. In the presence of a water-soluble Pd pre-catalyst (Na2PdCl4, 170 μM), aryl iodide reagents (l, Fig. 2) were shown to react with a series of alkene-bearing templates (e.g., maleimide, acrylamide, vinyl sulfone, cinnamamide) at pH 5.0 and 25 °C, although in moderate yields only.12 Pd(II)-catalyzed cross coupling was also reported between aryl boronic acids and either alkenyl, alkinyl and heteroaromatic (furan) derivatives (m, Fig. 2).35
In 2001, Ni2+ or Mn2+-catalysed reactions between two salicylaldehyde-functionalised DNA strands brought together by a complementary template were shown to yield DNA–metallosalen conjugates in the presence of ethylene diamine (n, Fig. 2).36 Few years later the same OTR was applied to the assembly of linear and branched nanostructures.37
In 2008, Franzini and Kool reported a mercury-catalysed OT organometallic reaction between a p-mercuriobenzoato probe and a Rhodamine B phenylthiosemicarbazide (Rhops) masked fluorophore.38 When brought into proximity, the mercury ion Hg(II) catalyses the semicarbazide cyclization to yield an oxadiazole product.38 This transformation unlocks the rhodamine spirolactam ring, switching-on a fluorescent signal (OFF–ON system, see section on DNA/RNA sensing).38
Functional group transformation
Whilst OT “coupling reactions” are most widespread, those leading to the transformation of functional groups broaden the spectrum of applications OTRs have in the fields of drug delivery39,40 and nucleic acid sensing.41Ester hydrolysis: in a pioneering work from 2007, Ma and Taylor described the first system in which a nucleic acid triggers the catalytic release of a drug. The DNA-templated and imidazole-catalysed hydrolysis of a p-nitrophenyl ester led to the formation of a carboxylic acid with concomitant release of p-nitrophenol (a, Fig. 3).39 Considering the large number of cytotoxic drugs that contain a phenoxy group (e.g. daunorubicin, phenol mustard or fluorouracil), this strategy can potentially be applied to the intracellular delivery of any phenoxy-containing drugs from hydroxymethylphenyl-based prodrugs.42 The same principle was subsequently extended to Cu(II)-catalysed hydrolysis of aryl esters by Kraemer and co-workers.43
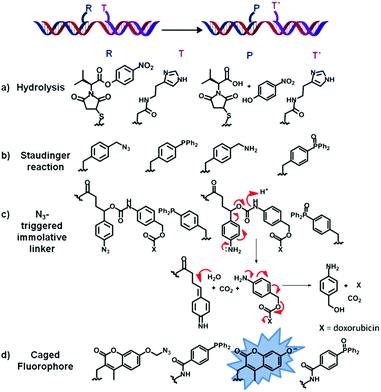 | ||
| Fig. 3 List of four examples (a–d) of chemical reactions of functional group transformation successfully applied to the concept of OTC. Briefly, two oligonucleotides are functionalised with two reactive moieties R and T (here represented in blue and purple, respectively). Upon simultaneous binding of both functionalised oligonucleotides to a single complementary strand (represented here in red), R reacts with T to form product P and T′. | ||
Staudinger reaction: in 2005, Liu and co-workers reported the first successful example of OT Staudinger reaction between a tertiary phosphine and an organic azide DNA-linked template (pH 10, 25–37 °C, Fig. 3b).44 A few years later, Winssinger and co-workers developed a novel and versatile design for the release of different functional molecules, including the cytotoxic doxorubicin, based on an azide-reduction triggered immolative linker (c, Fig. 3).45 The Staudinger reaction between the PNA–phosphine and the PNA–aryl azide, in presence of an RNA template, led to the rapid cleavage of the carbamoyl-linker affording p-hydroxymethyl aniline and free doxorubicin. The relevance of this procedure to drug delivery is highlighted by the bioorthogonality of the azido group with a cellular environment and the biocompatibility of the Staudinger reaction.46
A modification of the Staudinger reaction, developed by Bertozzi and co-workers,47 affords an amide product via an intramolecular cyclization between an ester group on the phosphine and the aza-ylide intermediate formed upon azide reduction. This Staudinger ligation47 was exploited by Taylor and co-workers in an OT manner, using the monoalkylated fluorescein ester of 2-carboxy triphenylphosphine (TPP) and an α-azido acetic acid conjugated to a PNA probe.48 Hydrolysis of the monoalkylated fluorescein ester, with concomitant amide bond formation, resulted in an increased fluorescence. Although the reaction efficiency proved to be sequence-dependent, the Staudinger ligation did not proceed to completion, likely due to the formation of oxidized phosphines.48
Several fluorogenic systems have been reported to date exploiting nucleic acid-triggered fluorescent probe activation by Staudinger reaction. Azido-rhodamine49 and 7-azidocoumarin50 have been extensively used as masked fluorophores, thanks to the possibility of restoring their fluorescence properties upon OT-reduction of the azide moiety into the corresponding amine by action of triphenyl51,52 or trialkyl53 phosphine and dithiothreitol (DTT)49 oligonucleotide conjugates. Another approach of OT Staudinger reaction used azido-based protecting group functionalities for the design of a phosphine-sensitive pro-fluorophore. This “caging methodology” has been applied to 7-azidomethoxy-coumarin (d, Fig. 3)54 and bis-azidomethyl-protected fluorescein probes.55 The OT-reduction of the azido moiety, by TPP, generated an amino hemiacetal, which was quickly hydrolysed thus yielding an unmasked phenol.54,55
OTRs applied to DNA/RNA sensing: a fluorescence-based readout
The ability of OTC to direct product formation in a sequence specific manner and in the presence of a complementary template can be applied to the detection of specific nucleic acid sequences (typically used as a template). Designing OTRs for sensing applications requires the development of reactions highly sequence-selective and that, preferentially, do not require any additional reactants other than the oligonucleotide probes themselves. Typically, such reagent-free reactions offer the advantages of being faster, more biocompatible and more selective than those requiring extra catalyst(s). The need for additional reactants is particularly detrimental for in vivo sensing applications, mainly because of delivery issues and because of the cytotoxicity of the catalysts employed (e.g., Cu(I), Pd(II), Hg(II)…).56Early examples of OT auto-ligations from the groups of Kool and Letsinger6,8 suffered mainly from analytical drawbacks, the determination of the OTR efficiency requiring the isolation (e.g., gel electrophoresis, HPLC) and characterisation of the ligated oligonucleotide.8 The lack of an easily detectable readout of reaction outcome precluded the application of this type of self-ligations for the detection of nucleic acids in vivo.
Although the OTRs exploited for sensing purposes still remain limited, several strategies have been developed in order to overcome the previously cited limitation. These novel approaches can be divided into three main groups: (i) OT-fluorescent energy transfer, (ii) OT-restoration of a fluorescent signal and (iii) OT–fluorogenic reactions (Fig. 4). In all three cases, reaction efficiency is directly correlated to the sensing of a nucleic acid target of interest and can be easily and rapidly monitored in situ using a spectroscopic (UV/fluorescence) readout.
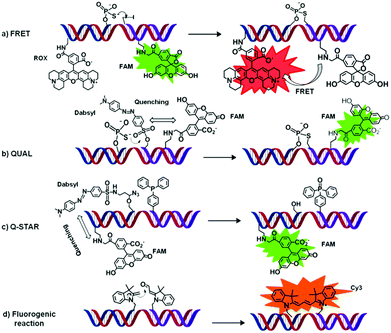 | ||
| Fig. 4 Four examples (a–d) of OTRs applied to DNA (or RNA) sensing. All sensing strategies are based on a fluorescent readout. Briefly, upon binding of two functionalised oligonucleotide probes (here represented in blue and purple) to a single oligonucleotide of interest (i.e. that needs to be sensed, here represented in red) a chemical reaction occurs that leads to the appearance of a characteristic fluorescence signal. | ||
Fluorescent energy transfer (FRET) probes. An OT–SN2 reaction (auto-ligation) was employed to covalently link two oligonucleotides bearing respectively a donor and an acceptor fluorophore generating a unique oligonucleotide product.57 The proximity of the FRET donor and acceptor generates a new specific signal (a, Fig. 4). This approach was first reported by Kool and co-workers who achieved highly specific nucleic acid sensing via an OT-ligation process (SN2 reaction) using a 3′-end phosphorothioate as a nucleophile and a 5′-end iodide oligonucleotide as electrophile, labelled at their other ends with rhodamine (ROX, acting as an acceptor) and 5-carboxyfluorescein (FAM, acting as a donor), respectively. Using this kind of approach, differentiation in a complex mixture of complementary (matched) and non-complementary (mismatched) RNA and DNA sequences was achieved.57 Although there exist only very few examples of OTRs applied to nucleic acid sensing and based exclusively on FRET, more recent developments of FRET-based probes (e.g. QUAL-FRET) combining FRET and fluorescence quenching will be discussed in the section below.
Restoration of a fluorescent signal. A different approach relies on the restoration/release of a fluorescent signal in response to an OT-reaction (b and c, Fig. 4). In such systems, the OTR results either in a loss (e.g. via hydrolysis) of fluorescence quencher, or in an electronic and/or conformational modification of the reaction product that enables recovery of the fluorescent signal.
Quenched auto-ligation (QUAL) probes are probably the most representative examples in this category.58 Fluorescent enhancement relies on a resonance energy transfer (RET) between a fluorophore and a quencher, a mechanism somewhat similar to that observed with Molecular Beacons (MBs). Typically, QUAL probes consist of (1) an unlabeled oligonucleotide modified with a nucleophilic phosphorothioate moiety at its 3′-end (b, Fig. 4), and (2) a dual-labelled electrophilic oligonucleotide containing a dabsyl group (quencher, directly attached at the 5′-hydroxy terminus), and a fluorescent dye.58 Simultaneous hybridization of both probes to a complementary DNA (or RNA) target results in the quencher loss (via a SN2 mechanism) and subsequent release of the fluorophore emission (ON-switch). QUAL probes have been exploited first in a single colour format, using a combination of fluorescein with dansyl quencher.58 Subsequently, Kool and co-workers demonstrated that single nucleotide differences in a target oligonucleotide could be easily distinguished using QUAL multicolour detection.59 Later improvements on QUAL probes include the introduction/modification of the linker (e.g. 5′-propane or butane-diol) between the oligonucleotide and the probe-head (i.e. the reactive moiety).60 When compared to the “first generation”, sulphur-bridged products, an increased flexibility within the ligated product is shown. This led to a reduced affinity for the oligonucleotide target/template, thus reducing significantly the product inhibition observed with previous QUAL models and increasing the target turnover, which resulted in a greater signal amplification.60
One of the major drawbacks of QUAL probes arises from a relatively high background signal, which can be ascribed to an incomplete initial quenching and/or to unspecific hydrolysis of the quencher. To overcome these intrinsic limitations, different nucleophiles have been tested to compare their efficiency and their effect on the reaction rates.61 Further improvements were achieved by the introduction of a second quencher unit. In this optimised system using sandwich probes, nucleic acid sensing required a double displacement process to fully “unquench” the fluorophore, which drastically increased the signal-to-noise ratio.62,63 Another variation of QUAL was obtained by replacing the quencher with a fluorescence acceptor and was named QUAL-FRET.64,65 QUAL-FRET probes conjugate the characteristic of QUAL with the possibility to observe FRET from a fluorescence donor (e.g., FAM) to a fluorescence acceptor (e.g., Cy564 or TAMRA65). The acceptor dye is located on the phosphorothioate probe, avoiding the possibility to detect unspecific FRET signal due to unwanted hydrolysis of the quencher group. Only when the OT–SN2 reaction takes place, thus displacing the dabsyl group, the donor and the acceptor are close enough to generate a FRET signal.
By combining the quencher displacement of QUAL probes with the rapid kinetic and bioorthogonality of the “caged fluorophore methodology”, Franzini and Kool introduced the concept of a novel and versatile system named Q-STAR (c, Fig. 4).66 The reported quenched Staudinger-triggered α-azido ether release (Q-STAR) methodology consists in oligonucleotide probes containing a fluorophore (fluorescein) and a quencher (Dabsyl) attached through an α-azido ether linker.66 In the presence of an oligonucleotide template the Q-STAR probes hybridize in close proximity to a TTP-oligonucleotide. The reduction of the azido unit triggers the cleavage of the linker and the release of the quencher, thus restoring the fluorescence of the dye.66 Q-STAR methodology has been applied to the detection of double-stranded DNA,67 using Ψ-cytosine modified probes, whereas a double displacement of two quenchers (2-STAR)68 and the development of new quenchers for red and NIR-fluorophore, as Cy5 derivatives, was also shown (NIR-STAR).69 Take altogether these features make Q-STAR probes highly versatile enabling their application as quenched probe in combination with different fluorophore, emitting from blue to near-infrared.
Other groups have also developed systems where binding to specific DNA or RNA sequences leads to the conversion of a (non-emitting) pro-fluorophore into a (emitting) fluorophore. For instance Ito and co-workers reported in 2009 the OT transformation of a non-fluorescent dinitro-benzenesulfonyl-amino-coumarin into a bright amino-coumarin via a SNAr mechanism involving a phosphorothioate-modified oligonucleotide.70 Upon transfer of the sulfonyl group from the coumarin to the phosphorothioate, a free amino-coumarin was generated which could be detected by the appearance a characteristic fluorescent signal.
Fluorogenic reactions. OT fluorogenic reactions typically occur between two oligonucleotide probes, both conjugated to non-fluorescent moieties (d, Fig. 4) that can form a fluorescent product when they covalently react with each other. These biosensors potentially offer the advantage (over more traditional sensors) of a low (or absence of) background fluorescence, hence a significantly improved S/N ratio. To date however, only few examples of OT fluorogenic reactions have been reported, likely due to the small number of suitable (i.e., biocompatible) chemical reactions of this type.71–74 In 2009, an OT–Cu(I) Huisgen cycloaddition (click reaction) has been reported.71 It involved reaction, in the presence of a Cu(I)-complex (Cu+-THPTA), between two dark oligonucleotides functionalised with either a 4-ethynyl-1,8-naphtalenimide or a benzylazide affording a 4-(1,2,3-triazol-4-yl)-1,8-naphtalenimide fluorescent product.71 Although this probe demonstrated high sensitivity and sequence selectivity, the copper complex partially quenches the product fluorescence, requiring post-reaction modification to restore the signal.71
One year earlier, Huang and Coull72 reported an OT-aldol-type condensation between heterocyclic quaternary ammonium salts bearing an active methylene group and an aryl aldehyde affording a fluorescent hemicyanine dye. The main drawback of this OT-fluorogenic reaction was the requirement of external additives such as cyclic (e.g. pyrrolidines) or acyclic amines to catalyse the hemicyanine synthesis through Schiff's base formation.72 This limitation was overcome by Ladame and co-workers in 2010 who reported the OT-fluorogenic synthesis of symmetrical and unsymmetrical trimethine cyanine dyes, by an aldolisation-elimination reaction that did not require any additives and could proceed smoothly under near physiological conditions of salt concentration and pH (d, Fig. 4).73
A guanine-rich DNA sequence found in the promoter region of the c-kit proto-oncogene and capable of folding into a G-quadruplex structure (namely ckit21T)75 was chosen as a DNA-template. Two PNA probes were designed to hybridize with five nucleobases upstream and downstream (flanking regions) of the G-quadruplex structure. Both PNA sequences were conjugated at their C-terminus or N-terminus with a methylene indoline and a Fisher's base aldehyde, respectively.73 The folding of the G-rich sequence in a parallel G-4 structure brought the two probes into enough close proximity allowing the formation of the fluorescent trimethine cyanine dye product in a “sequence + structure”-specific manner. However, when the DNA cannot fold into a quadruplex or when the PNA probes cannot fully hybridise to the quadruplex flanking arms then the probe heads are kept separated from each other and no reaction occurs (hence, the absence of emitted fluorescence).73 This study highlights the possibility for OTC to be applied not only for DNA or RNA sequence-specific sensing, but also as probes for sensing the formation of oligonucleotide secondary structures (e.g. quadruplex, hairpins…).76
Despite the increasing number of OTRs reported in the recent literature, only a small portion can be applied to oligonucleotide sensing. The reason could be ascribed to the necessity of conducting the OTR in water, under physiological (or near physiological) conditions of pH, temperature and salt concentration for in vivo application. This raises the question of the biocompatibility of OTRs, especially for those methods that pretend to be used in cellulo. Ideally, this OTR will require no additional catalyst (such as metals or bases) because of any potential delivery problems or cytotoxicity of the added reactants. These features drastically limit the number of chemical reactions that can be used for the OT-sensing methodology while such limitations do not apply to OT-synthetic methodology.
DNA-templated organic synthesis: a way towards new molecules and new reactions
Although the number of OTRs is still limited compared to the broad range of chemical reactions used in organic or inorganic synthesis in solution, it offers the advantage to enable synthetic reactions that cannot be realized using traditional synthetic methods. Moreover, the oligonucleotides linked to the synthetic products of OTR can be used as a “barcode” in an in vitro selection-amplification protocol enabling to reveal bioactive molecules for a target of interest and to discover new chemical reactions performed in a DNA-templated manner.77DNA-templated synthesis of small-molecule and polymer libraries
Ten years ago, Liu and co-workers demonstrated that the concept of DNA-templated synthesis (DTS) could be applied to chemical reactions with no structural similarity to natural compounds, high specificity for match sequences and distance-independence between templated reactive groups.28 Shortly afterwards, the first multistep synthesis of a small molecule “programmed” by DTS has been described through the syntheses of a tripeptide and a branched thioether by using as a template a 30-mer and amine-terminated DNA and three building blocks each conjugated to a unique 10-mer DNA oligonucleotide.78 Two years later, Gartner et al. synthesized a 65-membered library of macrocyclic fumarides.21a In this work, a library of 65 “DNA-tagged” reagents has been generated and translated into corresponding macrocycles by three successive DNA-templated amine acylations and one Wittig olefination reaction (a, Fig. 5). In 2008, novel advances in DNA-templated library synthesis led to a 13000-membered small molecule macrocycles library.21b Here, the templates were generated by split-pool DNA synthesis and the structural diversity of the library was obtained thanks to a set of 36 building blocks and eight different scaffolds.
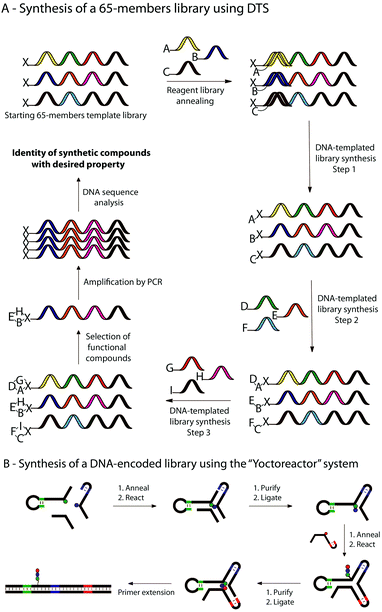 | ||
| Fig. 5 Selected strategies using OTC to synthesise libraries of small molecules and/or polymers. (A) Example of multi-step DNA-templated synthesis of a small library of 65 macrocyclic molecules by three successive DNA-templated reactions. (B) Multi-step synthesis of a DNA-encoded library within a DNA three-way junction reactor (so-called “Yoctoreactor”). | ||
The concept of DTS has also been applied to polymer chemistry. In the last decade, several researches have been performed to produce libraries of DNA-encoded synthetic polymers.13,79 In 2003, Rosenbaum et al. described a sequence-specific DNA-templated polymerization of peptide nucleic acid (PNA) tetramers,11 followed few years later by the synthesis and selection of a library of DNA-encoded synthetic PNAs.80 More recently, Hansen et al. reported the successful use of a yoctoliter-scale DNA reactor constituted of a DNA three-way junction (so-called “Yoctoreactor”) for the synthesis of DNA-encoded libraries of up to 100 different DNA-encoded compounds (b, Fig. 5).81
All of these studies aimed to generate libraries of DNA-encoded molecules with a potential biological interest and could be engaged in iterated cycles of translation, in vitro selection, and amplification.21,79–82 Different ways of in vitro selection have been already developed.82,83 For example, in vitro selection for affinity uses binding selection to an immobilized target.81 The non-binding library members are eliminated while the active library members stay on the solid support and can be amplified by PCR (a, Fig. 6). Successive rounds of in vitro selection can then be performed to enrich the most active library member. Another example is the interaction dependent PCR (IDPCR) based on the melting temperature (Tm) difference between duplex DNAs formed intramolecularly versus those formed intermolecularly (b, Fig. 6).83 In this method, active library members bind to a DNA-linked target capable of initiating primer on the DNA part of the active compounds and are extended by action of a DNA polymerase to form a DNA “hairpin” which will be preferentially amplified by PCR over the intermolecular duplex formed by non-reacting DNA-encoded library members. IDPCR is particularly interesting since it allows simultaneous evaluation of a combination of DNA-encoded molecules library and DNA-encoded targets in a single experiment.
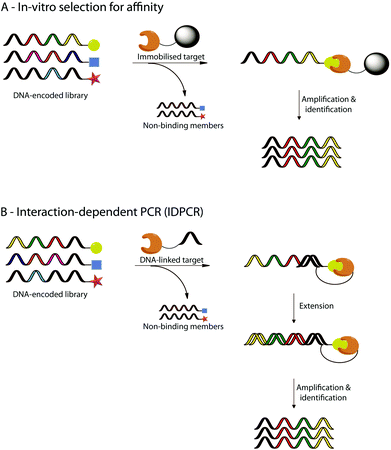 | ||
| Fig. 6 Selected examples of in vitro selections using DNA-encoded libraries. The selection process relies either (A) on the affinity of specific members of the DNA-encoded library for an immobilised target, or (B) on interaction-dependent PCR (IDPCR) after binding of specific members of the DNA encoded library to a DNA-linked target. | ||
Reaction discovery using DTS
The in vitro selection-amplification protocol has also been applied to the discovery of new reactions,83 and few new mild reactions have already been developed. A novel organometallic reaction of carbon–carbon bond formation was discovered via the first generation reaction discovery system (RDS) that uses DNA-hybridization to direct substrates combination. In the presence of a water-soluble Pd(II) catalyst (i.e., Na2PdCl4 500 μM), an enone was generated from reaction between a terminal alkyne and a terminal alkene.35 In order to demonstrate the feasibility of this newly-discovered reaction in solution, it was then successively conducted in a non-OT format in organic solvents to afford macrocyclic35 and linear trans-α,β-unsaturated ketones.84 A second-generation system, which does not rely on DNA hybridization to organize substrates into pair wise combinations, was recently developed.85,86 It proved compatible with harsher reaction conditions, expanding the scope of RDS to those reactions that do not support DNA hybridization, including organic solvents and elevated temperatures. Exploiting this methodology, two new reactions have been discovered. The first one is a mild method for the selective Markovnikov-type hydro-arylation of olefins with indoles, in the presence of a catalytic amount of AuCl3.85 The same reaction has been investigated in a non DNA-linked manner in organic solvents, using either aryl- and alkyl-trisubstituted olefins with N-phenyl sulfonyl-(Bs)-protected indole.85 It afforded, in the presence of a catalytic amount of triflic acid (TfOH 5 mol%, DCM, 25 °C), indole hydroarylated products with high regio-selectivity, efficiency and milder conditions compared to previous reported Friedel–Crafts methodology.Applying the second RDS, a novel Ru(II)-catalysed azide-reduction reaction induced by visible light has been recently discovered.86 The azide-reduction is highly efficient and can be conducted both in organic and aqueous solvents, open to the air, at room temperature and under neutral conditions. It exhibited a remarkable chemo-selectivity, in contrast to existing azide-reduction methods, and is compatible with a large range of functional groups enabling its application on oligonucleotide and oligosaccharide substrates, or in presence of protein enzymes, without compromising the structure or the enzymatic activity (conditions: 1 mM Ru(bpy)3Cl2, 50 mM of sodium ascorbate in aqueous 200 mM Tris, pH 7.4).86
Conclusions
In recent years, copying nature has become a trendy approach, one that many scientists have taken in their research. When it comes to find efficient ways to synthesise molecules, turning to Nature for inspiration is often a sensible and rewarding approach. Using DNA as a template to increase the efficiency of chemical reactions was first applied to the synthesis of oligonucleotide or oligonucleotide analogues and has notably given rise to the concept of PCR. Since then, a large diversity of OTRs have been developed that go well beyond the formation of phosphodiester bonds (as in oligonucleotides) and range from the Staudinger reaction to the reactions of carbon–carbon bond formation (e.g. Henry, Michael, Wittig…). But how far can we push the boundaries of OTRs? Not all chemical reactions are compatible with the concept of OTC. First, reactions must occur under conditions (of pH, temperature, solvent…) where the oligonucleotide (serving either as a template or as a coding agent) remains stable for the duration of the experiment. To overcome this intrinsic limitation of oligonucleotides chemical instability, more stable analogues such as PNA or LNA have been used as an alternative to natural oligonucleotides. Their ability to form highly stable heteroduplexes with either DNA or RNA, their greater chemical and enzymatic stability than DNA, and their ability to be easily functionalised make PNAs a very valuable tool for OTRs. Not only they allow the use of harsher conditions (e.g. for DTS or RDS) but they will also prove extremely valuable for in vivo sensing application.The large majority of OTRs relies on the hybridization of two (or more) oligonucleotides. It is therefore essential that the chemical reaction occurs under conditions (of salt concentration and pH) compatible with DNA (or RNA) hybridisation. Only recently, Liu and co-workers have developed strategies using OTC to discover new reactions that do not rely on oligonucleotides hybridisation. This allows the use of significantly harsher conditions, thus expanding the range of chemical reactions potentially suitable to OT.
These examples, among others, demonstrate that there are yet more reactions to be discovered that can be catalysed using an oligonucleotide template. Increasing the diversity of OTRs will also broaden the range of their applications. There has already been some successful studies (mainly from the group of E.T. Kool) demonstrating that oligonucleotide sensing can be achieved using OTRs both in vitro and in vivo.87 Most sensing strategies rely on OTR leading to the formation of a fluorescent product that can easily be detected even in the context of a cell.
Although the community of chemists interested in OTC remains relatively small, the recent advances made during the last two decades have led to the discovery of new reactions and the development of new OT strategies. But OTC is not only a conceptually elegant concept. It is also a powerful concept that has many applications in modern bioresearch and biotechnology. However, nearly 30 years after PCR was invented, the discovery of a new biotechnological breakthrough based on this concept is yet to come.
Notes and references
- K. Mullis, Sci. Am., 1990, 262, 56 CrossRef CAS; R. K. Saiki, S. Scharf, F. Faloona, K. B. Mullis, G. T. Horn, H. A. Erlich and N. Arnheim, Science, 1985, 230, 1350 Search PubMed.
- R. Naylor and P. T. Gilham, Biochemistry, 1966, 5, 2722 CrossRef CAS.
- G. Von Kiedrowski, Angew. Chem., 1986, 25, 932 CrossRef.
- P. E. Nielsen, M. Egholm, R. H. Berg and O. Buchardt, Science, 1991, 254, 1497 Search PubMed; M. Egholm, O. Buchardt, L. Christensen, C. Behrens, S. M. Freier, D. A. Driver, R. H. Berg, S. K. Kim, B. Norden and P. E. Nielsen, Nature, 1993, 365, 566 CrossRef CAS.
- T. Wu and L. E. Orgel, J. Am. Chem. Soc., 1992, 114, 7963 CrossRef CAS; T. Wu and L. E. Orgel, J. Am. Chem. Soc., 1992, 114, 5496 CrossRef; L. E. Orgel, Acc. Chem. Res., 1995, 28, 109 CrossRef.
- S. H. Wang and E. T. Kool, Nucleic Acids Res., 1994, 22, 2326 CrossRef CAS; E. Rubin, S. Rumney, S. H. Wang and E. T. Kool, Nucleic Acids Res., 1995, 23, 3547 CrossRef.
- Z. A. Shabarova, I. N. Merenkova, T. S. Oretskaya, N. I. Sokolova, E. A. Skripkin, E. V. Alexeyeva, A. G. Balakin and A. A. Bogdanov, Nucleic Acids Res., 1991, 19, 4247 CrossRef CAS.
- Y. Xu and E. T. Kool, Nucleic Acids Res., 1999, 27, 875 CrossRef CAS; S. M. Gryaznov, R. Schultz, S. K. Chaturvedi and R. L. Letsinger, Nucleic Acids Res., 1994, 22, 2366 CrossRef; M. K. Herrlein, J. S. Nelson and R. L. Letsinger, J. Am. Chem. Soc., 1995, 117, 10151 CrossRef.
- Y. Xu and E. T. Kool, J. Am. Chem. Soc., 2000, 122, 9040 CrossRef CAS.
- X. Li, Z. Y. Zhan, R. Knipe and D. G. Lynn, J. Am. Chem. Soc., 2002, 124, 746 CrossRef CAS; Y. Gat and D. G. Lynn, Biopolymers, 1998, 48, 19 CrossRef.
- D. M. Rosenbaum and D. R. Liu, J. Am. Chem. Soc., 2003, 125, 13924 CrossRef CAS.
- Z. J. Gartner, M. W. Kanan and D. R. Liu, Angew. Chem., Int. Ed., 2002, 41, 1796 CrossRef CAS.
- J. G. Schmidt, L. Christensen, P. E. Nielsen and L. E. Orgel, Nucleic Acids Res., 1997, 25, 4792 CrossRef CAS.
- C. Bohler, P. E. Nielsen and L. E. Orgel, Nature, 1995, 376, 578 CrossRef CAS.
- R. K. Bruick, P. E. Dawson, S. B. Kent, N. Usman and G. F. Joyce, Chem. Biol., 1996, 3, 49 CrossRef CAS.
- R. J. Lewis and P. C. Hanawalt, Nature, 1982, 298, 393 CrossRef CAS.
- J. Woo and P. B. Hopkins, J. Am. Chem. Soc., 1991, 113, 5457 CrossRef CAS.
- R. L. Letsinger, T. Wu and R. Elghanian, J. Am. Chem. Soc., 1994, 116, 811 CrossRef CAS; F. D. Lewis, T. Wu, E. L. Burch, D. M. Bassani, J.-S. Yang, S. Schneider, W. J. Sger and R. L. Letsinger, J. Am. Chem. Soc., 1995, 117, 8785 CrossRef.
- K. Fujimoto, S. Matsuda, N. Takahashi and I. Saito, J. Am. Chem. Soc., 2000, 122, 5646 CrossRef CAS.
- X. Li and D. R. Liu, Angew. Chem., Int. Ed., 2004, 43, 4848 CrossRef CAS.
- (a) Z. J. Gartner, B. N. Tse, R. Grubina, J. B. Doyon, T. M. Snyder and D. R. Liu, Science, 2004, 305, 1601 CrossRef CAS; (b) B. N. Tse, T. M. Snyder, Y. Shen and D. R. Liu, J. Am. Chem. Soc., 2008, 130, 15611 CrossRef CAS.
- Y. He and D. R. Liu, J. Am. Chem. Soc., 2011, 133, 9972 CrossRef CAS.
- T. M. Snyder and D. R. Liu, Angew. Chem., Int. Ed., 2005, 44, 7379 CrossRef CAS.
- X. Li, Z. J. Gartner, B. N. Tse and D. R. Liu, J. Am. Chem. Soc., 2004, 126, 5090 CrossRef CAS.
- S. Ficht, A. Mattes and O. Seitz, J. Am. Chem. Soc., 2004, 126, 9970 CrossRef CAS.
- T. N. Grossmann and O. Seitz, J. Am. Chem. Soc., 2006, 128, 15596 CrossRef CAS.
- M. L. McKee, A. C. Evans, S. R. Gerrard, R. K. O'Reilly, A. J. Turberfield and E. Stulz, Org. Biomol. Chem., 2011, 9, 1661 CAS.
- Z. J. Gartner and D. R. Liu, J. Am. Chem. Soc., 2001, 123, 6961 CrossRef CAS.
- M. L. McKee, P. J. Milnes, J. Bath, E. Stulz, A. J. Turberfield and R. K. O'Reilly, Angew. Chem., Int. Ed., 2010, 49, 7948 CrossRef CAS.
- M. L. McKee, P. J. Milnes, J. Bath, E. Stulz, R. K. O'Reilly and A. J. Turberfield, J. Am. Chem. Soc., 2012, 134, 1446 CrossRef CAS.
- H. C. Kolb, M. G. Finn and K. B. Sharpless, Angew. Chem., Int. Ed., 2001, 40, 2004 CrossRef CAS.
- Z. J. Gartner, R. Grubina, C. T. Calderone and D. R. Liu, Angew. Chem., Int. Ed., 2003, 42, 1370 CrossRef CAS.
- R. Kumar, A. El-Sagheer, J. Tumpane, P. Lincoln, L. M. Wilhelmsson and T. Brown, J. Am. Chem. Soc., 2007, 129, 6859 CrossRef CAS.
- X. Peng, H. Li and M. Seidman, Eur. J. Org. Chem., 2010, 4194 CrossRef CAS.
- M. W. Kanan, M. M. Rozenman, K. Sakurai, T. M. Snyder and D. R. Liu, Nature, 2004, 431, 545 CrossRef CAS.
- J. L. Czlapinski and T. L. Sheppard, J. Am. Chem. Soc., 2001, 123, 8618 CrossRef CAS.
- K. V. Gothelf, A. Thomsen, M. Nielsen, E. Clo and R. S. Brown, J. Am. Chem. Soc., 2004, 126, 1044 CrossRef CAS; K. V. Gothelf and R. S. Brown, Chem.–Eur. J., 2005, 11, 1062 CrossRef.
- R. M. Franzini and E. T. Kool, Org. Lett., 2008, 10, 2935 CrossRef CAS.
- Z. Ma and J. S. Taylor, Proc. Natl. Acad. Sci. U. S. A., 2007, 97, 11159 CrossRef.
- Z. Ma and J. S. Taylor, Bioorg. Med. Chem., 2001, 9, 2501 CrossRef CAS; Z. Ma and J. S. Taylor, Bioconjugate Chem., 2003, 14, 679 CrossRef.
- J. Cai, X. Li, X. Yue and J. S. Taylor, J. Am. Chem. Soc., 2004, 126, 16324 CrossRef CAS.
- J. P. Gesson, J. C. Jacquesy, M. Mondon, P. Petit, B. Renoux, S. Andrianomenjanahary, H. Dufat-Trinh Van, M. Koch, S. Michel, F. Tillequin, J. C. Florent, C. Monneret, K. Bosslet, J. Czech and D. Hoffmann, Anti-Cancer Drug Des., 1994, 9, 409 CAS; R. Lougerstay Madec, J. C. Florent, C. Monneret, F. Nemati and M. F. Poupon, Anti-Cancer Drug Des., 1998, 13, 995 Search PubMed.
- J. Brunner, A. Mokhir and R. Kräemer, J. Am. Chem. Soc., 2003, 125, 12410 CrossRef CAS; F. H. Zelder, J. Brunner and R. Kräemer, Chem. Commun., 2004, 902 RSC.
- K. Sakurai, T. M. Snyder and D. R. Liu, J. Am. Chem. Soc., 2005, 127, 1660 CrossRef CAS.
- K. Gorska, A. Manicardi, S. Barluenga and N. Wissinger, Chem. Commun., 2011, 47, 4364 RSC.
- E. M. Sletten and C. R. Bertozzi, Angew. Chem., Int. Ed., 2009, 48, 6974 CrossRef CAS.
- E. Saxon and C. R. Bertozzi, Science, 2000, 287, 2007 CrossRef CAS.
- J. Cai, X. Li, X. Yue and J. S. Taylor, J. Am. Chem. Soc., 2004, 126, 16324 CrossRef CAS.
- H. Abe, J. Wang, K. Furukawa, K. Oki, M. Uda, S. Tsuneda and Y. Ito, Bioconjug. Chem., 2008, 19, 1219 CrossRef CAS.
- Z. L. Pianowski and N. Winssinger, Chem. Commun., 2007, 3820 RSC.
- K. Furukawa, H. Abe, J. Wang, M. Uda, H. Koshino, S. Tsuneda and Y. Ito, Org. Biomol. Chem., 2009, 7, 671 CAS.
- M. Stoop and C. J. Leumann, Chem. Commun., 2011, 47, 7494 RSC.
- Z. L. Pianowski, K. Gorska, L. Oswald, C. A. Merten and N. Winssinger, J. Am. Chem. Soc., 2009, 131, 6492 CrossRef CAS.
- R. M. Franzini and E. T. Kool, ChemBioChem, 2008, 9, 2981 CrossRef CAS.
- A. Shibata, H. Abe, K. Furukawa, S. Tsuneda and Y. Ito, Chem. Pharm. Bull., 2009, 11, 1123 Search PubMed.
- A. P. Silverman and E. T. Kool, Chem. Rev., 2006, 106, 3775 CrossRef CAS.
- Y. Xu, N. B. Karalkar and E. T. Kool, Nat. Biotechnol., 2001, 19, 148 CrossRef CAS.
- S. Sando and E. T. Kool, J. Am. Chem. Soc., 2002, 124, 2096 CrossRef CAS; S. Sando and E. T. Kool, J. Am. Chem. Soc., 2002, 124, 9686 CrossRef.
- S. Sando, H. Abe and E. T. Kool, J. Am. Chem. Soc., 2004, 126, 1081 CrossRef CAS.
- H. Abe and E. T. Kool, J. Am. Chem. Soc., 2004, 126, 13980 CrossRef CAS.
- G. P. Miller, A. P. Silverman and E. T. Kool, Biorg. Med. Chem., 2008, 16, 56 CrossRef CAS.
- D. J. Kleinbaum, G. P. Miller and E. T. Kool, Bioconjug. Chem., 2010, 21, 1115 CrossRef CAS.
- D. J. Kleinbaum and E. T. Kool, Chem. Commun., 2010, 46, 8154 RSC.
- H. Abe and E. K. Kool, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 263 CrossRef CAS.
- A. P. Silverman, E. J. Baron and E. T. Kool, ChemBioChem, 2006, 7, 1890 CrossRef CAS.
- R. M. Franzini and E. T. Kool, J. Am. Chem. Soc., 2009, 131, 16021 CrossRef CAS.
- H. Li, R. M. Franzini, C. Bruner and E. T. Kool, ChemBioChem, 2010, 11, 2132 CrossRef CAS.
- R. M. Franzini and E. T. Kool, Chem.–Eur. J., 2011, 17, 2168 CrossRef CAS.
- R. M. Franzini and E. T. Kool, Bioconjug. Chem., 2001, 22, 1869 CrossRef.
- A. Shibata, H. Abe, M. Ito, Y. Kondo, S. Shimizu, K. Aikawa and Y. Ito, Chem. Commun., 2009, 6586 RSC.
- E. Jentzsch and A. Mokhir, Inorg. Chem., 2009, 48, 9593 CrossRef CAS.
- Y. Huang and J. M. Coull, J. Am. Chem. Soc., 2008, 130, 3238 CrossRef CAS.
- K. Meguellati, G. Koripelly and S. Ladame, Angew. Chem., Int. Ed., 2010, 49, 2738 CrossRef CAS; G. Koripelly, K. Meguellati and S. Ladame, Bioconjug. Chem., 2010, 21, 2103 CrossRef.
- D. K. Prusty and A. Hermann, J. Am. Chem. Soc., 2010, 132, 12197 CrossRef CAS.
- S. Rankin, A. P. Reszka, J. Huppert, M. Zloh, G. N. Parkinson, A. K. Todd, S. Ladame, S. Balasubramanian and S. Neidle, J. Am. Chem. Soc., 2005, 127, 10584 CrossRef CAS; H. Fernando, A. P. Reszka, J. Huppert, S. Ladame, S. Rankin, A. R. Venkitaraman, S. Neidle and S. Balasubramanian, Biochemistry, 2006, 45, 7854 CrossRef; S. T. Hsu, P. Varnai, A. Bugaut, A. P. Reszka, S. Neidle and S. Balasubramanian, J. Am. Chem. Soc., 2009, 131, 13399 CrossRef.
- M. Di Antonio, R. Rodriguez and S. Balasubramaniam, Methods, 2012, 57, 84 CrossRef CAS.
- R. E. Kleiner, C. E. Dumelinz and D. R. Liu, Chem. Soc. Rev., 2011, 40, 5707 RSC.
- Z. J. Gartner, M. W. Kanan and D. R. Liu, J. Am. Chem. Soc., 2002, 124, 10304 CrossRef CAS.
- B. G. Bag and G. von Keidrowski, Pure Appl. Chem., 1996, 68, 2145 CrossRef CAS; J. G. Schmidt, P. E. Nielsen and L. E. Orgel, Nucleic Acids Res., 1997, 25, 4797 CrossRef; I. A. Kozlov, M. Zielinski, B. Allart, L. Kerremans, A. Van Aerschot, R. Busson, P. Herdewijn and L. E. Orgel, Chem.–Eur. J., 2000, 6, 151 CrossRef; J. C. Chaput and J. W. Szostak, J. Am. Chem. Soc., 2003, 125, 9274 CrossRef.
- Y. Brudno, M. E. Birnbaum, R. E. Kleiner and D. R. Liu, Nat. Chem. Biol., 2010, 6, 148 CrossRef CAS.
- M. H. Hansen, P. Blakskjaer, L. K. Petersen, T. H. Hansen, J. W. Hojfeldt, K. V. Gothelf and N. J. V. Hansen, J. Am. Chem. Soc., 2009, 131, 1322 CrossRef CAS.
- J. B. Doyon, T. M. Snyder and D. R. Liu, J. Am. Chem. Soc., 2003, 125, 12372 CrossRef CAS.
- D. J. Gorin, A. S. Kamlet and D. R. Liu, J. Am. Chem. Soc., 2009, 131, 9189 CrossRef CAS; L. M. McGregor, D. J. Gorin, C. E. Dumelin and D. R. Liu, J. Am. Chem. Soc., 2010, 132, 15522 CrossRef.
- N. Momiyama, M. W. Kanan and D. R. Liu, J. Am. Chem. Soc., 2007, 129, 2230 CrossRef CAS.
- M. M. Rozenman, M. W. Kanan and D. R. Liu, J. Am. Chem. Soc., 2007, 129, 14933 CrossRef CAS.
- Y. Chen, A. S. Kamlet, J. B. Steinman and D. R. Liu, Nat. Chem., 2011, 3, 146 CrossRef CAS.
- For a recent review, see A. Shibata, H. Abe and Y. Ito, Molecules, 2012, 17, 2446 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2013 |