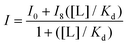Positional effects of click cyclization on β-hairpin structure, stability, and function†
Received
24th July 2012
, Accepted 11th September 2012
First published on 15th October 2012
Abstract
The use of the copper(I)-assisted azide–alkyne cycloaddition (CuAAC, or “click” reaction) as a method of β-hairpin stabilization was investigated at several different positions to determine the impact on hairpin structure and function, including hydrogen bonded sites, non-hydrogen bonded sites, and at the peptide termini. The role of the turn sequence in the peptide and the chain length of the azide were also investigated. It was determined that the CuAAC reaction was a suitable method for locking in β-hairpin structure in peptides possessing either the type I′ turn, VNGO and the type II′ turn, VpGO. Moreover, all cyclic variants exhibited improved thermal stability and resistance to proteolysis as compared to the non-cyclic peptides, regardless of the position in the strand. Additionally, the function of the CuAAC cyclized peptides was not altered as exhibited by similar binding affinities for ATP as the WKWK peptide. These studies provide a comprehensive investigation of CuAAC cyclization of β-hairpin peptides, which could further be applied to the inhibition of protein–protein and protein–nucleic acid interactions.
Introduction
While small molecules are optimal for inhibition of enzymes, they have been less successful at the inhibition of interactions between two biomolecules, such as protein–protein, protein–DNA, and protein–RNA interactions. These interactions play a critical role in many disease states, such as the p53-HDM2 interaction in cancer,1 sirtuin1 and PGC1-α in diabetes,2 and CRP and PINK1 in atherosclerosis3 as examples of dysregulated protein–protein interactions, and the binding of TAR RNA by the TAT protein interaction as an example of a medicinally relevant protein–RNA interaction. For this reason, researchers have sought to mimic the interface of protein–protein interactions (PPIs) and protein–nucleic acid interactions to disrupt the protein contacts, thereby inhibiting or slowing the progression of disease. One approach to mimic the interface is to excise the hotspot of a protein, the group of amino acids at the intermolecular interface that contribute to most of the binding energy.4 However, it is well known that isolation of a small portion of a protein often leads to an unfolded peptide, and random coil peptides are susceptible to rapid enzymatic degradation. In addition, the lack of structure often renders the peptide nonfunctional. To this end, there is a necessity to provide methods of stabilization of the structure as well as to minimize proteolytic degradation and maintain functionality.
A significant amount of research has focused on stabilizing peptide secondary structural motifs. Several groups report the use of chemical methods that are high-yielding to stabilize α-helices through intramolecular side-chain reactions.5 Moellering et al. utilized ring closing metathesis (RCM) on the dnMAML1 to stabilize its α-helical structure and prove its ability to inhibit the Notch signaling pathway,6 while Arora's group employed RCM to afford a hydrogen bond surrogate in the backbone of an α-helical portion of HIF-1α to inhibit p300.7 Klaveness and Bong have used copper(I)-assisted azide–alkyne cycloaddition (CuAAC or “click” cyclization)8 to stabilize 3,10-helices9 and α-helices,5b respectively. More recently CuAAC has been used as a method to replace disulfide linkages in an antibiotic, Tachyplesin I, which retained biological activity.10 In addition, Celentano et al. have performed studies with a TrpZip peptide to incorporate the 1,4-triazole linkage in the non-hydrogen bonded site of a β-hairpin.11
A common method for β-hairpin stabilization is cyclization via amide bond formation through the backbone or side chains.12 However, these approaches are labor intensive and often low yielding. In cases where a library of cyclic peptides is warranted, as in Robinson's approach to disrupting protein–RNA and protein–protein interactions with cyclic β-hairpins,13 an alternate method of cyclization would be advantageous.
Herein, we report a comprehensive study of the impact of “click” cyclization on β-hairpin structure and function through variation of the position of triazole incorporation in the hairpin, the relative positions of azide and alkyne in the N- and C-terminal strands of the hairpin, the turn sequence, and the chain length of the azido amino acid. We find that while there are some variations in stability, click cyclization is a general method to stabilize and in some cases enhance the structure of β-hairpins with the incorporation of a 1,4-triazole, regardless of position in the peptide, turn, or chain length. We chose to utilize mutants of a well-studied β-hairpin peptide, WKWK (Fig. 1), to investigate the effect of click cyclization on both structure and function, as this peptide has been shown to bind ATP.14 This study not only demonstrates the structural advantages of the CuAAC in β-hairpins but also the preservation of function and enhanced protection to proteolysis. Thus, CuAAC cyclization15 of β-hairpins is a general and promising approach to the disruption of protein–protein and protein–nucleic acid interactions.
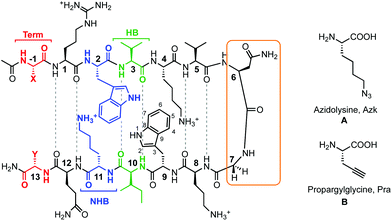 |
| | Fig. 1 Peptides investigated for CuAAC hairpin cyclization. The functional amino acids for CuAAC, A and B, are varied along terminal (Term, red), non-hydrogen bonded (NHB, blue), and hydrogen bonded (HB, green) positions. The turn residues (orange) can be one of two sequences, VNGO (shown) or VpGO, where p = dPro. Dashed lines represent hydrogen bonds. | |
Results
System design
A series of β-hairpin peptides were designed based on the WKWK peptide to determine the effects of cyclizing via CuAAC on structure and function (Fig. 1). The WKWK peptide was chosen as it has been thoroughly characterized and has been shown to be well folded. Additionally, it is known to bind purine bases, specifically ATP with a Kd of 170 μM (Fig. 2).14 Thus, click cyclization of variants of this sequence will allow us to investigate the impact on function as well as structure. The two unnatural amino acids used for the CuAAC cyclization were azidolysine (Azk) and propargylglycine (Pra). Azidolysine is easily synthetically accessible and allows for flexibility in the cyclic hairpin. These residues were incorporated cross-strand from each other, with Azk in the N-terminal strand and Pra in the C-terminal strand, along the β-hairpin in the hydrogen bonded (HB) positions, non-hydrogen bonded (NHB) positions, and terminal (Term) positions (Fig. 1, Table 1) to investigate positional effects of the click cyclization on β-hairpins. In the HB position, Azk and Pra make two cross-strand hydrogen bonds which rigidify these positions. In the NHB positions, the Azk and Pra residues do not form any hydrogen bonds, giving the side-chains more flexibility. Additionally the positions of Azk and Pra were reversed, such that Pra was in the N-terminal strand and Azk was in the C-terminal strand, to determine any changes in structure. These peptides are denoted with the suffix “rev” (Table 1). The effect of the turn sequence was also investigated for all positions by replacing the type I′ VNGO turn in WKWK with the type II′ VpGO turn. Each peptide was characterized by TOCSY and NOESY 2D NMR and circular dichroism (CD). Peptide stability was evaluated via variable temperature CD studies and protease degradation. Binding was assessed via fluorescence quenching of the Trp residues for applicable peptides.
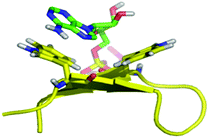 |
| | Fig. 2 Molecular dynamics simulation of WKWK (yellow) with the Trp shown in stick form bound to ATP (green).16 The peptide structure is based on an NMR structure. | |
| Peptide |
Sequence |
| Residues involved in turn sequence are underlined. Residues involved in potential ATP recognition are in bold. Prefixes of “U” and “C” denote linear unclicked and cyclic clicked peptides. p = dPro. |
| WKWK |
Ac-RWVK![[V with combining low line]](https://www.rsc.org/images/entities/char_0056_0332.gif) ![[N with combining low line]](https://www.rsc.org/images/entities/char_004e_0332.gif) ![[G with combining low line]](https://www.rsc.org/images/entities/char_0047_0332.gif) ![[O with combining low line]](https://www.rsc.org/images/entities/char_004f_0332.gif) WIKQ-NH2 WIKQ-NH2 |
| Term |
Ac-(Azk)RWVKWIKQ(Pra)-NH2 |
| Term-rev |
Ac-(Pra)RWVKWIKQ(Azk)-NH2 |
| NHB |
Ac-R(Azk)VKWI(Pra)Q-NH2 |
| NHB-rev |
Ac-R(Pra)VKWI(Azk)Q-NH2 |
| HB |
Ac-RW(Azk)KW(Pra)KQ-NH2 |
| HB-rev |
Ac-RW(Pra)KW(Azk)KQ-NH2 |
| Term-pG |
Ac-(Azk)RWVK![[p with combining low line]](https://www.rsc.org/images/entities/char_0070_0332.gif) WIKQ(Pra)-NH2 WIKQ(Pra)-NH2 |
| NHB-pG |
Ac-R(Azk)VKWI(Pra)Q-NH2 |
| HB-pG |
Ac-RW(Azk)KW(Pra)KQ-NH2 |
Structural studies of the clicked β-hairpins
Circular dichroism and 2-D NMR studies.
Structural studies were performed on all uncyclized (denoted by the suffix “U”) and cyclic peptides (denoted by the suffix “C”) via circular dichroism (CD) and 2-D NMR techniques including TOCSY and NOESY, to assess the influence of cyclization by CuAAC on structure. The β-sheet structure is characterized by a global minimum between 210 nm and 215 nm with a corresponding maximum at 195 nm. A random coil peptide is expected to have a global minimum at 195 nm. 2-D TOCSY and NOESY NMR spectra were acquired on all peptide variants. Downfield shifting of ≥0.1 ppm of the α-hydrogens (Hα) along the peptide backbone relative to unfolded values indicates a β-sheet conformation. The NOESY spectra were used to determine proper folding registry in the hairpin and additional interactions between residues.
Terminal positions for cyclization.
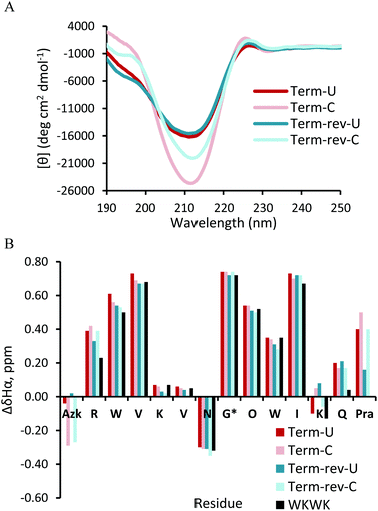
The CD spectrum (Fig. 3A) of Term-U and Term-rev-U show a minimum at 212 nm and maximum at 195 nm, distinctive of a global β-hairpin conformation. The cyclic counterparts, Term-C and Term-rev-C exhibit a more intense minimum at the same wavelength suggesting an overall more structured hairpin. Additional confirmation of proper structure is seen with the exciton coupling peak at 225 nm stemming from diagonal Trp interactions which is also seen in the WKWK parent peptide.14 The NMR data show that non-cyclic and cyclic Term and Term-rev do not display a distinct change in Hα shifts (Fig. 3B). The extent of splitting of the diastereotopic Hα's of Gly correlates with hairpin folding at the turn.17 Both non-cyclic and cyclic peptides of Term and Term-rev have a glycine splitting value of 0.74 ppm, indicating no measurable change in hairpin folding. This lack in difference is most likely due to the position of the CuAAC. There are several aspects to note in comparing the Hα shifts of the non-cyclic and cyclic peptides. Azk, at position −1 or 14, and Pra, at position 13 or −1, are more downfield shifted in the Term-C and Term-rev-C, respectively, suggesting the ends are pulled closer together. Other terminal positions are also stabilized as seen by the downfield shifts of Arg1Hα and Gln12Hα compared to the parent WKWK. Interestingly there is a slight difference in the reversal of the azide and alkyne (Term-rev) which is notable in the shift of Lys11Hα. In WKWK, the Lys11Hα is upfield shifted stemming from ring current effects from the cross-strand Trp2. This same trend is seen in Term-U, however upon cyclization Lys11Hα is upfield shifted, whereas the reverse trend is seen in Term-rev. This suggests that there are slight nuances in choosing which strand the CuAAC functional groups are placed. Residues surrounding the CuAAC functional groups can certainly impact the structure as is supported by the NOE data (Fig. 4). One of the Lys11Hγ of Term-C has a strong NOE with the triazole proton indicating that the presence of the triazole aromatic ring allows Lys11 to have another interaction. This conformation could be inaccessible in the Term-rev-C peptide, thus the downfield shifting is seen as in WKWK.
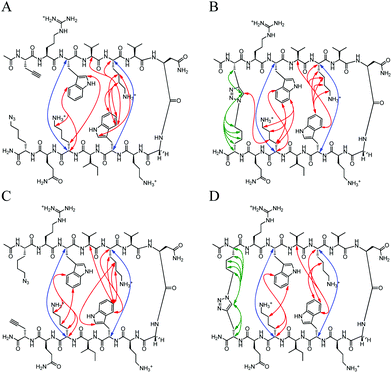 |
| | Fig. 4 Unambiguous NOEs seen for (A) Term-U, (B) Term-C, (C) Term-rev-U, (D) Term-rev-C shown are cross-strand interactions (blue) confirming registry, triazole interactions (green), and residue interactions (red). NOESY spectra were acquired with peptide concentration around 1 mM in 50 mM KD2PO4, pH 7 buffer at 20 °C. | |
There are strong NOEs between the Lys4Hα and Trp9Hα, and Lys11Hα and Trp2Hα confirming the correct registry for the β-hairpin. Additional NOE contacts are seen between the side chains of Lys4 and Lys11 and their cross-strand Trp residues, Trp2 and Trp9. However, there is not a significant increase in the contacts between strands upon CuAAC hairpin cyclization. Similar trends were observed in the peptides with a type II′ turn sequence, Term-pG (ESI Fig. S3 and S4†). Overall cyclization at the terminal position does not appear to enhance β-hairpin formation dramatically. In addition, there does not appear to be a marked difference in decreasing the number of methylene spacers in the azide residue, from 4 to 3 to 2 to 1, making the loop smaller (ESI Fig. S5†).
Non-hydrogen bonded position for cyclization.
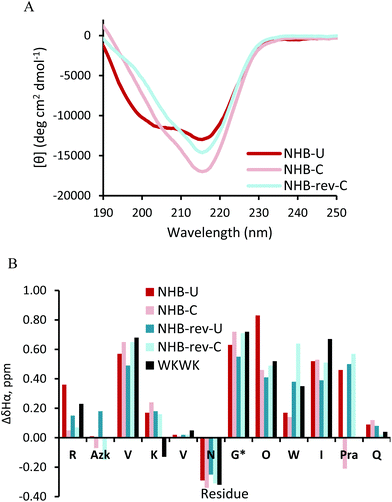
Replacement of the Trp2 and Lys11 cross-strand pair in a non-hydrogen bonded position deters β-hairpin folding since one of the stabilizing Trp-Lys pairs seen in WKWK is removed. This is also apparent in the CD of NHB-U (Fig. 5A). The CD spectrum of NHB-U has two minima, one at 205 nm and the other at 215 nm with a maximum at 195 nm suggesting a partially folded structure. NHB-rev-U was not fully soluble and formed a gel-like substance under the same conditions (50 mM KD2PO4, pH 7, 20 °C) and required a much lower pH to attain solubility. Thus a CD spectrum was not acquired. This again supports the fact that position of the CuAAC functional groups is important. NHB-C and NHB-rev-C both show greater hairpin conformation as seen by the sharp decrease in ellipticity at 215 nm. There is a slight shoulder at 205 nm suggesting some population of an unknown structure as seen in NHB-U. The difference in Hα shifts (Fig. 5B) agrees with the CD data. Most of the residues in the cyclic peptides display greater downfield shifting than that of the non-cyclic peptides. The glycine splitting values are also far greater for the cyclic peptides at 0.72 ppm and 0.71 ppm relative to 0.63 ppm and 0.55 ppm for NHB and NHB-rev. It appears that covalent linkage at the non-hydrogen bonded site restores stability provided by noncovalent interactions in WKWK. Additionally for NHB-rev, cyclizing restored solubility. The NOESY spectra (Fig. 6) better display the structural details of this peptide set. For NHB-U a strong NOE is observed between Lys4Hα and Trp9Hα confirming correct registry, however there is no NOE seen between Azk2 and Pra11. There are additional contacts between the side chains of Lys4 and Trp9. The NOE between Trp9H4 with Val3Hα implies that Trp9 is sandwiched Azk2 and Lys4. NHB-C is also in the correct registry thus showing that click cyclization in the non-hydrogen bonded position does not alter the type of hairpin formed. The sandwiching of Trp9 is far more pronounced in NHB-C as evidenced by NOEs present between Trp9H4 and the methylenes of Azk2. NHB-rev-U does not have any cross-strand backbone NOEs, suggesting that a β-hairpin is not formed. Instead there is a strong NOE between Azk11Hα and Trp9Hα. Even upon click cyclization no cross-strand NOEs are seen to confirm the correct registry (Fig. 6C and D). However, there are strong NOEs observed between Pra2Hα and Lys4Hα as well as between Trp9Hα and Azk11Hα. Structurally the observable NOEs imply a cradling of the hairpin where the terminal ends are sandwiched on top of the residues closer to the turn. NOEs between Trp9 and Lys4 as well as the triazole proton support this structure. Click cyclization at the NHB position of the peptide with the type II′ turn, NHB-pG, displayed similar results in the NOEs (ESI Fig. S7†). However, the difference between the CD and Hα shifts of the non-cyclic and cyclic NHB-pG were less pronounced than the type I′ turn peptides (ESI Fig. S6†). From these data we conclude that click cyclizing at the non-hydrogen bonded position does indeed stabilize hairpin structure, but with possible distortion.
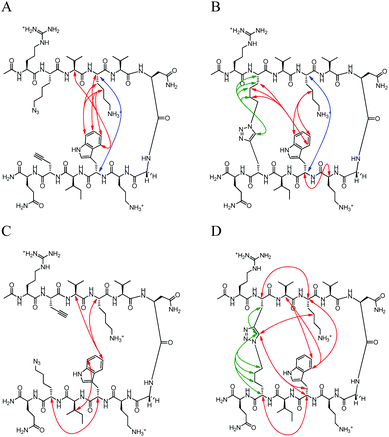 |
| | Fig. 6 Unambiguous NOEs seen for (A) NHB-U, (B) NHB-C, (C) NHB-rev-U, (D) NHB-rev-C shown are cross-strand interactions (blue) confirming registry, triazole interactions (green), and residue interactions (red). NOESY spectra were acquired with peptide concentration around 1 mM in 50 mM KD2PO4, pH 7 buffer at 20 °C with the exception of NHB-rev-U which was acquired in a 50 mM D-acetate buffer, pH 4. | |
Hydrogen bonded position for cyclization.
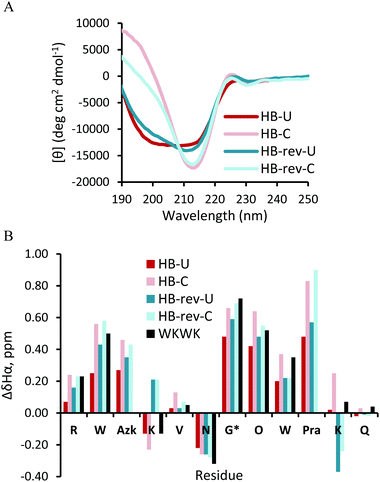
The CD spectra of the HB-U and HB-rev-U peptides (Fig. 7A) have a broad minimum spanning from 200 nm to 218 nm. Click cyclization for both HB and HB-rev stabilizes a β-hairpin structure as evidenced by a sharp minimum at 215 nm. Exciton coupling at 225 nm from the diagonal Trp2 and Trp9 interaction also confirms the formation of a β-hairpin. The difference in Hα shifts relative to random coil values agree with the CD as nearly all the Hα's in the cyclic peptides exhibit more downfield shifting than the non-cyclic peptides. Additionally the glycine splitting values for HB-U and HB-rev-U increase from 0.48 ppm and 0.59 ppm to 0.66 ppm to 0.69 ppm, respectively, upon triazole formation. This implies that the peptides become more well-folded. Again there are some nuances upon flipping of the click functional groups apparent in Lys4Hα and Lys11Hα (Fig. 7B). In the parent WKWK peptide the Lys4Hα is seen to be upfield shifted relative to random coil while Lys11Hα is downfield shifted. HB-U and HB-C both share the same trend as WKWK (Fig. 7); however, in HB-rev the trend is reversed where Lys4Hα is downfield shifted and Lys11Hα is upfield shifted. NOEs (Fig. 8) provide additional insight into the structural impacts of click cyclization. In both cases NOEs are seen between cross-strand Trp and Lys residues to indicate proper strand registry. HB-rev has several NOEs between cross-strand Trp and Lys pairs as well as additional contacts between Trp and other protons. There is a significant increase in the number of NOEs seen between side-chains upon click cyclization. As with the Term and NHB positions, substitution of the turn residues to the type II′ turn, dPro-Gly, sequence in the HB peptide lead to similar results (ESI Fig. S8 and S9†).
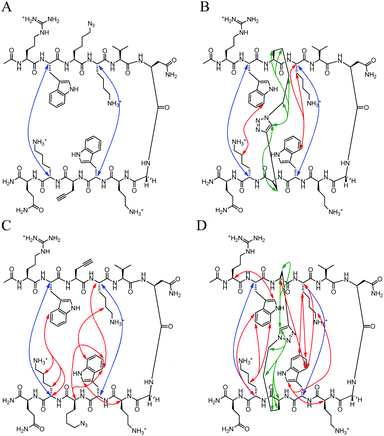 |
| | Fig. 8 Unambiguous NOEs seen for (A) HB-U, (B) HB-C, (C) HB-rev-U, (D) HB-rev-C shown are cross-strand interactions (blue) confirming registry, triazole interactions (green), and residue interactions (red). NOESY spectra were acquired with peptide concentration around 1 mM in 50 mM KD2PO4, pH 7 buffer at 20 °C. | |
Thermal stability of click cyclized β-hairpins
The thermal stability of the peptides was assessed by thermal denaturation monitored by circular dichroism. No direct measurement of melting temperature could be made as no standard S-curve was observed. Nonetheless, in all positions the cyclic peptide exhibited increased thermal stability compared to the non-cyclic peptides (ESI Fig. S1†) as expected. For all the type II′ turn peptides, an increase in thermal stability of the cyclic peptides was also observed (ESI Fig. S2†). From these results it can be confirmed that CuAAC cyclization of peptides affords stability to β-hairpins regardless of the position of the reaction.
Proteolytic stability of click cyclized β-hairpins
The triazole moiety is known to impart protease resistance as well as is cyclization of a peptide.18 Thus we sought to test the proteolytic stability of the non-cyclic and cyclic peptides by incubation with Pronase E, which is a mixture of non-specific endo- and exo-serine proteases. The type II′ turn sequences were chosen for experimental purposes as it is known that the dPro-Gly turn sequence enhances resistance to protease degradation, thus allowing for measurable half-lives under the conditions.19Table 2 summarizes the half-life of each peptide under the conditions described. The β-hairpin peptides were compared to a control unstructured peptide, Scramble, which consisted of a random order of the residues in Term-pG. The half-life of Scramble was found to be 3 min, while the non-cyclic peptides HB-pG-U, NHB-pG-U, and Term-pG-U had half-lives of 15, 20, and 60 min. Term-pG-U was found to be the least susceptible to degradation compared to the other non-cyclic peptides, most likely due to being the most well-folded.18 The CuAAC cyclization improved resistance to proteolytic degradation on all accounts, where HB-pG-C had a half-life 18-fold greater than HB-pG-U, NHB-pG-C showed no observable degradation, and Term-pG-C had a half-life at least 5-fold greater than Term-pG-U and 100-fold greater than Scramble. As the NHB-pG-C peptide did not degrade under the Pronase E conditions, its robustness was also tested using the protease trypsin which cleaves after the basic amino acids, Arg and Lys. Under trypsin degradation, NHB-pG-C had a half-life of 40 min which was 4 times greater than the non-cyclic peptide (ESI Fig. S17 and S18†). Additionally, the degradation products were analyzed by mass spectrometry which showed that only the termini were cleaved leaving an intact clicked cycle as the only product. Together these data demonstrate that click cyclization significantly increases protease resistance.
Table 2 Half-lives of type II′ turn peptides treated with Pronase Ea
| Peptide |
t
1/2 (min) |
| U |
C |
|
Reactions carried out in 10 mM sodium phosphate, 140 mM NaCl, pH 7.6 with 500 μM peptide at 37 °C.
Scrambled sequence: Ac-pQVIKRGOWW(Pra)(AzK)VK-NH2.
Over a 48 h time period.
|
| Scrambleb |
3 (±5) |
— |
| HB-pG |
15 (±5) |
280 (±5) |
| NHB-pG |
20 (±5) |
No observable degradationc |
| Term-pG |
60 (±5) |
>300 |
Assessing the ability of clicked β-hairpins to recognize ATP
WKWK binds ATP with a binding constant of 170 μM.14 The efficacy of the CuAAC peptides to recognize ATP was also assessed for Term and HB peptides as they still contain the necessary Trp-Lys cross-strand pairs for recognition. Term-C and Term-rev-C were determined to have binding constants of 109 μM and 141 μM respectively (Table 3, ESI Fig. S19†). These values do not differ significantly from WKWK. HB-C (Kd = 152 μM) and HB-rev-C (Kd = 267 μM) also bind ATP with comparable affinity. Thus, clicking in these positions has not altered the function of these peptides, providing further support that the overall β-hairpin structure is maintained.
Table 3 Binding constants of peptides for ATP
| Peptide |
K
d, μM (error) |
| WKWK |
170 |
| Term-C |
110 (30) |
| Term-rev-C |
140 (20) |
| HB-C |
150 (20) |
| HB-rev-C |
270 (20) |
Conclusions
We determined that placing residues compatible for CuAAC at the terminal ends of a well-folded β-hairpin maintains structure and affords resistance to proteolytic degradation and increased thermal stability. In addition, the function of the peptide is not eliminated and remains comparable to the parent peptide, WKWK. Decreasing the loop size in the cyclic peptide by shortening the azide chain for the terminal position does not impact folding significantly. However, work by Celentano et al. have shown there to be a difference in linker length when the CuAAC participating residues are in the NHB positions.11
Cyclizing via the formation of a 1,4-triazole functional group in the non-hydrogen bonded positions improves β-hairpin structure, as evidenced by CD and NMR data. Additionally it can improve solubility as is seen with NHB-rev. In this specific case, Trp9 is sandwiched between the triazole moiety and Lys4; however, this may not be standard to every β-hairpin sequence. The NOESY data clearly indicate that there are more established contacts between the two strands upon cyclization.
Moving the 1,4-triazole to the hydrogen bonded position is most effective in stabilizing the β hairpin structure. This position has intermediate protease stability (ESI Fig. S11†) most likely since there are more residues outside the macrocycle than the other peptides. As in the terminal position, no disruption in binding affinity to ATP is observed.
In summary, the use of the CuAAC reaction afforded an approach to synthesize cyclic β-hairpins in a robust, quick, and high yielding manner that display significant protease resistance while maintaining structure and function. This is a promising approach for producing cyclic β-hairpin peptides for inhibition of protein–protein and protein–nucleic acid interactions.
Experimental
Peptides were synthesized by automated solid-phase peptide synthesis on a Thuramed TetrasUI peptide synthesizer using Fmoc-protected amino acids on CLEAR-amide resin purchased from Peptides International. All natural Fmoc-[N]-protected amino acids were purchased from Advanced Chem Tech. Fmoc-[N]-L-propargyl glycine was purchased from Anaspec and Fmoc-[N]-azidolysine was synthesized from commercially available Fmoc-[N]-Lys-OH from NovaBiochem. Activation of amino acids was performed with HBTU and HOBt in the presence of DIPEA in DMF and NMP. Peptide deprotection was carried out in 2% DBU (1,8-diazobicyclo[5.4.0]undec-7-ene) and 2% piperidine in DMF for 2 cycles of approximately 15 min each. Each coupling step was performed twice for approximately 30 min. All peptides were acetylated at the N-terminus with 5% acetic anhydride and 6% 2,6-lutidine in DMF for 35 min. Cleavage of the peptide from the resin was performed in 95![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2.5:2.5 trifluoroacetic acid (TFA):triisopropylsilane (TIPS):water for 3 h. For peptides containing cysteine 94:2.5:2.5:1 trifluoroacetic acid (TFA):ethanedithiol (EDT):TIPS:water was used. TFA was evaporated and cleavage products were precipitated with cold ether. The peptide was extracted into water and lyophilized. It was then purified by reverse-phase HPLC using an Atlantis C-18 semi-preparative column and a gradient of 0 to 100%B over 45 min or 60 min, where solvent A was 95:5 water:acetonitrile with 0.1% TFA and solvent B was 95:5 acetonitrile:water with 0.1% TFA. After purification, the peptide was lyophilized to a powder and identified with ESI-TOF mass spectroscopy.
:2.5:2.5 trifluoroacetic acid (TFA):triisopropylsilane (TIPS):water for 3 h. For peptides containing cysteine 94:2.5:2.5:1 trifluoroacetic acid (TFA):ethanedithiol (EDT):TIPS:water was used. TFA was evaporated and cleavage products were precipitated with cold ether. The peptide was extracted into water and lyophilized. It was then purified by reverse-phase HPLC using an Atlantis C-18 semi-preparative column and a gradient of 0 to 100%B over 45 min or 60 min, where solvent A was 95:5 water:acetonitrile with 0.1% TFA and solvent B was 95:5 acetonitrile:water with 0.1% TFA. After purification, the peptide was lyophilized to a powder and identified with ESI-TOF mass spectroscopy.
Synthesis of α-N-Fmoc-ε-azido-L-Lysine20
Fmoc-Lysine-OH (2.7 mmol, 1 eq), potassium carbonate (5.5 mmol, 2 eq), and copper(II) sulfate pentahydrate (0.028 mmol, 1 mol%) were stirred in MeOH (15 mL) for 10 min. Imidazole-1-sulfonyl azide·HCl (3.3 mmol, 1.2 eq) was then added. [Safety precaution: imidazole-1-sulfonyl azide is potentially explosive. Aliquots of less than one gram were stored at 4 °C.] This was allowed to react overnight. The reaction mixture was diluted with water (2.5 mL) and the MeOH was removed via rotary evaporation. The reaction was diluted with more water (30 mL) and acidified with HCl (ca. 1 mL). Crude product was extracted into ethyl acetate (3 × 25 mL). The organic layers were combined and dried over MgSO4. Fmoc-Azidolysine-OH was purified by silica gel column chromatography (3–10% MeOH in CH2Cl2) to give the desired product as a white solid (80% yield). 1H NMR (400 MHz, CD3OD): δ 7.69 (d, J = 7.6, 2H), δ 7.60 (t, J = 8.0, 2H), δ 7.31 (t, J = 7.2, 2H), δ 7.24 (t, J = 7.2, 2H), δ 4.30 (d, J = 6.8, 2H), δ 4.19 (m, 1H), δ 4.13 (t, J = 6.8, 1H), δ 3.17 (m, 2H), δ 1.90–1.35 (m, 6H).
Synthesis of tris-tri(methylazolyl)amine ligand21
Methylbromoacetate (3.44 mmol, 1 eq) was dissolved in acetone (1.5 mL) and brought to 0 °C. Sodium azide (3.56 mmol, 1.03 eq) was dissolved in water (1.5 mL) and slowly added dropwise to the reaction mixture. The mixture was then allowed to warm to room temperature and then heated to 65 °C and stirred overnight. Azidomethylacetate was extracted using CH2Cl2 (3 × 10 mL). The organic layers were combined and dried over Na2SO4. The organics were then rotary evaporated to give azidomethylacetate. Tripropargylamine (0.44 mmol, 1 eq) was dissolved in CH3CN (1 mL) and cooled to −5 °C. To this was added azidomethylacetate (1.72 mmol, 3.9 eq) in CH3CN (1 mL) slowly over 5 min. 2,6-Lutidine (0.44 mmol, 1 eq) and Cu(MeCN)4PF6 (0.0088 mmol, 2 mol%) was added. The mixture was brought to room temperature then heated to 65 °C and allowed to stir for 3 days under a N2 atmosphere. The CH3CN was rotary evaporated to dryness. The product was purified by silica gel column chromatography (3% MeOH in CH2Cl2) to give the desired product as an off white solid (82% yield). 1H NMR (400 MHz, CDCl3): δ 7.84 (s, 1H), δ 5.18 (s, 2H), δ 3.79 (s, 2H), δ 3.78 (s, 3H). High-resolution ESI-MS: m/z calculated for C18H24N10O6 (M + Cs+), 609.0935; observed 609.0920.
General procedure for the cyclization of peptides using CuAAC
To a solution of peptide (1 mM) in 10 mM phosphate, pH 8.0 buffer was added a premixed solution of tris-tri(methylazolyl)amine ligand (2.7 eq) and [Cu(CH3CN)4][PF6] (1.8 eq) in acetonitrile. Sodium ascorbate (2.1 eq) dissolved in minimal buffer was added to the reaction. This solution was stirred overnight in the dark. The reaction mixture was quenched with %A HPLC solvent and lyophilized to a powder. The reaction was further desalted followed by HPLC purification. The yields were quantitative.
NMR spectroscopy
NMR samples were made to a concentration of at least 1 mM in D2O buffered to pD 7.0 (uncorrected) with 50 mM KPO4D2, 0.5 mM DSS. Samples were analyzed on a Varian Inova 600-MHz instrument. One dimensional spectra were collected using 32K data points and between 8 and 36 scans using 1.5 s presaturation. Two dimensional total correlation spectroscopy (TOCSY) and nuclear Overhauser spectroscopy (NOESY) experiments were carried out using the pulse sequences from the Chempack software. Scans in the TOCSY experiment were taken from 16 to 32 in the first dimension and from 64 to 128 in the second dimension. Scans in the NOESY experiments were taken from 32 to 64 in the first dimension and from 128 to 512 in the second dimension with mixing times of 200–500 ms. All spectra were analyzed using standard window functions (sinbell and Gaussian with shifting). Presaturation was used to suppress the water resonance. Assignments were made by using standard methods as described by Wüthrich.22 All experiments were run at 298.15 K.
CD spectroscopy was performed on an Applied Photophysics Chirscan Plus. Spectra were collected from 260 to 185 nm at 0.5 nm intervals at 25 °C. Melting temperature spectra were collected along the same wavelengths from 25 °C to 90 °C.
Peptidase concentration
Enzymes were purchase from Sigma-Aldrich, Pronase E from Streptomyces griseus (EC 3.4.24.31) as a solid and trypsin from bovine pancrease as a lyophilized powder containing lactose. Pronase E and trypsin were brought up in 10 mM sodium phosphate, 140 mM NaCl buffer, pH 7.6 to 0.3 mg mL−1.
Peptidase degradation reactions
The procedure for peptidase studies was adapted from Seebach et al.23 For each reaction, 0.25 mM peptide in the phosphate buffer (140 mM Na+/K+Cl− buffer pH 7.6) was reacted with the appropriate concentration of enzyme at 37 °C. A 75 μL aliquot was removed after 0, 5, 10, 15, 30, 45, 60, 90, 120, 190, 270, 360, 480, 600, 720, 840, 1100, 1260, 1440, 1680 min time points, quenched with 10 μL glacial acetic acid and 15 μL of buffer, brining the final volume to 100 μL. A 10 μL aliquot was injected on a Waters Alliance 2695 with an Atlantis C-18 column using a 0–40%B in 45 min gradient. The acetic acid peak was used as an internal control to account for differences in the injection volumes by the autosampler. The parent peak and the acetic acid peak were integrated using the accompanying software. To determine the fraction of full-length peptides remaining at each of the time points, the peak area for the full-length peptides was divided by the peak are at the zero time point.
Fluorescence quenching experiments with ATP
Peptide samples were prepared by dissolving purified peptide in 10 mM acetate buffer, pH 5.0. The concentrations of the peptide was determined in 5 M guanidine hydrochloride using the absorbance of the Trp residues at 280 nm (ε = 5690 M−1 cm−1). An ATP stock solution was prepared by dissolving in 10 mM acetate buffer, pH 5.0, and the concentration was determined using the absorbance at 260 nm (ε = 15400 M−1 cm−1). ATP concentrations were prepared via serial dilutions. Samples were prepared where the peptide host concentrations were held constant with increasing concentrations of guest. Fluorescence scans were obtained at 25 °C using an excitation wavelength of 297 nm for Trp. Emission was monitored between at 348 nm. The emission intensities of Trp at 348 nm as a function of nucleotide concentration were fit to this binding equation in
Kaleidagraph, where I is the observed fluorescence intensity, I0 is the initial intensity, I8 is the fluorescence intensity at binding saturation, [L] is the concentration of ATP added, and Kd is the dissociation constant.
Acknowledgements
This material is based in part upon work supported by the National Science Foundation under Grant No. CHE 1011794. Funding from the NIH is also gratefully acknowledged for partial support of this work (1R01EB011763-01).
Notes and references
-
(a) P. Chene, Nat. Rev. Cancer, 2003, 3, 102–109 CrossRef CAS;
(b) C. Klein and L. T. Vassilev, Br. J. Cancer, 2004, 91, 1415–1419 Search PubMed.
- P. Puigserver, Age, 2007, 29, 115–115 Search PubMed.
-
(a) J. T. Rodgers, C. Lerin, W. Haas, S. P. Gygi, B. M. Spiegelman and P. Puigserver, Nature, 2005, 434, 113–118 Search PubMed;
(b) F. Billia, L. Hauck, F. Konecny, V. Rao, J. Shen and T. W. Mak, Proc. Natl. Acad. Sci. U. S. A., 2011, 108, 9572–9577 Search PubMed;
(c) D. E. Manolov, W. Koenig, V. Hombach and J. Torzeski, Histol. Histopathol., 2003, 18, 1189–1193 Search PubMed.
-
(a) J. A. Robinson, ChemBioChem, 2009, 10, 971–973 CrossRef CAS;
(b) A. J. Wilson, Chem. Soc. Rev., 2009, 38, 3289–3300 RSC.
-
(a) L. D. Walensky, A. L. Kung, I. Escher, T. J. Malia, S. Barbuto, R. D. Wright, G. Wagner, G. L. Verdine and S. J. Korsmeyer, Science, 2004, 305, 1466–1470 CrossRef CAS;
(b) O. Torres, D. Yuksel, M. Bernardina, K. Kumar and D. Bong, ChemBioChem, 2008, 9, 1701–1705 CAS;
(c) M. Empting, O. Avrutina, R. Meusinger, S. Fabritz, M. Reinwarth, M. Biesalski, S. Voigt, G. Buntkowsky and H. Kolmar, Angew. Chem., Int. Ed., 2011, 50, 5207–5211 CrossRef CAS;
(d) M. Scrima, A. Le Chevalier-Isaad, P. Rovero, A. M. Papini, M. Chorev and A. M. D'Ursi, Eur. J. Org. Chem., 2010, 446–457 CrossRef CAS.
- R. E. Moellering, M. Cornejo, T. N. Davis, C. Del Bianco, J. C. Aster, A. L. K. Blacklow, D. G. Gilliland, G. L. Verdine and J. E. Bradner, Nature, 2009, 462, 182–190 CrossRef CAS.
- L. K. Henchey, S. Kushal, R. Dubey, R. N. Chapman, B. Z. Olenyuk and P. S. Arora, J. Am. Chem. Soc., 2010, 132, 941–943 CrossRef CAS.
-
(a) V. V. Rostovtsev, L. G. Green, V. V. Fokin and K. B. Sharpless, Angew. Chem., Int. Ed., 2002, 41, 2596–2599 CrossRef CAS;
(b) C. W. Tornoe, C. Christensen and M. Meldal, J. Org. Chem., 2002, 67, 3057–3064 CrossRef CAS.
- O. Jacobsen, H. Maekawa, N. H. Ge, C. H. Gorbitz, P. Rongved, O. P. Ottersen, M. Amiry-Moghaddam and J. Klaveness, J. Org. Chem., 2011, 76, 1228–1238 CrossRef CAS.
- K. Holland-Nell and M. Meldal, Angew. Chem., Int. Ed., 2011, 50, 5204–5206 CrossRef CAS.
- V. Celentano, D. Diana, L. De Rosa, A. Romanelli, R. Fattorusso and L. D. D'Andrea, Chem. Commun., 2012, 48, 762–764 RSC.
-
(a) C. J. White and A. K. Yudin, Nat. Chem., 2011, 3, 509–524 CrossRef CAS;
(b) P. Li, P. P. Roller and J. Xu, ChemInform, 2003, 34, 411–440 Search PubMed.
-
(a) R. Fasan, R. L. A. Dias, K. Moehle, O. Zerbe, D. Obrecht, P. R. E. Mittle, M. G. Grütter and J. A. Robinson, ChemBioChem, 2006, 7, 515–526 CrossRef CAS;
(b) K. Moehle, Z. Athanassiou, K. Patora, A. Davidson, G. Varani and J. A. Robinson, Angew. Chem., Int. Ed., 2007, 46, 9101–9108 CrossRef CAS.
-
(a) S. M. Butterfield and M. L. Waters, J. Am. Chem. Soc., 2003, 125, 9580–9581 CrossRef CAS;
(b) S. M. Butterfield, M. M. Sweeney and M. L. Waters, J. Org. Chem., 2005, 70, 1105–1114 CrossRef CAS.
-
(a) Y. L. Angell and K. Burgess, Chem. Soc. Rev., 2007, 36, 1674–1689 RSC;
(b) M. Empting, O. Avrutina, R. Meusinger, S. Fabritz, M. Reinwarth, M. Biesalski and S. Voigt, Angew. Chem., Int. Ed., 2011, 50, 5207–5211 CrossRef CAS;
(c) S. Ingale and P. E. Dawson, Org. Lett., 2011, 13, 2822–2825 Search PubMed.
-
R. M. Hughes, “Non-covalent interactions in β-hairpin peptides and small molecule model systems”. Doctoral dissertation, University of North Carolina, 2007.
- F. A. Syud, J. F. Espinosa and S. H. Gellman, J. Am. Chem. Soc., 1999, 121, 11577–11578 CrossRef.
- A. L. Jochim, S. E. Miller, N. G. Angelo and P. S. Arora, Bioorg. Med. Chem. Lett., 2009, 19, 6023–6026 CrossRef CAS.
- L. L. Cline and M. L. Waters, Pept. Sci., 2009, 92, 502–507 Search PubMed.
-
(a) Z. N. Mahmoud, S. B. Gunnoo, A. R. Thomson, J. M. Fletcher and D. N. Woolfson, Biomaterials, 2011, 32, 3712–3720 CrossRef CAS;
(b) A. L. C. Isaad, F. Barbetti, P. Rovero, A. M. D'Ursi, M. Chelli, M. Chorev and A. M. Papini, Eur. J. Org. Chem., 2008, 5308–5314 Search PubMed;
(c) S. Cantel, A. L. C. Isaad, M. Scrima, J. J. Levy, R. D. DiMarchi, P. Rovero, J. A. Halperin, A. M. D'ursi, A. M. Papini and M. J. Chorev, J. Org. Chem., 2008, 73, 5663–5674 CrossRef CAS.
-
(a) T. R. Chan, R. Hilgraf, K. B. Sharpless and V. V. Fokin, Org. Lett., 2004, 6, 2853–2855 CrossRef CAS;
(b) M. Juricek, K. Stout, P. H. J. Kouwer and A. E. Rowan, Org. Lett., 2011, 13, 3494–3497 Search PubMed.
-
K. Wüthrich, NMR of Proteins and Nucleic Acids, Wiley, New York, 1986 Search PubMed.
- J. Frackenpohl, P. I. Arvidsson, J. V. Schreiber and D. Seebach, ChemBioChem, 2001, 2, 445–455 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c2ob26445e |
|
| This journal is © The Royal Society of Chemistry 2013 |
Click here to see how this site uses Cookies. View our privacy policy here.