 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Doubly-bridged PNP-lariat ethers and their complexes with silver(I) ions in methanol solution
Natalia
Sołtys
a,
Piotr
Seliger
*a,
Grzegorz
Andrijewski
a and
Mariola
Siwy
b
aDepartment of Inorganic and Analytical Chemistry, University of Lodz, Tamka 12, 91-403 Lodz, Poland. E-mail: pitsel@chemia.uni.lodz.pl
bPolish Academy of Sciences, Centre of Polymer and Carbon Materials, Marii Curie-Skłodowskiej 34, 41-800 Zabrze, Poland
First published on 21st October 2013
Abstract
We report the preparation of four new double-bridged PNP-lariat ether derivatives L1–L4 synthesized from precursor 1 by the subsequent substitution of chlorine atoms. The process consists of two steps. In the first one two reactive chlorine atoms of the –PCl2 exocyclic moiety are substituted with respective nucleophiles: 2,2′-thiodiethanethiol 2 and 2,2′-oxydiethanethiol 3. In the second step the other chlorine atoms are substituted by aziridynyl 6 and pyrrolidinyl 7 groups, respectively. Their structures were established by MS, 1H NMR, 31P NMR, 13C NMR spectroscopy. Furthermore, this work includes the investigation of complexation of silver(I) ions with the new ligands L1–L4 in methanol solution. The formation of the novel tetra-nuclear silver(I) complexes were observed. Stability constants were determined using a simple potentiometric titration. 1H NMR, 31P NMR, 13C NMR spectra were recorded to investigate how the complexation of silver(I) ions by the investigated macrocyclic ligands occurs.
Introduction
Lariat ethers are group of compounds still considered to be attractive research objects.1–4 The term “lariat ether” refers to a crown ether having a side chain attached to the crown moiety by the so-called pivot atoms (C, N or P). The side arms contain atoms or groups with a lone pair of electrons that can cooperate with the electrons of the macrocycle ring heteroatoms, thus providing a three-dimensional coordination of the guest cation.5–7 We know three types of lariat ethers depending on the atom to which side arm is attached. When the bonding of side arm is realised through carbon we obtain C-pivot lariat ethers,8,9 nitrogen – N-pivot lariat ethers10,11 and through phosphorus atom – P-pivot lariat ethers.12,13The subjects of this publication are the P-pivot lariat ether derivatives. This is the class of reactive crown ethers which was formed by cyclocondensation of hexachlorocyclotriphosphazene with tetraethylene glycol in the presence of sodium hydride.14,15 The closure of the ring was assisted by the sodium cation which served as a template.16,17 The interaction with five oxygen donors of the polyether substrate which wrapped itself around the alkali metal cation satisfies its coordination requirements, thereby favouring ring closure to form the macrocycle.18 This type of lariat ethers may be regarded as “intelligent substrate” because it possesses reactive sites capable for spontaneous self assembling with molecular complementary reagents. This compound has the reactive chloride substituents, capable of undergoing various substitution reaction, in particular with sodium arylates19–21 and different amines.22–24
Due to the presence of binding sites in the form of both oxygen and nitrogen donor atoms, these ligands form stable complexes with cations of alkali metals and alkaline earth metals as well as transition metals.25–27
PNP-lariat ethers and their complexes are proved antitumor activity28 and anti-AIDS activity.29 This antiproliferative activity may be modified by the formation of complexes with metal ions. The thio-substituted macrocyclic derivatives of cyclotriphospazenes are commonly used for removing highly toxic, heavy metals pollutants and for recovering of some valuable metal ions (e.g. silver) from the industrial wastes.30
Complexing abilities of these compounds give the possibility for modelling of biological systems, in which sulfur-organic compounds are involved.31 Reaction of the PNP-lariat ethers with diamines represents interesting type of supramolecular control. We observe the regioselective involvement of the chlorine atoms adjacent to macrocyclic skeleton into the interactions with diamines, while chlorine situated in more distant positions from macrocyclic ring are neglected.32–34 They form transannular cyclophosphazene derivatives rather than the thermodynamically most favourable spiro-cyclosubstituted products, which are usually formed in the reaction of chlorocyclophosphazenes with short-chain diamines.35–38 In this type of reaction in general we always obtain the mixture of three types of structures, namely: intramolecular ansa diamino-bridged PNP-cryptands, as well as mono (bino) and doubly-bridged, bis-PNP-crown (bis-bino) derivatives, in which diamines span the positions adjacent to the macrocycles (Scheme 1).39
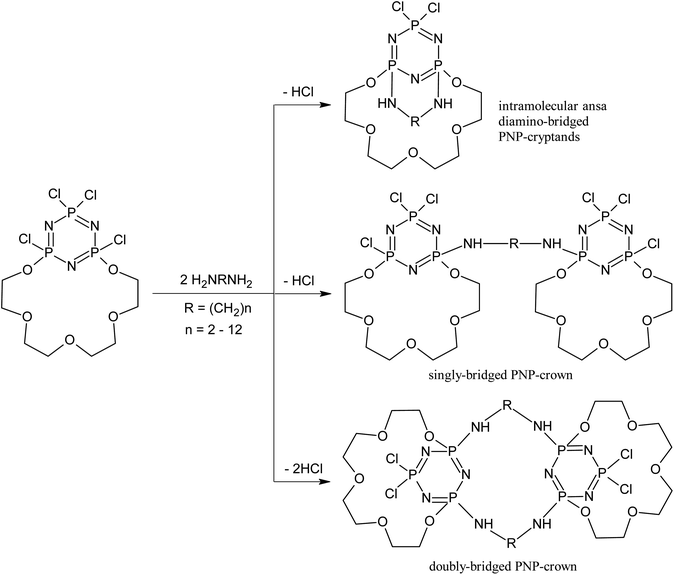 | ||
| Scheme 1 Polymacrocyclic structures formed in the reaction of chlorocyclophosphazenes with diamines. | ||
Depending on the reaction conditions, in particular the molar ratio of the reagents, their concentration in reaction mixture, as well as the reaction temperature and the nature of the solvent – all three polymacrocyclic structures are formed, as was mentioned above, but the yields of the separated reaction products are different.
In this work we present the most efficient synthetic conditions leading to obtain four doubly-bridged PNP-lariat ethers and describe their complexes with silver(I) ions in methanol solution. All these macrocyclic derivatives of cyclotriphosphazene were synthesized in two subsequent steps (Scheme 2). For bridging two molecules of tetrachloro-PNP-lariat ethers 1 we used 2,2′-thiodiethanethiol 2 and 2,2′-oxydiethanethiol 3. In the first step of the reaction we obtained the spiro derivatives 4 and 5 unlike in the case of the reactions of the investigated derivatives with diamines.32 Then, in the second reaction step endocyclic phosphorus atoms were substituted with aziridine 6 and pyrrolidine 7 groups. In such a way we obtained derivatives L1, L2, L3 and L4.
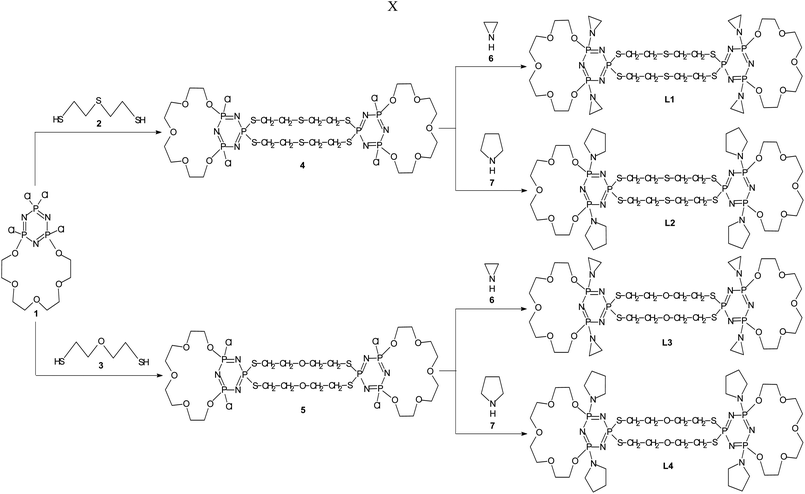 | ||
| Scheme 2 Syntheses of doubly-bridged PNP-lariat ethers. | ||
All ligands have several structural units which can potentially take part in binding of metal cations: the polyether oxygen donors of the macrocyclic PNP-crown skeleton, the endocyclic nitrogen atom of cyclophosphazene ring, the nitrogen atoms of the aziridynyl or pyrrolidynyl substituents, and the sulfur or oxygen donor atoms of the two bridging chains.
In this publication doubly-bridged derivatives of compound 1 have been synthesized and describe by NMR spectra as well as MS and DCS measurements.
Furthermore, this work covers the comparison of stability constants of complexes with silver(I) ions in methanol solutions.
Experimental sections
Materials
Sodium hydride, 60% dispersion in mineral oil, 2,2′-thiodiethanethiol (90%) and 2,2′-oxydiethanethiol (95%) from Aldrich were used as received. Aziridine (99%) from Fluka AG and pyrrolidine (99%) from Aldrich were distilled over KOH pellets under argon atmosphere before use.THF (POCh Gliwice) was distilled over cuprous chloride, then over calcium hydride, and finally twice over sodium–potassium alloy under an argon atmosphere, n-hexane (Merck) was used without purification. Methanol (POCh Gliwice) (99.8%) was used as received. For column chromatography silica gel 60 (100–200 mesh Merck) was used. The reactions were carried under a dry argon atmosphere.
Methods
TLC analyses were performed on Merck precoated silica gel 60 plates. Flash chromatography was carried out with silica gel (100–200 mesh Merck), with hexane–THF mixture as eluent. Electrospray mass spectrometric analyses (ESI-MS) were recorded on a Finnigan LCQ ion trap spectrometer (Finnigan, San Jose, CA, USA). The sample was dissolved in methanol and the solution was introduced into the ESI-MS source by continuous infusion by means of the instrument syringe pump at a rate of 3 μl min−1. The ESI-source was operated at 4.25 kV and the capillary heater was set to 200 °C. For fragmentation experiments, mass selected monoisotopic molecular ions were isolated in the ion trap and collisionally activated with 32% ejection RF-amplitude at standard He pressure. The experiments were performed in positive ion-mode.Differential scanning calorimetry (DSC) measurements were done using a DuPont 1090B apparatus with a heating rate of 20 °C min−1 under the nitrogen atmosphere.
In order to define the structure of compound L1–L41H NMR spectra were recorded on a Bruker Avance III 600 spectrometer using solutions in CDCl3. 31P NMR spectra were recorded on the same spectrometer operating at 242 MHz using solutions in CDCl3, and 85% H3PO4 as an external reference, with positive shifts recorded downfield from reference. In most cases both proton-coupled and proton-decoupled 31P NMR spectra were obtained. For 13C NMR measurements only proton-decoupled 151 MHz spectra were recorded.
Additionally, 1H NMR, 31P NMR, 13C NMR, HMBC and HMQC spectra of complexes were taken in order to study how the cation complexation of silver(I) ions by the investigated macrocyclic ligands occurs. All spectra in this part of work were recorded on a Bruker Avance III 600 spectrometer using solutions in CD3OD.
The potentiometric measurements were carried out with the use of Cerko microburette controlled by computer with silver electrode Ag0/0.01 M AgNO3 + 0.1 M (C2H5)4NNO3 in methanol solution as reference electrode. The activity of the uncomplexed silver ions was calculated from the emf values using the Nernst equation verified at the first step of the measurement procedure.27 The stability constants were calculated using HYPERQUAD 2003 program.40
Synthesis
The synthesis of all compound required two steps (1 and 2).L1 – 1,1′,3,3′ – bis(oxytetraethyleneoxy) – 5,5,5′,5′ – bis(2,2′-thiodiethanethiol) – 1,1′,3,3′ – tetra(aziridinylo)cyclotri-phosphazatriene.
Step 1. 0.469 g (1 mmol) of 1,3-(oxytetraethylenoxy)-1,3,5,5-tetrachlorocyclotriphosphazen 1, 0.3429 g (2 mmol) 2,2′-thiodiethenethiol 2 and 0.08 g (2 mmol) of 60% sodium hydride were dissolved in 20 ml of THF and placed in a 100 ml, 3-necked round-bottomed flask equipped with a mechanical stirrer and argon inlet. The reaction mixture was stirred under argon for 2 h in an ice bath then an ice bath was removed and synthesis was carried out at room temperature for 24 hours. The progress of the reaction was traced on TLC plates in hexane![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :THF = 1:5 as eluent.
:THF = 1:5 as eluent.
Step 2. The solution of the obtained in the first step intermediate product 4 and 0.344 g (8 mmol) of aziridine 6 and 8 pellets NaOH were dissolved in 20 ml hexane. The synthesis was carried out under argon at room temperature for 23 hours. The resulting mixture was filtered, the solvents were evaporated from the filtrate and crude product was isolated by flash chromatography on silica gel using hexane:THF = 1:2 as eluent. The yield of the obtained product was 21%.
31P NMR spin system A2B (242 MHz, CDCl3) δp [ppm], 25.2 (d, P(OCH2)Az), 54.2 (t, P(SCH2)2), JPP = 18.0 [Hz]; 1H NMR (600 MHz CDCl3) δH [ppm]: 4.18 (m, 8H, CH2OP), 3.72 (m, 24H, CH2OC), 3.07 (m, 16H, CH2S), 2.07 (m, 16H, CH2N); 13C NMR (151 MHz, CDCl3) δc [ppm]: 71.0, 70.9 (d, 2JPC = 3.1 Hz), 70.1 (d, 3JPC = 1.4 Hz), 65.1 (d, 2JPC = 2.3 Hz), 38.7 (d, 3JPC = 1.1 Hz), 32.7 (d, 2JPC = 1.9 Hz), 32.4 (d, 2JPC = 2.9 Hz), 24.2 (d, 2JPC = 1.6 Hz); mp = 106.8 °C, differential scanning calorimetry (DSC); ESI-MS m/z = 1127.3 (M + H+), 1151.2 (M + Na+).
L2 – 1,1′,3,3′ – bis(oxytetraethyleneoxy) – 5,5,5′,5′ – bis(2,2′-thiodiethanethiol) – 1,1′,3,3′ – tetra(pyrrolidinylo)cyclotri-phosphazatriene.
Step 1. 0.469 g (1 mmol) of 1,3-(oxytetraethylenoxy)-1,3,5,5-tetrachlorocyclotriphosphazen 1, 0.3429 g (2 mmol) 2,2′-thiodiethenethiol 2 and 0.08 g (2 mmol) of 60% sodium hydride were dissolved in 20 ml of THF and placed in a 100 ml, 3-necked round-bottomed flask equipped with a mechanical stirrer and argon inlet. The reaction mixture was stirred under argon for 2 h in an ice bath then an ice bath was removed and synthesis was carried out at room temperature for 24 hours. The progress of the reaction was traced on TLC plates in hexane:THF = 1:1 as eluent.
Step 2. The solution of the obtained in the first step intermediate product 4 and 0.5747 g (8 mmol) of pyrrolidine 7 were dissolved in 20 ml hexane. The synthesis was carried out at room temperature for 21 hours. The resulting mixture was filtered, the solvents were evaporated from the filtrate and product was isolated by flash chromatography on silica gel using hexane:THF = 3:1, then 2:1, 1:1, THF and finally methanol as eluent. The yield of the obtained product was 34%.
31P NMR spin system A2B (242 MHz, CDCl3) δp [ppm], 16.8 (d, P(OCH2)Pir), 52.0 (t, P(SCH2)2), JPP = 19.2 [Hz]; 1H NMR (600 MHz CDCl3) δH [ppm]: 4.06 (m, 8H, CH2OP), 3.70 (m, 24H, CH2OC), 3.15 (m, 16H, CH2N), 3.00 (m, 16H, CH2S), 1.82 (m, 16H, CH2C); 13C NMR (151 MHz, CDCl3) δc [ppm]: 71.0, 70.8 (d, 2JPC = 3.2 Hz), 70.1 (d, 3JPC = 1.1 Hz), 64.3 (d, 2JPC = 2.2 Hz), 46.6 (d, 2JPC = 2.1 Hz) 38.7 (d, 3JPC = 1.2 Hz), 32.6 (d, 2JPC = 2.4 Hz), 31.6 (d, 2JPC = 2.3 Hz), 26.5 (d, 3JPC = 2.1 Hz); mp = 114.2 °C, differential scanning calorimetry (DSC); ESI-MS m/z = 1239.6 (M + H+), 1261.6 (M + Na+).
L3 – 1,1′,3,3′ – bis(oxytetraethyleneoxy) – 5,5,5′,5′ – bis(2,2′-oxydiethanethiol) – 1,1′,3,3′ – tetra(aziridinylo)cyclotri-phosphazatriene.
Step 1. 0.469 g (1 mmol) of 1,3-(oxytetraethylenoxy)-1,3,5,5-tetrachlorocyclotriphosphazen 1, 0.3072 g (2 mmol) 2,2′-oxydiethenethiol 3 and 0.08 g (2 mmol) of 60% sodium hydride were dissolved in 20 ml of THF and placed in 100 ml 3-necked round-bottomed flask equipped with a mechanical stirrer and argon inlet. The reaction mixture was stirred under argon for 2 h in an ice bath then an ice bath was removed and synthesis was carried out at room temperature for 24 hours. The progress of the reaction was traced on TLC plates with hexane:THF = 1:5 as eluent.
Step 2. The solution of the obtained in the first step intermediate product 5 and 0.344 g (8 mmol) of aziridine 6 and 8 pellets of NaOH were dissolved in 20 ml hexane. The synthesis was carried out at room temperature for 23 hours. The resulting mixture was filtered, the solvents were evaporated from the filtrate and product was isolated by flash chromatography on silica gel using hexane:THF = 1:1, then 2:3, 1:2, 1:3, THF and finally methanol as eluent. The yield of the obtained product was 25%.
31P NMR spin system A2B (242 MHz, CDCl3) δp [ppm], 25.4 (d, P(OCH2)Az), 55.2 (t, P(SCH2)2), JPP = 14.1 [Hz]; 1H NMR (600 MHz CDCl3) δH [ppm]: 4.17 (m, 8H, CH2OP), 3.77 (m, 8H, CH2OCH2CH2S), 3.70 (m, 24H, CH2OC), 3.09 (m, 8H, CH2S), 2.06 (m, 16H, CH2N); 13C NMR (151 MHz, CDCl3) δc [ppm]: 71.1, 70.8 (d, 2JPC = 3.3 Hz), 70.1 (d, 3JPC = 2.2 Hz), 69.1 (d, 3JPC = 3.6 Hz), 65.0 (d, 2JPC = 2.4 Hz), 31.9 (d, 2JPC = 1.3 Hz), 21.1 (d, 2JPC = 1.2 Hz); mp = 77.0 °C, differential scanning calorimetry (DSC); ESI-MS m/z = 1095.1 (M + H+), 1117.3 (M + Na+).
L4 – 1,1′,3,3′ – bis(oxytetraethyleneoxy) – 5,5,5′,5′ – bis(2,2′-oxydiethanethiol) – 1,1′,3,3′ – tetra(pyrrolidinylo)cyclotri-phosphazatriene.
Step 1. 0.469 g (1 mmol) of 1,3-(oxytetraethylenoxy)-1,3,5,5-tetrachlorocyclotriphosphazen 1, 0.3072 g (2 mmol) 2,2′-oxydiethenethiol 3 and 0.08 g (2 mmol) of 60% sodium hydride were dissolved in 20 ml of THF and placed in 100 ml 3-necked round-bottomed flask equipped with a mechanical stirrer and argon inlet. The reaction mixture was stirred under argon for 2 h in an ice bath then an ice bath was removed and synthesis was carried out at room temperature for 24 hours. The progress of the reaction was traced on TLC plates with hexane:THF = 1:1 as eluent.
Step 2. The solution of the obtained in the first step intermediate 5 product and 0.5747 g (8 mmol) of pyrrolidine 7 were dissolved in 20 ml hexane. The synthesis was carried out at room temperature for 21 hours. The resulting mixture was filtered, the solvents were evaporated from the filtrate and product was isolated by flash chromatography on silica gel using hexane:THF = 3:1, then 2:1, 1:1, THF and finally methanol as eluent. The yield of the obtained product was 40%.
31P NMR spin system A2B (242 MHz, CDCl3) δp [ppm], 17.1 (d, P(OCH2)Pir), 52.9 (t, P(SCH2)2), JPP = 14.8 [Hz]; 1H NMR (600 MHz CDCl3) δH [ppm]: 4.09 (m, 8H, CH2OP), 3.78 (m, 8H, CH2OCH2CH2S), 3.68 (m, 24H, CH2OC), 3.17 (m, 16H, CH2N), 3.10 (m, 8H, CH2S), 1.81 (m, 16H, CH2C); 13C NMR (151 MHz, CDCl3) δc [ppm]: 71.0, 70.9 (d, 2JPC = 3.2 Hz), 70.2 (d, 3JPC = 1.1 Hz), 69.3 (d, 3JPC = 3.1 Hz), 64.3 (d, 2JPC = 2.2 Hz), 46.6 (d, 2JPC = 2.7 Hz), 31.9 (d, 2JPC = 2.3 Hz), 26.4 (d, 3JPC = 2.1 Hz); mp = 83.2 °C, differential scanning calorimetry (DSC); ESI-MS m/z = 1207.4 (M + H+), 1229.5 (M + Na+).
Results and discussion
The investigated ligands form several complex products with silver(I) ions. The obtained stepwise stability constants were collected in Table 1. Additionally the results of two previously described derivatives (with only one macrocyclic chain) presented on Fig. 1 were attached in order to compare the obtained values.| Complex form |
L1 logβ |
L2 logβ |
L3 logβ |
L4 logβ |
L5 logβ |
L6a logβ |
|---|---|---|---|---|---|---|
| a Values taken from the previous paper.26 | ||||||
| Ag+ + L → AgL+ | 5.42 ± 0.03 | 6.35 ± 0.03 | 4.16 ± 0.03 | 5.07 ± 0.03 | 3.33 ± 0.01 | 4.38 ± 0.01 |
| AgL+ + L → AgL2+ | 3.42 ± 0.03 | 3.24 ± 0.03 | 2.91 ± 0.03 | 2.06 ± 0.03 | 2.26 ± 0.01 | 3.14 ± 0.03 |
| AgL2+ + Ag+ → Ag2L+ | 3.55 ± 0.03 | 2.97 ± 0.03 | 3.53 ± 0.03 | 3.98 ± 0.03 | 2.71 ± 0.02 | |
| Ag2L+ + Ag+ → Ag3L+ | 2.75 ± 0.03 | 3.83 ± 0.03 | ||||
| Ag3L++ Ag+ → Ag4L+ | 2.89 ± 0.03 | 3.17 ± 0.03 | ∼3 ± 0.03 | ∼3 ± 0.03 | ||
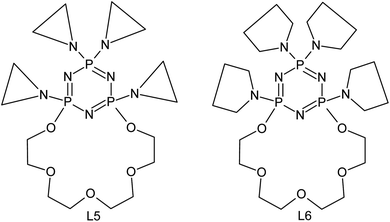 | ||
| Fig. 1 Aziridine substituted PNP – lariat ether (L5)24 and pyrrolidine substituted PNP – lariat ether (L6).24,26 | ||
In case of aziridine substituted derivative (L5), two forms of complexes were observed AgL (1:1) and AgL2 (1:2) and the preferred is 1:1 adduct. Such behaviour is probably connected with lowering of the electrodonor properties of the endocyclic cyclotriphosphazene N atom, arose from the presence of aziridine substituents. The weaker N–Ag+ interaction gives the possibility of stronger binding of the cation by oxygen donor atoms from crown ether moiety of the ligand. The silver ion is strongly accommodated inside the ligand cavity what makes the formation of sandwich type complex AgL2 more difficult. Hence the lower stability constant of this system is observed.
The formation of Ag2L do not occurred due to the deficiency of the binding places in the macrocyclic ring, what was confirmed by the potentiometric measurement results. The second Ag+ ion is usually bound to the oxygen atoms from the ether moiety of the ring. Such situation was observed in case of the derivative with pyrrolidinyl substituents (L6). Aziridinyl substituents substantially decrease the stability of the obtained complexes in comparison with the pyrrolidinyl ones. For L6 complexes the stepwise stability constants are logβ = 4.38 (AgL) and logβ = 3.14 for AgL and AgL2 respectively, and for L5 ligand logβ = 3.33 and logβ = 2.62 for AgL and AgL2 respectively. Furthermore for L6 derivatives one more stability constant is observed for Ag2L complexed form which is equal logβ = 2.67.
Such behaviour is possible due to shorter N–Ag+ binding and therefore availability of oxygen atoms from macrocyclic ether ring. Comparing the stability constant of 15C5 crown ether in methanol solutions presented in the literature (3.07),43 we noticed that in the case of the tetrapyrrolidinyl as well as tetraaziridinyl derivatives, the obtained complexes are much more stable. Furthermore in the cited paper there were no other forms of complexes observed, which suggests that in the case of the simple crown ether the silver cation is nested inside the cavity which makes the formation of other types of complexes impossible. The complexes with mono aza 15C5 crown derivative due to the presence of a nitrogen atom in the ligand molecule show much stronger coordination of silver ions 5.52 in methanol.44 Furthermore the presence of sandwich complexes was also observed.
In case of investigated ligands L1–L4 their common characteristic is that their structure consist of three macrocyclic systems. Two of them are formed from the crown ether moieties attached to the cyclotriphosphazene ring and the third one is formed by two bridges connecting two PNP-lariat ether systems. In case of L1 and L2 ligands, the additional macrocyclic ring formed through two 2,2′-thiodiethanothiol moieties, gives six sulfur atoms able to bind silver(I) ions. The bridges of the ligands L3 and L4 formed through two 2,2′-oxydiethanothiol moieties, possess four sulfur and two oxygen atoms theoretically able to bind metal ions.
Replacing of oxygen donor atoms in crown ethers with soft sulfur or nitrogen donor atoms causes the increase in stability constants of the obtained silver ion complexes.41,42 The main reason for such effect is connected with more covalent character of the S–Ag+ and N–Ag+ bonding. Such increase in stability constants is observed within the L1–L4 compounds.
In the case of AgL system the much higher values of stability constants suggest that the stabilization of the system is realized through six or four sulfur atoms (for L1, L2 or L3, L4 ligand respectively) from the macrocyclic moiety situated in the centre of the investigated molecules. Stability constants values for complexes with L1 and L2 ligands are much higher than for complexes with L3 and L4 ligands. This fact is probably connected with the replacing of two oxygen atoms with two sulfur atoms in the bridge fragments of the investigated compounds.
NMR measurements
In order to determine the way of silver(I) ions complexation by investigated ligands, the NMR spectra in methanol of the L1 and L4 compounds were done as well as the spectra of their complexes – after adding of silver ions with the amounts corresponding to suitable molar Ag:L ratio. All data were collected in Table 2 and Table 3 for ligand L1 and L4 respectively.
:L molar ratio 0.5:1, 1:1, 2:1, 3:1 and 4:1
| L1 | L1 + 0.5 Ag | L1 + 1 Ag | L1 + 2 Ag | L1 + 3 Ag | L1 + 4 Ag | |
|---|---|---|---|---|---|---|
| 1H NMR | 4.18 (m, 8H, CH2OP) | 4.20 (m, 8H, CH2OP) | 4.19 (m, 8H, CH2OP) | 4.21 (m, 8H, CH2OP) | 4.10 (m, 8H, CH2OP) | 4.21 (m, 8H, CH2OP) |
| 3.69 (m, 24H, CH2OC) | 3.70 (m, 24H, CH2OC) | 3.70 (m, 24H, CH2OC) | 3.71 (m, 24H, CH2OC) | 3.70 (m, 24H, CH2OC) | 3.71 (m, 24H, CH2OC) | |
| 3.09 (m, 16H, CH2S) | 3.16 (m, 16H, CH2S) | 3.23 (m, 16H, CH2S) | 3.29 (m, 16H, CH2S) | 3.31 (m, 16H, CH2S) | 3.29 (m, 16H, CH2S) | |
| 2.08 (m, 16H, CH2N) | 2.09 (m, 16H, CH2N) | 2.10 (m, 16H, CH2N) | 2.11 (m, 16H, CH2N) | 2.10 (m, 16H, CH2N) | 2.12 (m, 16H, CH2N) | |
| 31P NMR | 16.7 (m, P(MES)2); | 17.0 (m, P(MES)2); | ||||
| 25.5; 25.4 (d, P(OCH2)Az | 25.3; 25.2 (d, P(OCH2)Az | 25.1 (d, P(OCH2)Az | 25.0; 24.9 (d, P(OCH2)Az | 25.1; 24.9 (d, P(OCH2)Az | 24.9; 24.8 (d, P(OCH2)Az | |
| 54.7 (t, P(SCH2)2) | 53.6; 53.4 (t, P(SCH2)2) | 53.3 (t, P(SCH2)2) | 52.3; 52.2 (t, P(SCH2)2) | 53.0 (t, P(SCH2)2) | 52.3; 52.1 (t, P(SCH2)2) |
:L molar ratio 0.5:1, 1:1, 2:1, 3:1 and 4:1
| L4 | L4 + 0.5 Ag | L4 + 1 Ag | L4 + 2 Ag | L4 + 3 Ag | L4 + 4 Ag | |
|---|---|---|---|---|---|---|
| 1H NMR | 4.11 (m, 8H, CH2OP) | 4.23 (m, 8H, CH2OP) | 4.24 (m, 8H, CH2OP) | 4.21 (m, 8H, CH2OP) | ||
| 4.09 (m, 8H, CH2OP) | 3.85 (m, 8H, CH2OCH2CH2S) | 3.49 (m, 8H, CH2OCH2CH2S) | 3.48 (m, 8H, CH2OCH2CH2S) | 3.40 (m, 8H, CH2OCH2CH2S) | ||
| 4.05 (m, 8H, CH2OP) | 3.83(m, 8H, CH2OCH2CH2S) | 3.77 (m, 24H, CH2OC) | 3.80 (m, 24H, CH2OC) | 3.79 (m, 24H, CH2OC) | 3.74 (m, 24H, CH2OC) | |
| 3.84 (m, 8H, CH2OCH2CH2S) | 3.75 (m, 12H, CH2OC) | 3.73 (m, 12H, CH2OC) | 3.75 (m, 12H, CH2OC) | 3.74 (m, 12H, CH2OC) | 3.72 (m, 12H, CH2OC) | |
| 3.75 (m, 24H, CH2OC) | 3.72 (m, 12H, CH2OC) | 3.20 (m, 16H, CH2N) | 3.22 (m, 16H, CH2N) | 3.23 (m, 16H, CH2N) | 3.23 (m, 16H, CH2N) | |
| 3.16 (m, 16H, CH2N) | 3.18 (m, 16H, CH2N) | 3.24 (m, 4H,CH2S) | 3.25 (m, 4H,CH2S) | 3.25 (m, 4H,CH2S) | 3.30 (m, 4H,CH2S) | |
| 3.14 (m, 8H, CH2S) | 3.22 (m, 8H, CH2S) | 3.13 (m, 4H, CH2S) | 3.14 (m, 4H, CH2S) | 3.14 (m, 4H, CH2S) | 3.22 (m, 4H, CH2S) | |
| 1.84 (m, 16H, CH2C) | 1.86 (m, 16H, CH2C) | 1.88 (m, 16H, CH2C) | 1.93 (m, 16H, CH2C) | 1.92 (m, 16H, CH2C) | 1.91 (m, 16H, CH2C) | |
| 31P NMR | 17.3 (d, P(OCH2)Pir | 16.8; 16.7 (d, P(OCH2)Pir | 16.1; 15.6 (d, P(OCH2)Pir | 13.8; 13.6 (d, P(OCH2)Pir | 13.8; 13.6 (d, P(OCH2)Pir | 14.4; 14.2 (d, P(OCH2)Pir |
| 53.7 (t, P(SCH2)2) | 54.9; 54.7 (t, P(SCH2)2) | 41.4 (m, P(SCH2)2) | 37.4 (m, P(SCH2)2) | 37.5 (m, P(SCH2)2) | 39.5 (m, P(SCH2)2) | |
| 54.9; 54.6 (t, P(SCH2)2) | 54.1; 53.4 (t, P(SCH2)2) | 54.2; 53.4 (t, P(SCH2)2) | 51.3 (t, P(SCH2)2) |
For better visualization of the obtained results 1H NMR spectra were presented on Fig 2 and Fig. 3 for ligand L1 and L4 respectively.
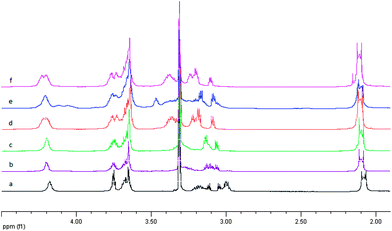 | ||
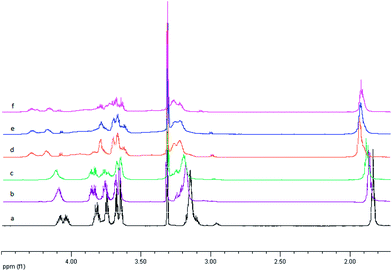
| Fig. 2
1H NMR spectra in methanol for uncomplexed ligand L1 – (a) and after adding silver ion with the Ag:L molar ratio 0.5:1, 1:1, 2:1, 3:1 and 4:1 – spectrum b, c, d, e, and f respectively. | ||
 | ||
| Fig. 3
1H NMR spectra in methanol for uncomplexed ligand L4 (a) and after adding silver ion with the Ag:L molar ratio 0.5:1, 1:1, 2:1, 3:1 and 4:1 – spectrum b, c, d, e, and f respectively. | ||
On the basis of 1H NMR analysis for ligand L1 and its complexes, after adding of the first silver ion portion (Ag:L = 0.5:1) the significant shift (0.07 ppm) of the signal within the CH2S group originated from the 2,2′-thiodiethanothiol bridges was observed. This can support the fact that silver ion is strongly bound by sulfur atoms of the third macrocyclic ring placed in the center of the molecule. 31P NMR measurements show that in this case probably two such system exist – forming sandwich complexes Fig. 4a with slight shift between two ligand molecules Fig. 5 (two triplets at 53.6 ppm and 53.4 ppm and two doublets at 25.3 and 25.2 ppm).
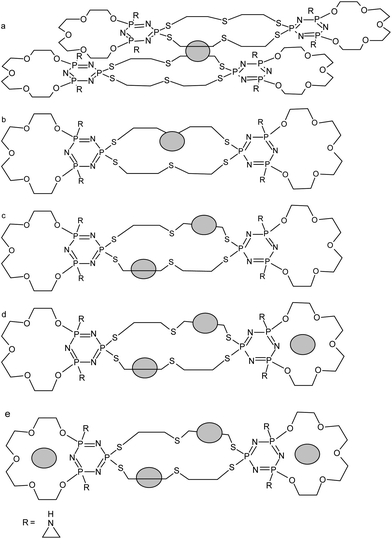 | ||
| Fig. 4 Schematic way of silver ions binding for L1. | ||
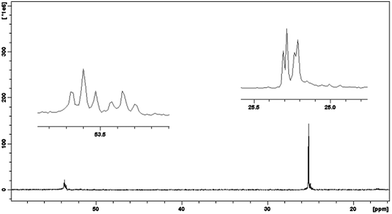 | ||
| Fig. 5 31P NMR spectrum for AgL2 system of ligand L1 in methanol. | ||
31P NMR for the AgL system show that the structure of this form of complex is relatively stiff – only one triplet at 53.3 ppm from exocyclic phosphorous atoms as well as one doublet at 25.4 ppm from the endocyclic phosphorous atom is seen on the spectrum. Furthermore on 1H NMR spectrum further shift (of about 0.07 ppm) within the CH2S protons region is observed – what can be explained by the deeper allocation of silver ion within the ligand cavity (Fig. 4b).
In case of Ag2L system there is further downfield shift of CH2S group protons what suggests the addition of the second silver ion is realized through the same part of the centered macrocyclic ring (Fig. 4c). This hypothesis is confirmed by 1H–31P NMR spectrum on which the multiplet assigned to phosphorous from the bridging part of the ligand is slightly splitted into two parts (multiplet 3.38 ppm and 52.2 ppm) (Fig. 6). This splitting is probably caused by unequal location of two silver ions toward donor sulfur atoms from the bridging moieties of the ligand.
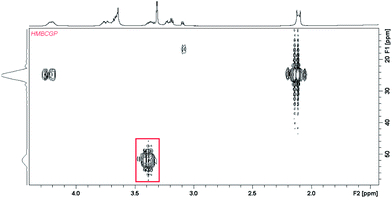 | ||
| Fig. 6 1H–31P HMBC spectrum for ligand L1 in methanol, the signal connected with the protons of CH2S group connected with silver ions binding in Ag2L system was marked by rectangle. | ||
In case of triple excess of silver ions over the ligand – Ag3L, quite significant separation of the signals originated from the CH2OP group is observed. This fact is probably connected with the binding of additional silver ion by endocyclic nitrogen atom from one of the terminal macrocyclic part of the ligand (Fig. 4d). Such effect was observed in cases of monomacrocyclic PNP-lariat ethers.27 After adding next portion of silver ion to the ligand the splitting of the signal of the abovementioned protons almost disappear what makes the binding of the fourth silver ion to the nitrogen of the second terminal macrocyclic ring very probable (Fig. 4e). The relatively lower values of the stability constants Ag3L (logβ = 2.75) and Ag4L (logβ = 2.89) in comparison with Ag2L system (logβ = 3.55) is strictly connected with the withdrawal of the electrons from the terminal macrocyclic rings by silver ions bound by sulfur donor atoms from the bridging parts of the ligand. Aziridine substituents are rather not involved in enhancing the metal ion binding – there are no shifts within the region of CH2–N protons. In case of the second derivative with pyrrolidine moieties L2, again one can observe the tendency toward the supporting of the metal ion binding when forming Ag3L and Ag4L systems. Such behaviour result in the higher stability constants for Ag3L and Ag4L – logβ = 3.83 and 3.17 respectively.
On the basis of the spectrometric results for L4 derivative and its complexes with silver ion we can conclude that the way of complexation especially when forming of AgL, Ag2L and Ag4L system, is slightly different. In the case of AgL complex formation the silver ion is not situated at the centre of the molecule but is rather shifted toward two sulfur donor atoms originated from the bridging moiety of the ligand. Hence this cation is situated asymmetrically, closer to the one of the cyclotriphosphazene subunit. This hypothesis is confirmed by the signal which appears on the 1H–31P HMBC spectrum (3.13–41.4) (Fig. 7). This signal is assigned to CH2S groups attached to the phosphorous atom from one of the cyclotriphosphazene ring. Proton signals from these groups are shifted of about 0.1 ppm. The additional confirmation of such binding might be also the difference between stability constants of this system for ligands L2 – logβ = 6.35 in which the binding occurs in the centre of the bridging moieties of the molecule and ligand L4 – logβ = 5.07.
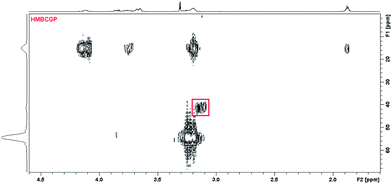 | ||
| Fig. 7 1H – 31P HMBC spectrum for ligand L4 in methanol, the signal connected with the protons of CH2S group connected with silver ions binding in AgL system was marked by rectangle. | ||
On the spectrum of Ag2L complex the significant shifts (0.18 ppm) of the signals assigned to protons from CH2OP group are observed. Such behaviour is connected with different way of binding of the second silver ion to the investigated ligand, as it was observed in case of the ligand L1. The shifting of the spectrum in this region is usually connected with the binding of silver ion by endocyclic nitrogen atom from cyclotriphosphazene ring.
The lack of the determined stability constant for Ag3L doesn't prove that for ligands L3 and L4 these forms of complexes do not exist. Such situation may appear when the constant value is very low and therefore couldn't be determined by the selected potentiometric method.
The very low value of the stability constant may be connected with destabilization of the part of bridged moiety of the ligand to which the first silver ion was connected. Hence the withdrawal of electrons from the cyclotriphosphazene unit is observed thus the second N–Ag+ binding is very weak.
After adding the last portion of silver ions, on the 1H NMR spectrum there is a split of one of the two multiplets of protons from the CH2OP group observed, which may be caused by attaching of two silver ions to one of the side macrocyclic part of the ligand.
Conclusions
Complexes of silver ions with the bridged ligands L1–L4 show diversity of possible structures which can be formed. The way of binding and the form of the coordination observed depend on the number of preferred donor atoms within the ligand molecule and the steric aspect of the investigated compounds. In case of both presented groups – the bridged ligands L1–L4 as well as the simpler forms L5 and L6, the most stable form of the complexes with silver ions is the one with stoichiometry equal 1:1 (AgL). Despite of different ways of binding the first silver ion – all these systems are characterized by the highest values of stepwise stability constants. The ligands with six sulfur atoms within the structure form more stable systems (for AgL system for L1 and L2 logβ equals 5.42 and 6.35, respectively) in comparison to those with only four sulfur donor atoms (for AgL system for L3 and L4 logβ equals 4.16 and 5.07, respectively). The influence of pyrrolidinyl and aziridynyl substituent groups on the stepwise stability constants is also observed but it depends on the form of the obtained system. In the case of the sandwich type systems the bigger the substituent is, the lower value of the stepwise stability constant of the obtained complexes is observed. In other investigated forms of the complexes the pyrrolidinyl substituents enhanced the stability of the observed systems in comparison to the aziridinyl derivatives. Due to the possibility of complexation of four silver ions within one molecule and relatively high stability constants of the obtained systems, the synthesized new ligands could be good medium for silver recovering from the industrial wastes. Furthermore the investigated complexes especially with L5 and L6 ligands should strengthened the previously described antitumor28 properties of uncomplexed compounds.
Notes and references
- A. H. M. Elwahy and A. A. Abbas, J. Heterocycl. Chem., 2009, 46(6), 1035–1079 CrossRef.
- A. H. M. Elwahy and A. A. Abbas, J. Heterocycl. Chem., 2008, 45(1), 1–65 CrossRef CAS.
- G. W. Gokel, W. M. Leevy and M. E. Weber, Macrocyclic Chemistry: Current trends and future perspectives, Springer, 2005, pp. 253–265 Search PubMed.
- G. W. Gokel, W. M. Leevy and M. E. Weber, Chem. Rev., 2004, 104, 2723–2750 CrossRef CAS PubMed.
- G. W. Gokel, Chem. Soc. Rev., 1992, 39, 39–47 RSC.
- G. W. Gokel, Crown ethers and cryptand, University of Miami Coral Gables, Florida, USA, 1991 Search PubMed.
- K. Brandt, I. Porwolik and M. Siwy, Wiad. Chem., 1997, 91–117 CAS.
- D. M. Goli, D. M. Dishing, C. J. Diamond and G. W. Gokel, Tetrahedron Lett., 1982, 23(50), 5243–5246 CrossRef CAS.
- D. B. White, K. A. Arnold and G. W. Gokel, Tetrahedron Lett., 1987, 28(16), 1749–1752 CrossRef.
- D. M. Dishing, C. J. Diamond, M. I. Cinoman and G. W. Gokel, J. Am. Chem. Soc., 1983, 105, 586 CrossRef.
- R. A. Schultz, D. B. White, D. M. Dishing, K. A. Arnold and G. W. Gokel, J. Am. Chem. Soc., 1985, 107, 6659 CrossRef CAS.
- K. Brandt, T. Kupka, J. Drozd, J. C. van de Grampel, A. Meetsma and A. P. Jekel, Inorg. Chim. Acta, 1995, 228(2), 187–192 CrossRef CAS.
- K. Brandt, I. Porwolik, T. Kupka, R. A. Shaw and D. B. Davies, J. Org. Chem., 1995, 60, 7433–7438 CrossRef CAS.
- K. Brandt, T. Kupka, J. Drozd, J. C. van de Grampel, A. Meetsma and A. P. Jekel, Inorg. Chim. Acta, 1995, 228(2), 187–192 CrossRef CAS.
- K. Brandt, I. Porwolik, T. Kupka, R. A. Shaw and D. B. Davies, J. Org. Chem., 1995, 60, 7433–7438 CrossRef CAS.
- R. Cacciapaglia and L. Mandolini, Chem. Soc. Rev., 1993, 221 RSC.
- R. Hoss and F. Vogtle, Angew. Chem., Int. Ed. Engl., 1994, 33, 375 CrossRef.
- K. Brandt, Unique synthetic opportunities on the borderline of cyclophosphazene chemistry and supramolecular chemistry, Nova Science Publisher, Inc., 2004, pp. 93–132 Search PubMed.
- R. A. Bartsch, E. K. Lee, S. Chun, N. Elkarim, K. Brandt, I. Porwolik-Czomperlink, M. Siwy, D. Lach and J. Silberring, J. Chem. Soc., Perkin Trans. 1, 2002, 2, 442–448 Search PubMed.
- K. Brandt, I. Porwolik, T. Kupka, A. Olejnik, R. A. Shaw and D. B. Davies, J. Org. Chem., 1995, 60, 7433–7438 CrossRef CAS.
- K. Brandt, I. Porwolik, M. Siwy, T. Kupka, R. A. Shaw, D. B. Davies, M. B. Hursthouse and G. D. Sykara, J. Am. Chem. Soc., 1997, 119, 12432–12440 CrossRef CAS.
- K. Brandt, I. Porwolik, M. Siwy, R. A. Shaw, D. B. Davies and M. B. Hursthouse, Phosphorus Res. Bull., 1996, 6, 17–20 CAS.
- S. Beşli, Inorg. Chem. Commun., 2005, 8, 449–452 CrossRef PubMed.
- K. Brandt, I. Porwolik-Czomperlink, M. Siwy, T. Kupka, R. A. Shaw, D. B. Davies and R. A. Bartsch, J. Inclusion Phenom. Macrocyclic Chem., 1999, 35, 281–289 CrossRef CAS.
- K. Brandt, P. Seliger, A. Grzejdziak, T. J. Bartczak, R. Kruszyński, D. Lach and J. Silberring, Inorg. Chem., 2001, 40, 3704–3710 CrossRef CAS PubMed.
- P. Seliger, G. Andrijewski, M. Siwy and D. Sęk, Pol. J. Chem., 2009, 83, 581–588 CAS.
- P. Seliger, N. Sołtys, G. Andrijewski and M. Siwy, New J. Chem., 2012, 36, 2607–2612 RSC.
- M. Siwy, D. Sęk, B. Kaczmarczyk, I. Jaroszewicz, A. Nasulewicz, M. Pelczyńska, D. Nevozhay and A. Opolski, J. Med. Chem., 2006, 49, 806–810 CrossRef CAS PubMed.
- K. Brandt, R. Kruszyński, T. J. Bartczak and I. Porwolik-Czomperlink, Inorg. Chim. Acta, 2001, 322, 138–144 CrossRef CAS.
- R. Kruszyński, M. Siwy, I. Porwolik-Czomperlink and A. Trzesowska, Inorg. Chim. Acta, 2006, 359, 649–658 CrossRef PubMed.
- T. Nabeshima, Coord. Chem. Rev., 1996, 148, 151 CrossRef CAS; V. V. Litvinova and A. V. Anisimov, Khim. Geterotsikl. Soedin., 1999, 1587 Search PubMed.
- K. Brandt, I. Porwolik-Czomperlik, M. Siwy, T. Kupka, R. A. Shaw, S. Ture, A. Clayton, D. B. Davies, M. B. Hursthouse and G. D. Sykara, J. Org. Chem., 1999, 64, 7299 CrossRef CAS.
- D. B. Davies, T. A. Clayton, R. J. Eaton, R. A. Shaw, A. Egan, M. B. Hursthouse, G. D. Sykara, I. Porwolik-Czomperlik, M. Siwy and K. Brandt, J. Am. Chem. Soc., 2000, 122, 12447–12457 CrossRef CAS.
- I. Porwolik-Czomperlink, K. Brandt, T. A. Clayton, D. B. Davies, R. J. Eaton and R. A. Shaw, Inorg. Chem., 2002, 41(19), 4944–4951 CrossRef PubMed.
- H. R. Allcock, M. L. Turner and K. B. Visscher, Inorg. Chem., 1992, 31, 4354–4364 CrossRef CAS.
- R. A. Shaw, Phosphorus, Sulfur Silicon Relat. Elem., 1989, 13, 840–844 Search PubMed.
- P. R. Dave, F. Forohar, T. Axenrod, C. D. Bedfort, M. Chaykovsky, M. K. Rho, R. Gilardi and C. George, Phosphorus, Sulfur Silicon Relat. Elem., 1994, 90, 1–4 CrossRef.
- J. F. Labarre, Top. Curr. Chem., 1985, 129, 173–230 CrossRef CAS.
- K. Brandt, I. Porwolik, M. Siwy, T. Kupka, R. A. Shaw, D. B. Davies, M. B. Hursthouse and G. D. Sykara, J. Am. Chem. Soc., 1997, 119, 1143–1144 CrossRef CAS.
- P. Gans, A. Sabatini and A. Vacca, Talanta, 1996, 43, 1739 CrossRef CAS.
- H. K. Frensdorff, J. Am. Chem. Soc., 1971, 93, 600–606 CrossRef CAS.
- R. M. Izatt, R. E. Terry, L. D. Hause, A. G. Avondet, J. S. Bradshaw, N. K. Dalley, T. E. Jensen, B. L. Haymore and J. J. Christensen, Inorg. Chim. Acta, 1978, 30, 1–8 CrossRef CAS.
- H. J. Buschmann, Chem. Ber., 1985, 118, 4297–4302 CrossRef CAS.
- T. Ossowski, R. Trokowski, D. Rogowska and J. Kira, Pol. J. Chem., 2001, 75, 345–353 CAS.
| This journal is © The Royal Society of Chemistry 2013 |