Indole synthesis – something old, something new
Martyn
Inman
and
Christopher J.
Moody
*
School of Chemistry, University of Nottingham, University Park, Nottingham NG7 2RD, UK. E-mail: c.j.moody@nottingham.ac.uk
First published on 6th September 2012
Abstract
Indoles, both naturally occurring and synthetic, exhibit wide-ranging biological activity. Unusual and complex molecular architectures occur among their natural derivatives. As a result, this important ring system continues to attract attention from the international chemical community, and new methodologies for the construction of this ever relevant heteroaromatic ring continue to be developed. Unfortunately, many methods frequently start from ortho-substituted anilines, thereby greatly restricting the availability of starting materials. A more general approach would start from a mono-functionalized arene such as an aniline or halobenzene, followed by cyclization with C–C or C–N bond formation to an unactivated C–H bond. Such methods are the subject of this perspective.
1. Introduction
The indole ring system represents one of the most abundant and important heterocycles in nature. Found in a hugely diverse array of biologically significant natural compounds, from simple derivatives such as the neurotransmitter serotonin to complex alkaloids such as the clinically used anticancer agents vinblastine and mitomycin C, and the antihypertensive alkaloid reserpine (Fig. 1), the importance of indoles to biological chemistry cannot be overstated. Additionally, a number of important synthetic drugs contain an indole motif, including sumatriptan, tadalafil, rizatriptan and fluvastatin (Fig. 2), accounting for a total of $3.2bn in sales in 2010.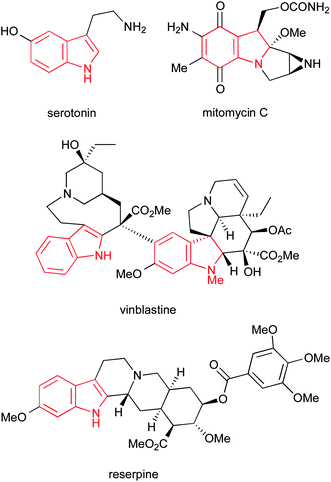 | ||
| Fig. 1 Some naturally occurring indoles. | ||
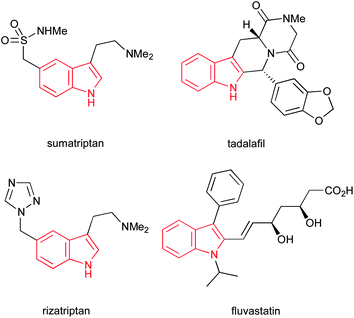 | ||
| Fig. 2 Four clinically used indoles accounting for a total of $3.2bn in sales in 2010. | ||
The continued development of routes towards indoles has been a central theme in organic synthesis over the last century, in keeping with their importance.1 However, there are still limitations on the chemical space which is easily accessible; this can be readily observed by comparison of the naturally occurring indole drugs with their synthetic counterparts. In particular, the substitution pattern around the six-membered ring is notably less complex in synthetic indoles than in naturally occurring ones. To our knowledge, there are no synthetic indoles bearing substituents at more than one of the benzenoid ring positions currently in clinical use. We attribute this observation not to any perceived pharmacological disadvantages of highly substituted indoles, but rather to their relative synthetic intractability. Hence, there is still room for improvement in the field of indole synthesis.
With a few exceptions, such as those proceeding through functionalization of pyrroles,2 and methods that form both rings simultaneously,3 indole syntheses almost universally involve annelation of the five-membered ring to an existing benzene ring bearing the appropriate functionality. These can be divided into those in which two substituents in an ortho-relationship are connected together (Scheme 1A), and those in which a single substituent is cyclized directly onto the aromatic ring (Scheme 1B).1
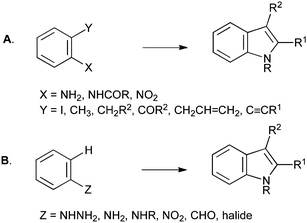 | ||
| Scheme 1 General methods for indole synthesis. | ||
Most approaches to indole synthesis, including many of the modern catalytic methods, require an ortho-disubstituted arene precursor (Scheme 1A). These reactions can be further divided into two broad classes: those utilizing transition-metal catalysis and those using more classical methods. Examples of the former include the Larock (Scheme 2A), Castro (Scheme 2B), Hegedus (Scheme 2C), Mori–Ban (Scheme 2D), Ma (Scheme 2E) and Cacchi (Scheme 2F) indole syntheses.1
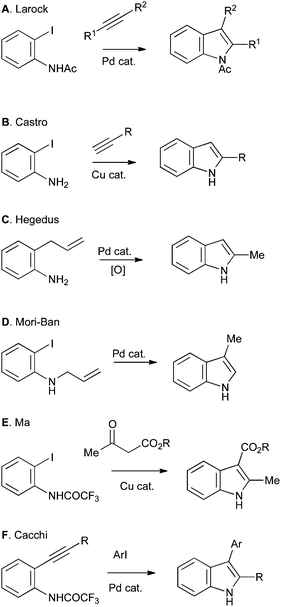 | ||
| Scheme 2 Examples of indole synthesis from ortho-substituted anilines mediated by transition-metal catalysis. | ||
Non-catalytic methods include the more classical Reissert (Scheme 3A), Madelung (Scheme 3B), Leimgruber–Batcho (Scheme 3C) and Fürstner (Scheme 3D) indole syntheses.1
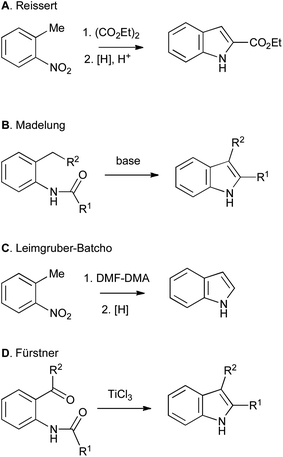 | ||
| Scheme 3 Classical routes to indoles from ortho-substituted anilines. | ||
Although the use of such bifunctional precursors allows regiospecific closure of the indole ring, it imposes an additional level of constraint, complexity and synthetic inaccessibility on the required starting material, and often leads to a circuitous synthetic route overall. However, such routes remain important and valuable contributions to the field, and are extremely useful when applied judiciously, particularly when the six-membered ring of the indole bears few or no substituents. An excellent example can be found in Baran's 2008 synthesis of psychotrimine, in which one of the tryptamine units must be introduced via nucleophilic addition on nitrogen. As no preformed indole derivatives performed satisfactorily, inexpensive 2-iodoaniline was used as a surrogate, and an ensuing Larock synthesis then gave the required bis-indole derivative on multi-gram scale.4 Disubstituted arenes can also be used appropriately when their preparation is convenient and incurs no additional costs relative to their monosubstituted counterparts. A notable example is found in the Smith synthesis of penitrem D, wherein a complex ortho-toluidine was used in a daring Madelung reaction that proceeded smoothly to provide the pivotal heptacyclic indole.5 Nevertheless, despite some successes, even the modern methods, often transition-metal catalyzed, that start from ortho-substituted anilines (Schemes 1A–3), greatly restrict the availability of starting materials. A more general approach would start from a mono-functionalized arene, followed by cyclization with C–C or C–N bond formation to an unactivated C–H bond (Scheme 1B), and several classical and modern approaches both fall into this category. It is the latter class of reaction that forms the main subject of this perspective article.
2. Indoles from monosubstituted precursors
Although the connection of two substituents to form an indole gives complete regiospecificity, it can be extremely costly in terms of synthetic steps required to make the precursor. This is especially problematic when a large number of compounds are required, for example in the high-throughput synthesis of industrial compound libraries. In contrast, monosubstituted precursors are usually accessed much more readily; however, there can be issues of regioselectivity when the ring closure can occur in two different directions to give isomeric products. This obviously does not apply when the precursor is symmetrical, or when one ortho-position is blocked.2.1 The Fischer indole synthesis and modern variants
First reported in 1883, the Fischer reaction remains the pre-eminent method for the synthesis of indoles (Scheme 4).6 On heating under acidic conditions, an arylhydrazone tautomerizes to the enehydrazine, and undergoes a [3,3]-sigmatropic rearrangement that results in the functionalization of an unactivated aromatic C–H position. Further tautomerization and imine exchange give the desired indole, with ammonia eliminated as a side-product. Although maybe unfashionable, this celebrated reaction addresses all the requirements of a modern indole synthesis in its convenience and simplicity – coupling a mono-functionalized arene with a readily available aldehyde or ketone, the only drawback being the relatively small range of arylhydrazines that are commerically available.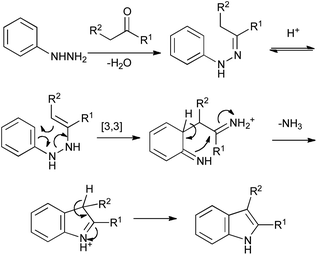 | ||
| Scheme 4 The Fischer indole synthesis. | ||
Applications of the Fischer indole synthesis abound in total synthesis; a recent example can be found in Garg's synthesis of aspidophylline (Scheme 5). Here, the intermediate indolenine 1 is prevented from aromatization by the absence of an α-proton; instead, the pendant alcohol released by lactone methanolysis can add to the imine, giving the required aminal 2.7
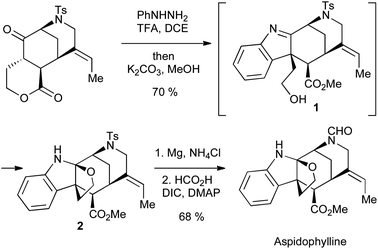 | ||
| Scheme 5 Garg's synthesis of aspidophylline using a Fischer indole approach. | ||
Perhaps the most impressive application of the Fischer reaction is in Fukuyama's synthesis of haplophytine, in which the structurally complex arylhydrazine 3 is condensed with tricyclic ketone 4. Heating in the presence of 4-toluenesulfonic acid gave a separable 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1.6 mixture of indole 5 and indolenine 6; the latter could be converted into the final target in five steps (Scheme 6).8 This example is remarkable not only for the extreme structural complexity of the substrates, but also because it gives a good yield of the desired product in spite of the well-documented incompatibility of ortho-alkoxyarylhydrazines with the Fischer reaction.9
:1.6 mixture of indole 5 and indolenine 6; the latter could be converted into the final target in five steps (Scheme 6).8 This example is remarkable not only for the extreme structural complexity of the substrates, but also because it gives a good yield of the desired product in spite of the well-documented incompatibility of ortho-alkoxyarylhydrazines with the Fischer reaction.9
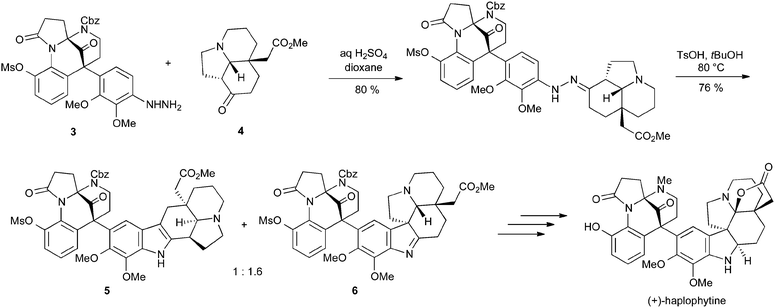 | ||
| Scheme 6 Fukuyama's synthesis of haplophytine using a Fischer indole approach. | ||
As already alluded to, the primary difficulty associated with the Fischer indole synthesis has been the inaccessibility and toxicity of the arylhydrazine precursors. Traditionally, these have been made by diazotization of the corresponding aniline and reduction of the diazonium salt; this often capricious reaction has a relatively poor substrate scope, and so there has been a considerable effort to find safer and more reliable alternatives.
The conditions most commonly employed to convert aryldiazonium salts into hydrazines have used either elemental tin or SnCl2 in acidic media; a recently rediscovered alternative is the use of ascorbic acid as a readily available and metal-free reductant.10 The reaction proceeds via a rearrangement to give an acid-labile oxalyl hydrazide which can be used directly in a Fischer reaction as a surrogate for the hydrazine; this procedure has been applied to the synthesis of eletriptan (Scheme 7), and the reduction step has also been demonstrated in flow.11
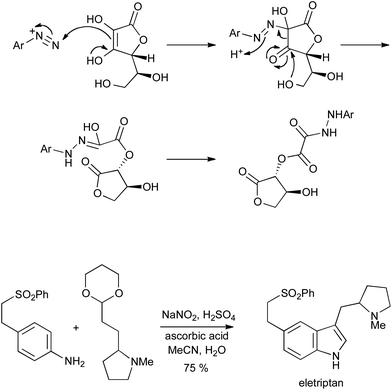 | ||
| Scheme 7 Reduction of aryldiazonium salts with ascorbic acid, and Fischer indolization of the resulting arylhydrazines. | ||
Alternatively, N-substituted arylhydrazines can be prepared by reduction of the N-nitroso derivative, although the toxicity associated with nitrosamines renders this far from ideal. This reduction can be effected with zinc dust in acetic acid12 or lithium aluminium hydride;13 sodium dithionite provides a milder alternative,14 and has been used on pilot plant scale in the synthesis of the 5HT2C receptor agonist 7 (Scheme 8).15
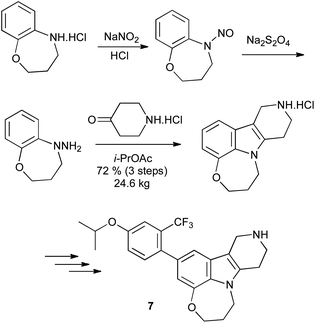 | ||
| Scheme 8 Arylhydrazines from reduction of N-nitrosoanilines. | ||
Hydroamination of alkynes represents another approach to the required enehydrazine intermediates.16 In the first reported example, heating hydrazidozirconocene 8 with alkynes gave the aminozirconocene 9, presumably by hydroamination and [3,3]-sigmatropic rearrangement.17 Treatment with acid gave the indole, although the reaction is limited to symmetrical alkynes and hence indoles bearing the same substituent at C-2 and C-3 (Scheme 9).
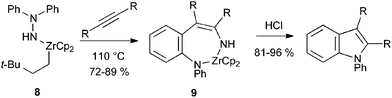 | ||
| Scheme 9 Alkyne hydroamination with hydrazine zirconium derivatives. | ||
Titanium catalysts such as 10 and 11 can perform hydroaminations of 1,1-disubstituted18 and monosubstituted19 hydrazines, respectively; these can be converted into protected or unprotected indoles by addition of ZnCl2 to the reaction mixture (Scheme 10). More recently, simple zinc salts such as ZnCl2 or Zn(OTf)2 have been used stoichiometrically to effect both the regioselective hydroamination of terminal alkynes and Fischer cyclization in a single step.20
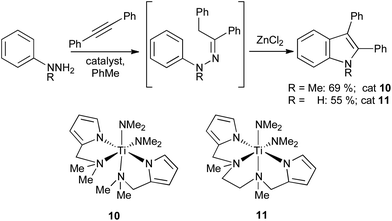 | ||
| Scheme 10 Titanium mediated hydrazination of alkynes. | ||
A number of new methods have been reported in the last decade for metal-catalyzed cross coupling of aryl halides with hydrazine derivatives. The earliest reports were by Buchwald et al. and Hartwig, who independently reported arylations of benzophenone hydrazone.21,22 Buchwald also demonstrated that the resulting arylhydrazones could undergo acid-catalyzed exchange with enolizable ketones and subsequent Fischer reaction, avoiding the need to isolate the arylhydrazine (Scheme 11).21 After extensive optimization of the catalyst, this cross coupling has proved extremely effective on multi-kilogram scale.23
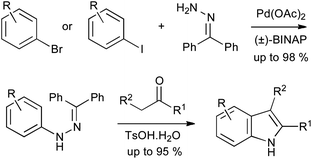 | ||
| Scheme 11 Buchwald–Hartwig Pd-catalyzed arylation of benzophenone hydrazone in indole synthesis. | ||
Many further catalytic arylation reactions of substituted hydrazines have been developed, several of which could in principle be applied to indole synthesis; in some cases this transformation has also been described. Buchwald reported on the copper-catalyzed arylation of tert-butyl carbazate with aryl iodides, finding that arylation occurred exclusively on the Boc-protected nitrogen in most cases (Scheme 12A).24 Conversely, the use of benzhydrazide gave arylation on the unprotected nitrogen under similar conditions. The copper iodide–4-hydroxyproline catalytic system has recently been demonstrated to perform the same transformation effectively and with broad substrate scope, using aryl iodides or bromides.25 The conversion of the products into indoles with concomitant deprotection of the Boc group has been demonstrated;26 similar chemistry utilizing benzyl carbazate has also been shown to yield Cbz-protected indoles (Scheme 12B),27 while the corresponding ethyl carbazates, derived in the same manner, have been subjected to tandem Fischer–Claisen reactions to give tricyclic indole derivatives (Scheme 12C).28
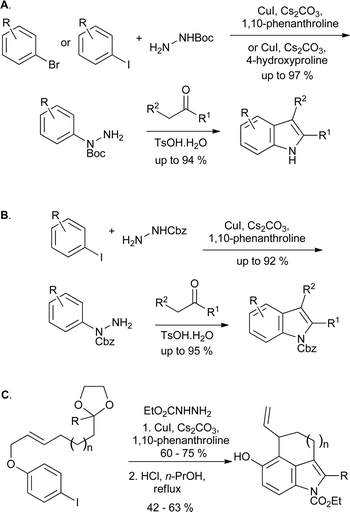 | ||
| Scheme 12 Copper catalyzed arylations of protected hydrazines in indole synthesis. | ||
The use of protected hydrazine nucleophiles has been necessary because hydrazine itself acts as a reductant in palladium-catalyzed reactions,29 giving simple dehalogenation rather than N-arylation. However, after an extensive catalyst screening programme, Stradiotto and Lundgren reported recently that [Pd(cinnamyl)Cl]2 and Mor-DalPhos 12 provide an effective catalyst system for the hydrazination of aryl chlorides and tosylates with hydrazine hydrate. To simplify isolation, the resulting arylhydrazines were converted into their benzaldehyde hydrazones (Scheme 13); although no examples were reported, the conversion of the hydrazones into indoles can be easily envisaged.30
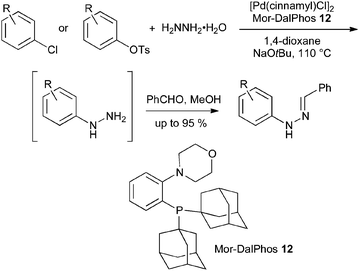 | ||
| Scheme 13 Palladium-catalyzed arylation of hydrazine hydrate. | ||
We have recently reported on the synthesis of indoles from aryl Grignard reagents and tert-butyl azodicarboxylate, proceeding via the double Boc-protected arylhydrazine 13 (Scheme 14A).31 The use of isopropyl- or phenyl-magnesium chloride for the metallation provides a mild method for conversion of aryl iodides and bromides into the hydrazine derivatives, with relatively broad substrate scope. On heating the protected hydrazine under acidic conditions in the presence of a ketone, the Boc groups were removed, and hydrazone formation and Fischer reaction occurred in one step. This method has been extended to the synthesis of azaindoles and other heterocyclic derivatives, whilst the use of ortho-directing groups can allow the direct synthesis of indoles from non-halogenated arenes (Scheme 14B).32
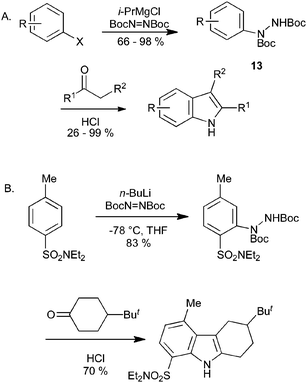 | ||
| Scheme 14 Indole synthesis by magnesiation (or lithiation) of halobenzenes. | ||
Alternative methods have been reported for the addition of aryl groups to azodicarboxylates to give doubly protected arylhydrazines similar to 13. These include Friedel–Crafts-type direct amination of electron rich arenes,33 and the palladium34 or copper35 catalyzed addition of areneboronic acids to azodicarboxylates.
The use of arylhydrazines can be obviated entirely by the Japp–Klingemann reaction, first reported in 1887.36 In this modification to the Fischer reaction, arylhydrazones are obtained by reaction of an aryldiazonium salt with a β-ketoester under basic conditions. The enolate of the ketoester attacks the terminal nitrogen of the diazonium salt, and is followed by hydroxide-mediated deacylation (Scheme 15). The resulting hydrazone can then be converted into an indole under acidic conditions, as in the Fischer reaction.
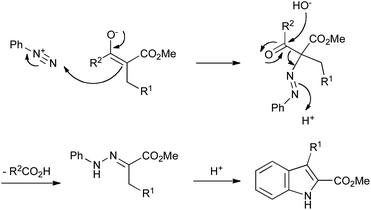 | ||
| Scheme 15 The Japp–Klingemann variation on the Fischer indole synthesis. | ||
The addition of organometallics to aryldiazonium salts provides an extension of the Japp–Klingemann synthesis, proceeding via tautomerization of the azo intermediate 14. Although known for over fifty years,37 it is only recently that this reaction has found application to the synthesis of indoles (Scheme 16).38 The reaction of alkylzinc bromides with aryldiazonium tetrafluoroborates gave the hydrazone intermediate; subjecting the reaction mixture to microwave irradiation in the presence of chlorotrimethylsilane gave the indole product in a single step from the diazonium salt. It is noteworthy that the reaction tolerated often-troublesome functionalities such as ketones, esters and nitro groups.
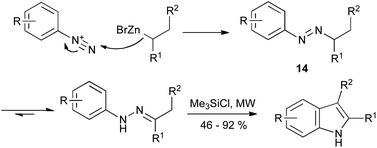 | ||
| Scheme 16 Modern variant on the Japp–Klingemann reaction using organozinc nucleophiles. | ||
Alternatively, the nitrogen functionality can be attached to the alkyl precursor. Thus, aryllithiums can add to α-diazoesters, giving the requisite azo compounds which can be transformed into indoles as described above (Scheme 17).39 As with the Japp–Klingemann reaction, the scope of this procedure is currently limited to indole 2-esters, but it proceeds with generally good yields in comparison to the classical method.
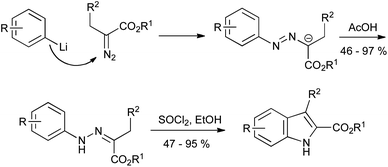 | ||
| Scheme 17 Hydrazones from α-diazoesters en route to indole 2-carboxylates. | ||
Upon first inspection, the Fischer reaction may not appear well suited to asymmetric catalysis; however, the use of carbonyl compounds with α-chiral centres leads via a prochiral enehydrazine to a potentially chiral indoline. A single example of asymmetric induction, using an excess of BINOL derived phosphoric acid 15, gave only 28% ee for the synthesis of a furoindoline using this method (Scheme 18).40
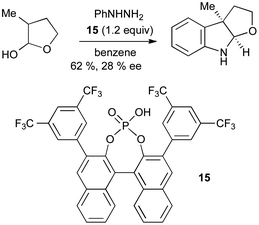 | ||
| Scheme 18 Asymmetric catalysis in Fischer indole synthesis. | ||
A more successful development has been the asymmetric synthesis of 3-substituted tetrahydrocarbazoles from prochiral hydrazones (Scheme 19). Again, a chiral phosphoric acid catalyst 18 is used, though the origin of stereoselectivity is different: rather than forcing a face-selective sigmatropic rearrangement, the catalyst effects a dynamic kinetic resolution of the two rapidly equilibrating enehydrazine enantiomers 16 and 17.41
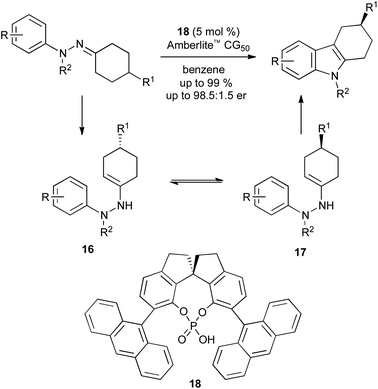 | ||
| Scheme 19 Asymmetric catalysis in Fischer indole synthesis. | ||
Hence despite its longevity, the Fischer indole reaction has stood the test of time, and with a range of modern variations, remains pre-eminent among indole syntheses.
2.2 The Bischler indole synthesis and modern variants
First reported in 1892, the Bischler indole synthesis involves alkylation of an aniline with an α-haloketone, followed by acid-catalyzed ring closure (Scheme 20).42 Although rather limited in scope due to the harsh reaction conditions required, it circumvents the problems associated with arylhydrazines, and has found widespread application in the synthesis of relatively simple indole derivatives. It can be complicated by rearrangements of the α-anilinoketone intermediate under acidic conditions if the aniline is not protected before cyclization, giving isomeric mixtures of products in some cases (Scheme 20).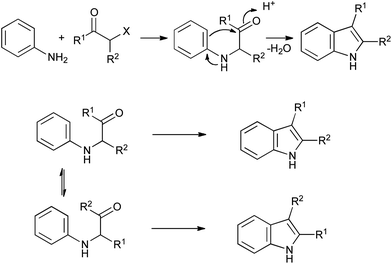 | ||
| Scheme 20 The Bischler indole synthesis. | ||
A useful variation on the Bischler reaction is the use of α-haloaldehyde acetals, which permits access to 3-unsubstituted indoles (Scheme 21).43
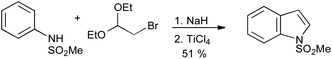 | ||
| Scheme 21 Nordlander and Sundberg modification of the Bischler indole synthesis. | ||
A further development in the Bischler synthesis has involved the use of N–H insertion reactions of diazo compounds and N-alkylanilines using rhodium or copper catalysis, providing a route to N-alkylindole-2-carboxylates (Scheme 22). N-Unsubstituted indoles could be accessed by use of the 2-ethoxycarbonylethyl or 2-phenylsulfonylethyl group as protection for the aniline nitrogen, which could be removed readily under mild basic conditions.44
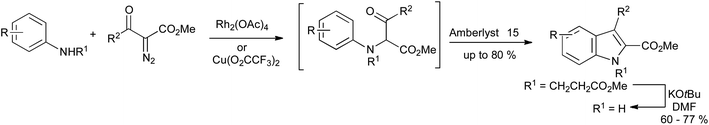 | ||
| Scheme 22 Rhodium or copper catalyzed carbene N–H insertion reactions in a variation of the Bischler indole synthesis. | ||
As with the Fischer reaction, the development of hydroamination reactions has extended the scope of the Bischler reaction from ketones and aldehydes to alkynes. Thus, treatment of a propargylic alcohol with an aniline and catalyst gives the hydroxyenamine, which can tautomerize to the required α-anilinoketone. Ru3(CO)12 (ref. 45) and Zn(OTf)2 (ref. 46) catalyze both the hydroamination and cyclization, giving the indole directly (Scheme 23). In both cases, the major indole product is that obtained after isomerization of the anilinoketone.
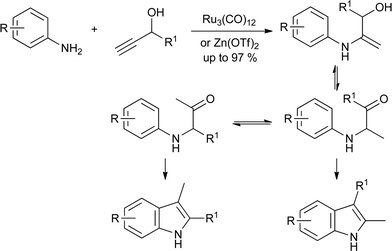 | ||
| Scheme 23 Alkyne hydroamination variant on the Bischler synthesis. | ||
2.3 The Hemetsberger indole synthesis
β-Styryl azides readily undergo thermal decomposition to the corresponding 2H-azirines, in equilibrium with the vinyl nitrene isomer, followed by electrocyclization onto the aromatic ring.47Unlike in the Fischer or Bischler indole syntheses, the C3–C3a bond is already present in the precursor for the Hemetsberger reaction; it is thus particularly suited to the regiospecific synthesis of 4- or 6-substituted indoles from ortho- or para-substituted benzaldehydes (Scheme 24). Since the reaction is effected simply by heating, it can be used in combination with other thermal processes such as the Claisen rearrangement,48 and the resulting products elaborated into carbazole alkaloids.49 It is also useful in the synthesis of a range of indole natural products such as coenzyme PQQ,50 and highly substituted indoles, as exemplified by the synthesis of the pyrroloindoles PDE-I and PDE-II, proceeding through two sequential Hemetsberger reactions (Scheme 25).51 Despite the potential hazards associated with the use of azides, the Hemetsberger reaction is amenable to scale up and has been carried on 90 g scale in Cook's synthesis of roeharmine.52
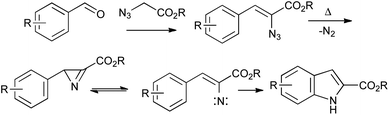 | ||
| Scheme 24 The Hemetsberger indole synthesis. | ||
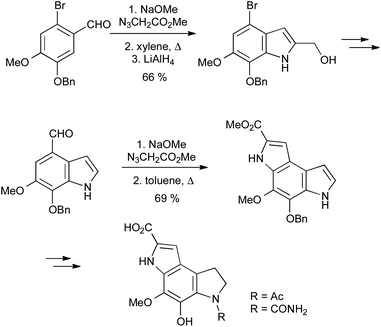 | ||
| Scheme 25 Synthesis of the pyrroloindole phosphodiesterase inhibitors PDE-I and PDE-II. | ||
The Hemetsberger reaction can be carried out under transition metal catalysis, using either the azides53 or azirines54 as starting materials. These reactions, using rhodium or iron catalysts, are presumed to occur via a metallonitrenoid species, and allow a significant reduction in reaction temperature.
As the azides are prepared by condensation of benzaldehydes with azidoacetate esters, the Hemetsberger reaction is limited to the synthesis of indole 2-carboxylates. However, alternative routes to the requisite 2H-azirines have been exploited for the synthesis of indoles. These include the Neber reaction of oximes (Scheme 26A)55 and the oxidation of enamines with diacetoxyiodobenzene (Scheme 26B).56
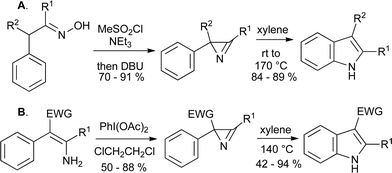
2.4 The Nenitzescu indole synthesis
5-Hydroxyindoles can be accessed in a convergent manner through the Nenitzescu reaction of enamines with quinones (Scheme 27).57 The regiochemistry is usually as shown, except where R3 is an electron-withdrawing group, in which case a 4-substituted indole is the main product. The Nenitzescu reaction has been plagued by poor yields, but its inexpensive starting materials and scalability have allowed it to be used in the large-scale synthesis of relatively simple indole building blocks. For example, a kilogram-scale Nenitzescu reaction was reported in the synthesis of the chemokine receptor antagonist 19 (Scheme 27).58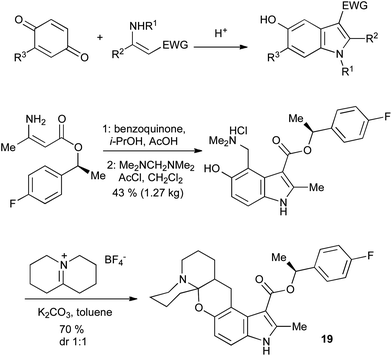 | ||
| Scheme 27 The Nenitzescu indole synthesis and its application. | ||
2.5 The Bartoli indole synthesis
The Bartoli reaction (Scheme 28) provides a very convenient and scalable method for the synthesis of indoles with minimal substitution on the 2- and 3-positions; a limitation imposed by the required excess of the Grignard reagent. It is, however, useful in the synthesis of indoles bearing numerous substituents on the benzenoid ring, as exemplified by the synthesis of 4,5,6,7-tetrasubstituted indoles (Scheme 28).59 Only ortho-substituted nitroarenes perform well, as without them nucleophilic attack occurs at the nitrogen rather than the oxygen atoms of the nitro group.60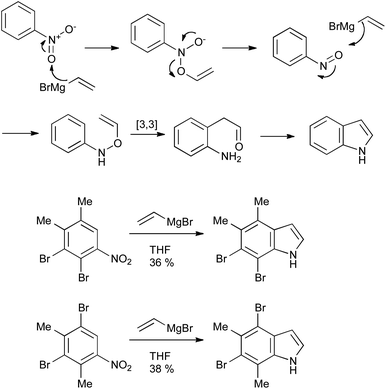 | ||
| Scheme 28 The Bartoli indole synthesis and its application. | ||
2.6 Oxidative enamine cyclizations
Of the many methods developed in recent years for the oxidative cyclization of N-aryl enamines, most have involved the use of transition-metal catalysis. The origins of such reactions lie in the synthesis of carbazoles from diarylamines (Scheme 29A),61 and of indoles from anilines and electron-poor alkynes (Scheme 29B),62 using stoichiometric palladium(II) salts which were concomitantly converted into palladium(0). It is presumed that the reactions proceed via successive electrophilic palladation of the enamine and arene, followed by reductive elimination to give the indole and a reduced palladium species (Scheme 30).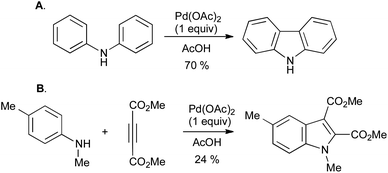 | ||
| Scheme 29 Oxidative cyclization of A, diarylamines, and B, N-aryl enamines using stoichiometric palladium(II). | ||
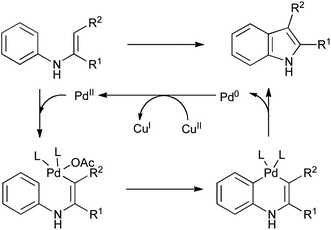 | ||
| Scheme 30 Mechanism of palladium(II) mediated cyclization of N-aryl enamines. | ||
The requirement for stoichiometric or excess palladium makes these cyclizations rather unattractive, even on relatively small scale. However, the use of additional oxidants such as copper(II) acetate to convert Pd(0) back into Pd(II) has rendered such processes much more viable,63,64 and has also been combined with inexpensive iron catalysts to effect the same transformation.65 Furthermore, the use of oxygen as a terminal oxidant has opened up the possibility of more environmentally benign processes,66–68 while a metal-free variation has been developed using bis(acetoxy)iodobenzene.69 A range of conditions is summarized in Scheme 31.
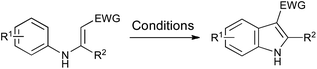 | ||
| Scheme 31 Oxidative cyclization of N-aryl enamines. Conditions: (a) Pd(OAc)2 (10 mol%), Cu(OAc)2 (3 equiv.), K2CO3 (3 equiv.), DMF, 140 °C, 24–86%;63 (b) Pd(OAc)2 (10 mol%), Cu(OAc)2 (10 mol%), O2, MeCN, reflux, 31%;64 (c) FeCl3 (10 mol%), Cu(OAc)2·CuCl2 (1.5 equiv.), K2CO3 (3 equiv.), DMF, 120 °C, 30–72%;65 (d) Pd(OAc)2 (5 mol%), O2, AcOH, 95 °C, 61–89%;66 (e) Pd(OAc)2 (10 mol%), O2, DMA–PivOH (4:1), 120 °C, 20–99%;67 (f) CuI (5 mol%), phen (17.5 mol%), Li2CO3 (2 equiv.), air, DMF, 100 °C, 51–84%;68 (g) PhI(OAc)2 (1.3 equiv.), (CH2Cl)2, 60 °C, 33–91%.69 | ||
The scope of these reactions has been limited by the requirement for an electron-withdrawing group to stabilize the enamine intermediate, but in a recent development, unstabilized imines have been converted into indoles using palladium catalysis under an oxygen atmosphere (Scheme 32A). The direct synthesis of indoles from anilines and ketones was also reported, although copper(II) acetate was required as a stoichiometric co-oxidant (Scheme 32B).70
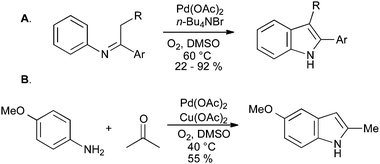 | ||
| Scheme 32 Oxidative cyclization of N-aryl imines into indoles. | ||
Oxidative cyclization of N-arylenamines can also take place readily when the enamine bears a halogen atom; such substrates can be accessed by simple addition of N-sulfonylanilines to bromoalkynes at elevated temperatures (Scheme 33).71
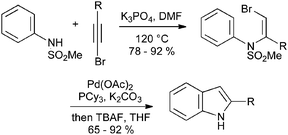 | ||
| Scheme 33 Oxidative cyclization of N-aryl bromoenamines. | ||
A mechanistically distinct alternative involves the use of catalytic ortho-metalation of a nitrogen functionality, followed by carbometalation of an alkyne. Reductive elimination of the resulting azametallacycle gives the indole product and a reduced metal species, from which the active catalyst can be regenerated by copper(II) acetate. The nitrogen functionality can be a 2-pyrimidylamino group72 or a simple acetamide (Scheme 34).73 In the latter case, the oxidant can also be used in catalytic quantities under an atmosphere of oxygen, allowing the reaction temperature to be reduced from 120 °C to 60 °C.74
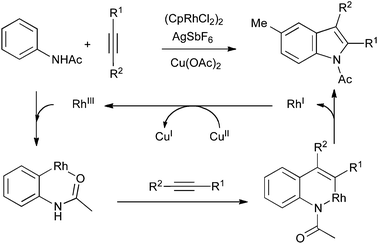 | ||
| Scheme 34 ortho-Metalation/carbometalation strategy. | ||
An interesting alternative to the oxidative enamine cyclization approach involves the formation of a C–N bond rather than a C–C bond, in a process analogous to the Hemetsberger reaction (Section 2.3); this transformation can be carried out with palladium catalysis on tosyl or Boc-protected enamines (Scheme 35A),75,76 or by treatment with bis(trifluoroacetoxy)iodobenzene at room temperature (Scheme 35B).77N-Aryl-2-alkylindoles can also be synthesized in a similar manner via enamine halogenation with N-chlorosuccinimide or N-bromosuccinimide followed by cyclization with zinc acetate.78
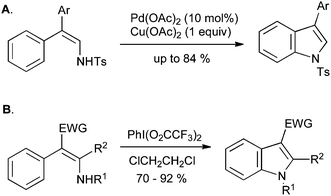 | ||
| Scheme 35 Enamine oxidative cyclization to indoles. | ||
The cyclization of benzyl oxime esters proceeds under palladium catalysis; as the initial step is an oxidative addition of palladium(0) into the N–O bond, this process is overall redox neutral and so no oxidant is required (Scheme 36).79
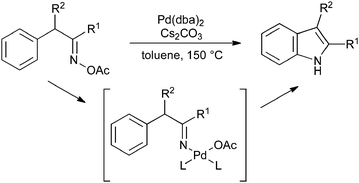 | ||
| Scheme 36 Cyclization of benzyl oxime esters. | ||
2.7 Miscellaneous methods
In addition to the important developments discussed above, numerous other methodologies have emerged for the synthesis of indoles in recent years. Of these, the Sugasawa et al. (Scheme 37A)80 and Gassman et al. (Scheme 37B)81 reactions have been used extensively, but require harsh reaction conditions.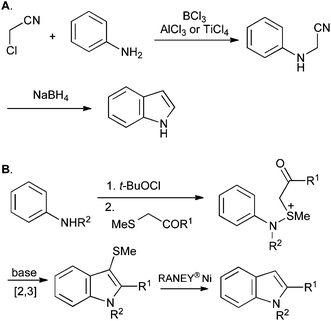
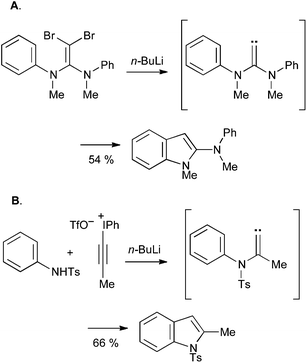
New methods continue to be developed, and have yet to find wider application: for example, vinylidene carbenes will readily insert into arene C–H bonds; the appropriate aminoalkylidenes can be accessed either by lithiation of dibromoenamines (Scheme 38A)82 or by addition of an arylsulfonamide to an alkynyl iodonium salt (Scheme 38B).83
Treatment of anilides with ethyl diazoacetate, trifluoromethanesulfonic anhydride and a mixture of 2-chloropyridine and 2,6-dichloropyridine gives, through activation of the amide to nucleophilic attack, the corresponding indole-3-ester (Scheme 39).84
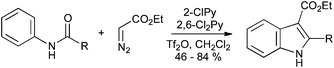 | ||
| Scheme 39 Synthesis of indole-3-carboxylates from anilides and ethyl diazoacetate. | ||
The manufacture of simple indoles from anilines and ethylene glycol is commercially attractive, but limited by its requirement for extremely high reaction temperatures. Recent developments in iridium and rhodium catalysis have allowed the use of substituted diols to give 2,3-disubstituted indoles (Scheme 40A),85 and the use of triethanolamine as a replacement for ethylene glycol (Scheme 40B).86 In both cases, some substituents are tolerated, but the requirement for elevated temperatures in these reactions has not yet been fully overcome.
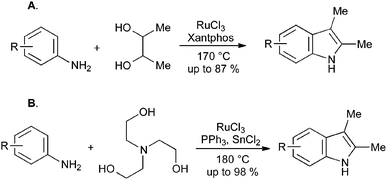
3. Conclusion
The indole ring system represents one of the most abundant and important heterocycles in nature, found in neurotransmitters such as serotonin and complex alkaloids such as the clinically used chemotherapeutic agent vinblastine. Additionally, a number of important synthetic drugs contain an indole ring, and hence this important ring system continues to fascinate chemists worldwide.Many approaches to indole synthesis, including several of the modern catalytic methods, require an ortho-disubstituted arene precursor, thereby greatly restricting the availability of starting materials by imposing an additional level of constraint, and, often, synthetic inaccessibility. A more general approach would start from a mono-functionalized arene such as an aniline or halobenzene, followed by cyclization to an unactivated C–H bond. As this perspective has demonstrated, several classical and modern methods fall into this category, and constitute extremely valuable approaches to access a wide range of indoles. The fact that so many of the literature references in this perspective are from the last decade attests to the continuing importance of indoles, and that there is always “something new” for their synthesis.
Acknowledgements
CJM's own research in indole synthesis has spanned three decades and has been supported by the University of Nottingham, the EPSRC (and its forerunner the SERC), Cancer Research UK, the Association for International Cancer Research, and many companies (some sadly no longer in existence) – May & Baker, Wyeth, Shell, Lilly, AstraZeneca, GlaxoSmithKline (and its forerunners), Tripos Receptor Research, AgrEvo, Morvus Technology. It has also involved a number of excellent graduate students and postdocs, as listed in the references.References
- R. J. Sundberg, The Chemistry of Indoles, Academic Press, New York, 1970 RSC; R. J. Sundberg, Indoles, Academic Press, London, 1996 RSC; G. W. Gribble, J. Chem. Soc., Perkin Trans. 1, 2000, 1045–1075 RSC; S. Cacchi and G. Fabrizi, Chem. Rev., 2005, 105, 2873–2920 CrossRef CAS; S. Cacchi and G. Fabrizi, Chem. Rev., 2011, 111, PR2215–PR2283 CrossRef; G. R. Humphrey and J. T. Kuethe, Chem. Rev., 2006, 106, 2875–2911 CrossRef; D. F. Taber and P. K. Tirunahari, Tetrahedron, 2011, 67, 7195–7210 CrossRef.
- P. M. Jackson and C. J. Moody, J. Chem. Soc., Perkin Trans. 1, 1990, 2156–2158 RSC.
- E. Vedejs and S. D. Monahan, J. Org. Chem., 1997, 62, 4763–4769 CrossRef CAS.
- T. Newhouse and P. S. Baran, J. Am. Chem. Soc., 2008, 130, 10886–10887 CrossRef CAS.
- A. B. Smith, N. Kanoh, H. Ishiyama, N. Minakawa, J. D. Rainier, R. A. Hartz, Y. S. Cho, H. Cui and W. H. Moser, J. Am. Chem. Soc., 2003, 125, 8228–8237 CrossRef CAS.
- E. Fischer and F. Jourdan, Ber. Dtsch. Chem. Ges., 1883, 16, 2241–2245 CrossRef; B. Robinson, The Fischer Indole Synthesis, John Wiley & Sons Inc., New York, 1982 Search PubMed.
- L. Zu, B. W. Boal and N. K. Garg, J. Am. Chem. Soc., 2011, 133, 8877–8879 CrossRef CAS.
- H. Ueda, H. Sato, K. Matsumoto, K. Sugimoto, T. Fukuyama and H. Tokuyama, Angew. Chem., Int. Ed., 2009, 48, 7600–7603 CrossRef CAS.
- Y. Murakami, Proc. Jpn. Acad., Ser. B, Phys. Biol. Sci., 2012, 88, 1–17 CrossRef CAS.
- T. Norris, C. Bezze, S. Z. Franz and M. Stivanello, Org. Process Res. Dev., 2009, 13, 354–357 CrossRef CAS; C. P. Ashcroft, P. Hellier, A. Pettman and S. Watkinson, Org. Process Res. Dev., 2011, 15, 98–103 CrossRef.
- D. L. Browne, I. R. Baxendale and S. V. Ley, Tetrahedron, 2011, 67, 10296–10303 CrossRef CAS.
- W. W. Hartman and L. J. Roll, Org. Synth., 1933, 13, 66 CAS.
- H. Ishii, Y. Murakami and T. Ishikawa, Chem. Pharm. Bull., 1990, 38, 597–604 CrossRef CAS.
- R. H. Poirier and F. Benington, J. Am. Chem. Soc., 1952, 74, 3192 CrossRef CAS.
- L. A. Hobson, W. A. Nugent, S. R. Anderson, S. S. Deshmukh, J. J. Haley, P. Liu, N. A. Magnus, P. Sheeran, J. P. Sherbine, B. R. P. Stone and J. Zhu, Org. Process Res. Dev., 2007, 11, 985–995 CrossRef CAS.
- K. Krüger, A. Tillack and M. Beller, Adv. Synth. Catal., 2008, 350, 2153–2167 CrossRef.
- P. J. Walsh, M. J. Carney and R. G. Bergman, J. Am. Chem. Soc., 1991, 113, 6343–6345 CrossRef CAS.
- C. Cao, Y. Shi and A. L. Odom, Org. Lett., 2002, 4, 2853–2856 CrossRef CAS.
- S. Banerjee, E. Barnea and A. L. Odom, Organometallics, 2008, 27, 1005–1014 CrossRef CAS.
- A. Tillack, H. Jiao, I. G. Castro, C. G. Hartung and M. Beller, Chem.–Eur. J., 2004, 10, 2409–2420 CrossRef CAS; K. Alex, A. Tillack, N. Schwarz and M. Beller, Angew. Chem., Int. Ed., 2008, 47, 2304–2307 CrossRef.
- S. Wagaw, B. H. Yang and S. L. Buchwald, J. Am. Chem. Soc., 1998, 120, 6621–6622 CrossRef CAS; S. Wagaw, B. H. Yang and S. L. Buchwald, J. Am. Chem. Soc., 1999, 121, 10251–10263 CrossRef.
- J. F. Hartwig, Angew. Chem., Int. Ed., 1998, 37, 2090–2093 CrossRef CAS.
- M. Wolter, A. Klapars and S. L. Buchwald, Org. Lett., 2001, 3, 3803–3805 CrossRef CAS.
- L. Jiang, X. Lu, H. Zhang, Y. Jiang and D. Ma, J. Org. Chem., 2009, 74, 4542–4546 CrossRef CAS.
- C. C. Mauger and G. A. Mignani, Org. Process Res. Dev., 2004, 8, 1065–1071 CrossRef CAS.
- Y.-K. Lim and C.-G. Cho, Tetrahedron Lett., 2004, 45, 1857–1859 CrossRef CAS.
- I.-K. Park, S.-E. Suh, B.-Y. Lim and C.-G. Cho, Org. Lett., 2009, 11, 5454–5456 CrossRef CAS.
- I.-K. Park, J. Park and C.-G. Cho, Angew. Chem., Int. Ed., 2012, 51, 2496–2499 CrossRef CAS.
- P. P. Cellier, J.-F. Spindler, M. Taillefer and H.-J. Cristau, Tetrahedron Lett., 2003, 44, 7191–7195 CrossRef CAS.
- R. L. Lundgren and M. Stradiotto, Angew. Chem., Int. Ed., 2010, 49, 8686–8690 CrossRef CAS.
- M. Inman and C. J. Moody, Chem. Commun., 2011, 47, 788–790 RSC.
- M. Inman, A. Carbone and C. J. Moody, J. Org. Chem., 2012, 77, 1217–1232 CrossRef CAS.
- H. Mitchell and Y. Leblanc, J. Org. Chem., 1994, 59, 682–687 CrossRef CAS; J. S. Yadav, B. V. S. Reddy, G. M. Kumar and C. Madan, Synlett, 2001, 1781–1783 CrossRef; L. Gu, B. S. Neo and Y. Zhang, Org. Lett., 2011, 13, 1872–1874 CrossRef.
- K. Muñiz and A. Iglesias, Angew. Chem., Int. Ed., 2007, 46, 6350–6353 CrossRef.
- T. Uemura and N. Chatani, J. Org. Chem., 2005, 70, 8631–8634 CrossRef CAS; K. Kisseljova, O. Tšubrik, R. Sillard, S. Mäeorg and U. Mäeorg, Org. Lett., 2006, 8, 43–45 CrossRef.
- F. R. Japp and F. Klingemann, Ber. Dtsch. Chem. Ges., 1887, 20, 2942–2944 CrossRef.
- D. Y. Curtin and J. A. Ursprung, J. Org. Chem., 1956, 21, 1221–1225 CrossRef CAS.
- B. A. Haag, Z.-G. Zhang, J.-S. Li and P. Knochel, Angew. Chem., Int. Ed., 2010, 49, 9513–9516 CrossRef CAS; Z.-G. Zhang, B. A. Haag, J.-S. Li and P. Knochel, Synthesis, 2011, 23–29 Search PubMed.
- E. Yasui, M. Wada and N. Takamura, Tetrahedron Lett., 2006, 47, 743–746 CrossRef CAS; E. Yasui, M. Wada and N. Takamura, Tetrahedron, 2009, 65, 461–468 CrossRef.
- A. W. Schammel, B. W. Boal, L. Zu, T. Mesganaw and N. K. Garg, Tetrahedron, 2010, 66, 4687–4695 CrossRef CAS.
- S. Müller, M. J. Webber and B. List, J. Am. Chem. Soc., 2011, 133, 18534–18537 CrossRef.
- A. Bischler and H. Brion, Ber. Dtsch. Chem. Ges., 1892, 25, 2860–2879 CrossRef; A. Bischler and P. Firemann, Ber. Dtsch. Chem. Ges., 1893, 26, 1336–1349 CrossRef.
- J. E. Nordlander, D. B. Catalane, K. D. Kotian, R. M. Stevens and J. E. Haky, J. Org. Chem., 1981, 46, 778–782 CrossRef CAS; R. J. Sundberg and J. P. Laurino, J. Org. Chem., 1984, 49, 249–254 CrossRef.
- C. J. Moody and E. Swann, Synlett, 1998, 135–136 CrossRef CAS; K. E. Bashford, A. L. Cooper, P. D. Kane, C. J. Moody, S. Muthusamy and E. Swann, J. Chem. Soc., Perkin Trans. 1, 2002, 1672–1687 RSC; M. A. Honey, A. J. Blake, I. B. Campbell, B. D. Judkins and C. J. Moody, Tetrahedron, 2009, 65, 8995–9001 CrossRef.
- M. Tokunaga, M. Ota, M.-A. Haga and Y. Wakatsuki, Tetrahedron Lett., 2001, 42, 3865–3868 CrossRef CAS.
- M. P. Kumar and R.-S. Liu, J. Org. Chem., 2006, 71, 4951–4955 CrossRef CAS.
- H. Hemetsberger and D. Knittel, Monatsh. Chem., 1972, 103, 194–204 CrossRef CAS.
- C. J. Moody, J. Chem. Soc., Perkin Trans. 1, 1984, 1333–1337 RSC.
- T. Martin and C. J. Moody, J. Chem. Soc., Perkin Trans. 1, 1988, 241–246 RSC.
- A. R. MacKenzie, C. J. Moody and C. W. Rees, J. Chem. Soc., Chem. Commun., 1983, 1372–1373 RSC.
- R. E. Bolton, C. J. Moody, C. W. Rees and G. Tojo, J. Chem. Soc., Perkin Trans. 1, 1987, 931–935 RSC.
- M. S. Reddy and J. M. Cook, Tetrahedron Lett., 1994, 35, 5413–5416 CrossRef CAS.
- J. Bonnamour and C. Bolm, Org. Lett., 2011, 13, 2012–2014 CrossRef CAS; B. J. Stokes, H. Dong, B. E. Leslie, A. L. Pumphrey and T. G. Driver, J. Am. Chem. Soc., 2007, 129, 7500–7501 CrossRef.
- S. Chiba, G. Hattori and K. Narasaka, Chem. Lett., 2007, 36, 52–53 CrossRef CAS; S. Jana, M. D. Clements, B. K. Sharp and N. Zheng, Org. Lett., 2010, 12, 3736–3739 CrossRef.
- D. F. Taber and W. Tian, J. Am. Chem. Soc., 2006, 128, 1058–1059 CrossRef CAS.
- X. Li, Y. Du, Z. Liang, X. Li, Y. Pan and K. Zhao, Org. Lett., 2009, 11, 2643–2646 CrossRef CAS.
- C. D. Nenitzescu, Bull. Soc. Chim. Romania, 1929, 11, 37–43 Search PubMed.
- M. Rönn, Q. McCubbin, S. Winter, M. K. Veige, N. Grimster, T. Alorati and L. Plamondon, Org. Process Res. Dev., 2007, 11, 241–245 CrossRef.
- K. R. Buszek, N. Brown and D. Luo, Org. Lett., 2009, 11, 201–204 CrossRef CAS; P. B. Huleatt, S. S. Choo, S. Chua and C. L. L. Chai, Tetrahedron Lett., 2008, 49, 5309–5311 CrossRef.
- M. Bosco, R. Dalpozzo, G. Bartoli, G. Palmieri and M. Petrini, J. Chem. Soc., Perkin Trans. 2, 1991, 651–655 Search PubMed.
- B. Åkermark, L. Eberson, E. Jonsson and E. Petterson, J. Org. Chem., 1975, 40, 1365–1367 CrossRef.
- T. Sakakibara, Y. Tanaka and S.-I. Yamasaki, Chem. Lett., 1986, 797–800 CrossRef CAS.
- S. Würtz, S. Rakshit, T. Dröge, J. J. Neumann and F. Glorius, Angew. Chem., Int. Ed., 2008, 47, 7230–7233 CrossRef CAS; J. J. Neumann, S. Rakshit, T. Dröge, S. Würtz and F. Glorius, Chem.–Eur. J., 2011, 17, 7298–7303 CrossRef.
- H. Iida, Y. Yuasa and C. Kibayashi, J. Org. Chem., 1980, 45, 2938–2942 CrossRef CAS.
- Z.-H. Guan, Z.-Y. Yan, Z.-H. Ren, X.-Y. Liu and Y.-M. Liang, Chem. Commun., 2010, 46, 2823–2825 RSC.
- H. Hagelin, J. D. Oslob and B. Åkermark, Chem.–Eur. J., 1999, 5, 2413–2416 CrossRef CAS.
- Z. Zhi, C. Zhang, S. Li, D. Pan, S. Ding, Y. Cui and N. Jiao, Angew. Chem., Int. Ed., 2009, 48, 4572–4576 CrossRef.
- R. Bernini, G. Fabrizi, A. Sferrazza and S. Cacchi, Angew. Chem., Int. Ed., 2009, 48, 8078–8081 CrossRef CAS.
- W. Yu, Y. Du and K. Zhao, Org. Lett., 2009, 11, 2417–2420 CrossRef CAS.
- Y. Wei, I. Deb and N. Yoshikai, J. Am. Chem. Soc., 2012, 134, 9098–9101 CrossRef CAS.
- M. Yamagishi, K. Nishigai, A. Ishii, T. Hata and H. Urabe, Angew. Chem., Int. Ed., 2012, 51, 1–5 CrossRef.
- L. Ackermann and A. V. Lygin, Org. Lett., 2012, 14, 764–767 CrossRef CAS.
- D. R. Stuart, M. Bertrand-Laperle, K. M. N. Burgess and K. Fagnou, J. Am. Chem. Soc., 2008, 130, 16474–16475 CrossRef CAS.
- D. R. Stuart, P. Alsabeh, M. Kuhn and K. Fagnou, J. Am. Chem. Soc., 2010, 132, 18326–18339 CrossRef CAS.
- A. S. Abreu, P. M. T. Ferreira, M.-J. R. P. Queiroz, I. C. F. R. Ferreira, R. C. Calhelha and L. M. Estevinho, Eur. J. Org. Chem., 2005, 2951–2957 CrossRef CAS.
- K. Inamoto, T. Saito, K. Hiroya and T. Doi, Synlett, 2008, 3157–3162 CrossRef CAS.
- Y. Du, R. Liu, G. Linn and K. Zhao, Org. Lett., 2006, 8, 5919–5922 CrossRef CAS.
- Q. Yan, J. Luo, D. Zhang-Negrerie, H. Li, X. Qi and K. Zhao, J. Org. Chem., 2011, 76, 8690–8697 CrossRef CAS.
- Y. Tan and J. Hartwig, J. Am. Chem. Soc., 2010, 132, 3676–3677 CrossRef CAS.
- T. Sugasawa, M. Adachi, K. Sasakura and A. Kitagawa, J. Org. Chem., 1979, 44, 578–586 CrossRef CAS.
- P. G. Gassman, T. J. van Bergen and G. Gruetzmacher, J. Am. Chem. Soc., 1973, 95, 6508–6509 CrossRef CAS.
- A. R. Petrov, C. G. Daniliuc, P. G. Jones and M. Tamm, Chem.–Eur. J., 2010, 16, 11804–11808 CrossRef CAS.
- K. S. Feldman, M. M. Bruendl and K. Schildknegt, J. Org. Chem., 1995, 60, 7722–7723 CrossRef CAS.
- S.-L. Cui, J. Wang and Y.-G. Wang, J. Am. Chem. Soc., 2008, 130, 13526–13527 CrossRef CAS.
- M. Tursky, L. L. R. Lorentz-Petersen, L. B. Olsen and R. Madsen, Org. Biomol. Chem., 2010, 8, 5576–5582 CAS.
- C. S. Cho, J. H. Kim and S. C. Shim, Tetrahedron Lett., 2000, 41, 1811–1814 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2013 |