σ–π-Diauration as an alternative binding mode for digold intermediates in gold(I) catalysis†
Dieter
Weber
and
Michel R.
Gagné
*
Caudill Laboratories, Department of Chemistry, University of North Carolina at Chapel Hill, NC 27599, USA. E-mail: mgagne@unc.edu
First published on 10th September 2012
Abstract
While investigating the gold(I)-catalyzed intramolecular hydroarylation of allenes, the structure of a digold-vinyl intermediate was verified. Instead of the previously proposed geminally diaurated binding mode for the digold when L = PPh3, an alternative σ–π-diauration mode was observed with the bulkier ligand L = P(o-Tol)3. Reactivity studies indicate the σ–π-mode has a disproportionate effect on protonolysis reactivity.
Introduction
Cationic gold complexes efficiently catalyze a broad variety of complex C–X (X = C, O, N) bond forming reactions, principally through the activation of C–C multiple bonds.1 Early studies in gold(I) catalysis have principally focused on methodology development, though a movement to underpin such studies with targeted mechanistic investigations is emerging.2,3We have recently reported that in gold(I)-catalyzed intramolecular allene hydroarylation and hydroalkoxylation reactions (10 mol% Ph3PAuNTf2 (4a)4 and (2-biphenyl)tBu2PAuOTs, respectively), most of the gold catalyst rests as a dinuclear species, the adduct of an expected monogold-vinyl intermediate and the cationic R3PAu+ catalyst (Scheme 1).5,6
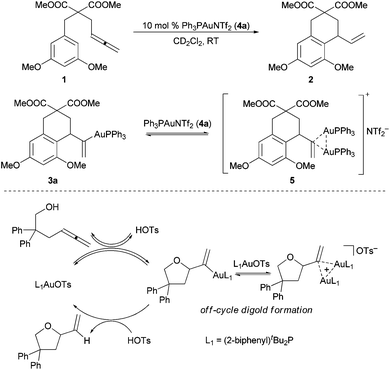 | ||
| Scheme 1 Digold resting states in the gold(I)-catalyzed intramolecular allene hydroarylation (top) and hydroalkoxylation (bottom) reactions. | ||
These species could easily be detected by in situ31P NMR as two sharp signals in a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 ratio, a consequence of the two diastereotopic P-ligands. In at least the hydroalkoxylation example, the digold intermediate was demonstrated to principally (but not exclusively) exist off-cycle.5b In the hydroarylation reaction, both gold-vinyl intermediates 3a and 5 were isolated and characterized by NMR; unfortunately, only 3a could be confirmed by X-ray analysis. Intermediate 5 was proposed to contain a vinyl-bridged Au
:1 ratio, a consequence of the two diastereotopic P-ligands. In at least the hydroalkoxylation example, the digold intermediate was demonstrated to principally (but not exclusively) exist off-cycle.5b In the hydroarylation reaction, both gold-vinyl intermediates 3a and 5 were isolated and characterized by NMR; unfortunately, only 3a could be confirmed by X-ray analysis. Intermediate 5 was proposed to contain a vinyl-bridged Au![[dash dash, graph caption]](https://www.rsc.org/images/entities/char_e091.gif) Au+ unit that benefited from an aurophilic closed shell interaction (5–10 kcal mol−1).5a This structural proposal was primarily based on characteristic NMR data and reactivity that paralleled the seminal geminally diaurated gold-aryl complexes of Grandberg, Nesmeyanov, and Schmidbaur.7
Au+ unit that benefited from an aurophilic closed shell interaction (5–10 kcal mol−1).5a This structural proposal was primarily based on characteristic NMR data and reactivity that paralleled the seminal geminally diaurated gold-aryl complexes of Grandberg, Nesmeyanov, and Schmidbaur.7
The viability of geminally diaurated vinyl intermediates in gold-catalyzed reactions was recently reinforced by Fürstner and co-workers, who synthesized model complexes and provided structural evidence for hyperconjugated vinylic C(sp2) atoms (Fig. 1).8 Additionally supportive are more recently characterized geminally diaurated vinyl and aryl catalytic intermediates by Hashmi et al. in gold(I)-catalyzed alkyne functionalization reactions.9 In each case the geminally diaurated products are symmetric in nature.
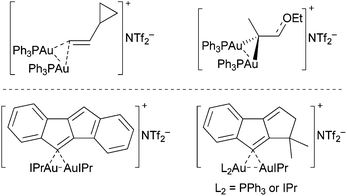 | ||
| Fig. 1 Structurally characterized geminally diaurated vinyl-complexes by Fürstner (top) and Hashmi (bottom). | ||
In this work, we report the structural elucidation of a digold intermediate of catalysis that suggests an alternative binding mode for digold-vinyl intermediates in gold(I) catalysis: σ–π-diauration. To the best of our knowledge this binding mode has not yet been reported in organogold-vinyl chemistry,10 though it has been recently described in gold(I)-acetylide chemistry by the groups of Widenhoefer, Russell, and Finze (Fig. 2).11
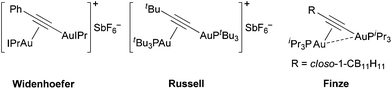 | ||
| Fig. 2 Representative examples of known σ–π-diaurated gold-acetylide complexes. | ||
Results and discussion
31P NMR spectroscopy proved to be an effective tool for investigating catalyst speciation during the conversion of allene 1 to 2 with various Ar3PAuNTf2 catalysts. As previously reported, most Ar3PAuNTf2 catalysts rested at a digold structure like 5, as suggested by two sharp resonances in a 1:1 ratio at δ ≈ 36 ppm in the 31P NMR spectrum.
However, when 10 mol% of the sterically demanding catalyst (o-tolyl)3PAuNTf2 (4b) was employed, 31P NMR data revealed three broad signals at 22.6, 16.1, and −0.7 ppm in a 1:1:0.6 ratio (Fig. 3) during the conversion,12 which contrasted previous observations. The signal at δ = −0.7 ppm corresponded to 4b. While the relative integration of the 22.6 and 16.1 ppm peaks suggested a digold structure, the broad asymmetric shape of the signals caused us to investigate this case more carefully.
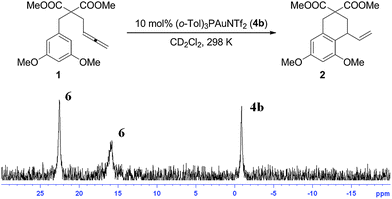 | ||
| Fig. 3 31P NMR data of the catalytic reaction of 1 with 10 mol% 4b. Two broad and asymmetrically shaped signals were detected at δ = 22.6 and 16.1 ppm. The signal at δ = −0.7 ppm for 4b did not disappear as catalysis progressed. | ||
The species 6 giving rise to the peaks at 22.6 and 16.1 ppm could be enriched by reacting 4b with 1 in the presence of 2,6-di-tert-butyl pyridine (DTBP), which inhibited protodemetallation.13 NMR data revealed that 2 equiv. of 4b were required for complete consumption of 1 and that the reaction with 4b (13–24 h) was significantly slower than with 4a (30 min). We presume this rate difference is attributable to the more basic or encumbered nature of (o-CH3–C6H4)3P versus PPh3.14
Slow evaporation of an enriched sample of 6 in CH2Cl2 layered with pentanes produced a mixture of single crystals suitable for X-ray diffraction analysis. NMR spectroscopy indicated that the crystals were a mixture of {(o-Tol)3P}2AuNTf2 and 6 (see ESI†).15 Although these crystals were not visibly differentiable, the third mounting provided a single crystal of 6 that could be solved (Fig. 4).
![ORTEP diagram of 6 with 50% probability ellipsoids; most hydrogen atoms, CH2Cl2, and the NTf2− anion are omitted for clarity. Key bond lengths [Å] include: Au1–Au2 [3.13563(18)], Au2–C2 [2.059(3)], Au1–C1 [2.275(3)], Au1–C2 [2.287(3)], and C1–C2 [1.369(5)].17](/image/article/2013/SC/c2sc21281a/c2sc21281a-f4.gif) | ||
| Fig. 4 ORTEP diagram of 6 with 50% probability ellipsoids; most hydrogen atoms, CH2Cl2, and the NTf2− anion are omitted for clarity. Key bond lengths [Å] include: Au1–Au2 [3.13563(18)], Au2–C2 [2.059(3)], Au1–C1 [2.275(3)], Au1–C2 [2.287(3)], and C1–C2 [1.369(5)].17 | ||
The structure of 6 revealed a geminal diauration binding mode different from that observed by Fürstner and Hashmi. Instead, the second gold cation interacted with the original gold-vinyl in a fashion best described as σ–π with the addition of a stabilizing aurophilic bond. The length of the Au2–C2 σ-bond [2.059(3) Å] (c.f. 2.050(2) Å for 3a) and the sum of bond angles at C2 (359.4° vs. 359.9° in 3a) indicated that the incorporation of a second gold cation did not significantly perturb the gold-vinyl component of the structure. Based on these structural parameters, the second gold atom (Au1 in Fig. 4) seemingly binds to the C1![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C2 double bond independent of the gold vinyl (Au2) and actually closely resembles the cationic gold(I) π-alkene complexes published by Widenhoefer et al.16 The distances between Au1–C1 [2.275(3) Å] and Au1–C2 [2.287(3) Å] match the distances in other π-coordinated olefins, which have ranged from 2.167 to 2.365 Å. While no bond angle changes were noted on π-complexation, the C1C2 double bond does become elongated in comparison to 3a (1.369(5) Å vs. 1.324(4) Å).
C2 double bond independent of the gold vinyl (Au2) and actually closely resembles the cationic gold(I) π-alkene complexes published by Widenhoefer et al.16 The distances between Au1–C1 [2.275(3) Å] and Au1–C2 [2.287(3) Å] match the distances in other π-coordinated olefins, which have ranged from 2.167 to 2.365 Å. While no bond angle changes were noted on π-complexation, the C1C2 double bond does become elongated in comparison to 3a (1.369(5) Å vs. 1.324(4) Å).
The distance between Au1 and Au2 [3.13563(18) Å] suggested a stabilizing aurophilic bond, the demonstrated range for which is quite large [2.8 Å–3.5 Å].18 The Au1–Au2 distance in 6, however, was intermediate between the short Fürstner and Hashmi geminally diaurated complexes [2.75 to 2.84 Å],8,9 and the longer Au–Au distances of the σ–π-diaurated acetylide complexes (3.41 to 3.62 Å).11 Worth noting in the analogous σ–π-diaurated acetylide chemistry was a rapid interconversion of the two gold units through a process proposed to involve a geminally diaurated acetylide.11c
Since crystallographic evidence for the structure of previously published digold 5 has not been forthcoming, the key 1H NMR signals (600 MHz) of 6 and 5 were examined to draw comparisons to the binding mode in 5. Specifically, protons H1syn, H1anti, and H3 revealed significant differences.
While proton H1syn appeared as a doublet in 5 (δ = 6.43 ppm, geminal coupling to H1anti with JHH = 3.0 Hz), it appeared in 6 as an apparent quartet at δ = 6.14 ppm due to coupling to three spin ½ nuclei (two JPH of 3.0 Hz each and one JHH of 3.0 Hz to H1anti). H1anti resonated in the spectrum of 5 at δ = 5.90 ppm as an apparent doublet of triplets due to coupling to two diastereotopic phosphorus atoms (6.0 Hz and 3.0 Hz) and the geminal proton H1syn (3.0 Hz). In contrast, proton H1anti in 6 appeared at δ = 5.81 ppm as a doublet of doublets with measurable coupling to only one phosphorus atom (9.6 Hz) and the geminal proton H1syn (3.0 Hz). The allylic proton H3 (δ = 4.50 ppm) in 5 appeared as a multiplet, but could be deconvoluted into equal couplings to the two diastereotopic phosphorus atoms and the two protons at C4 (each 7.8 Hz). In 6 H3 appeared as a quartet at δ = 4.21 ppm, coupling to only one phosphorus atom and the two protons at C4 (7.8 Hz).19 The σ–π-bonding mode thus creates an environment where one P-ligand is distinctly different as noted through JPH coupling to H1syn, H1anti, and H3, the σ-gold presumably being more coupled into the spin system than the π-gold.
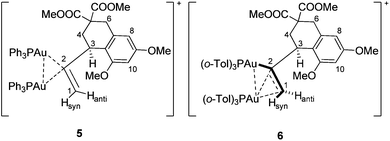
Reactivity similarities and differences between 5 and 6 were also noted.20 Like 5, compound 6 was found to have one gold unit readily abstracted by the addition of P(o-Tol)3 or NBu4Br to form gold-vinyl 3b21 and {(o-Tol)3P}2AuNTf2 or (o-Tol)3PAuBr, respectively. Similarly, conc. HCl led to immediate protodemetallation to 2, though like 5 digold 6 was acetic acid stable.
Where their reactivity diverged, however, was in the reaction with non-coordinating strong acids like HNTf2. For example, on reacting with ∼40 equiv. of HNTf25 reacted in ≤5 min to give 2, while complete consumption of 6 (to multiple decomposition products) with ∼60 equiv. HNTf2 took nearly 11 h. This difference in acid sensitivity indicated that the σ–π-binding mode significantly diminishes the propensity for protonolysis. The stability of 6 towards Brønsted acids is not entirely unreasonable given the direct engagement of the π-unit in coordinating the second (o-Tol)3PAu+ unit. The hypothesis that the protonolysis occurs by direct π attack onto the CC double bond is in line with reports by Blum22 and Fürstner,8 and related similar observations by Widenhoefer with σ–π-diaurated acetylene complexes.11c
Conclusions
In conclusion, a σ–π-binding mode was observed as a digold intermediate in the hydroarylation of allenes when a bulky P-ligand was utilized. Based on the broad and asymmetric signal shapes of 6 in the 31P NMR spectrum and the contrasting sharp resonances with less bulky catalysts, different coupling constants in the 1H NMR spectra, and most importantly different reactivity with excess HNTf2 it is proposed that the σ–π-diaurated structure of 6 does not resemble the binding mode of previously isolated 5, and that the latter better reflects the cyclopropyl-stabilized digold-vinyl structure of Fürstner and the examples by Hashmi. It does, however, represent another bonding scheme for how bone fide catalytic intermediates can become sequestered into polynuclear aggregates and affect catalysis efficiency and speciation.Financial support is gratefully acknowledged from the Fulbright Foreign Student Program (DW) and the National Institute of General Medicine (GM-60578).
Notes and references
- (a) N. Krause and C. Winter, Chem. Rev., 2011, 111, 1994 CrossRef CAS; (b) A. Corma, A. Leyva-Pérez and M. J. Sabater, Chem. Rev., 2011, 111, 1657 CrossRef CAS; (c) T. C. Boorman and I. Larrosa, Chem. Soc. Rev., 2011, 40, 1910 RSC; (d) C. Aubert, L. Fensterbank, P. Garcia, M. Malacria and A. Simonneau, Chem. Rev., 2011, 111, 1954 CrossRef CAS; (e) M. Bandini, Chem. Soc. Rev., 2011, 40, 1358 RSC; (f) A. Pradal, P. Y. Toullec and V. Michelet, Synthesis, 2011, 1501 CAS; (g) M. Rudolph and A. S. K. Hashmi, Chem. Commun., 2011, 47, 6536 RSC; (h) S. Sengupta and X. Shi, ChemCatChem, 2010, 2, 609 CrossRef CAS; (i) A. Fürstner, Chem. Soc. Rev., 2009, 38, 3208 RSC; (j) Z. Li, C. Brouwer and C. He, Chem. Rev., 2008, 108, 3239 CrossRef CAS; (k) R. A. Widenhoefer, Chem.–Eur. J., 2008, 14, 5382 CrossRef CAS; (l) A. S. K. Hashmi and M. Rudolph, Chem. Soc. Rev., 2008, 37, 1766 RSC; (m) D. J. Gorin, B. D. Sherry and F. D. Toste, Chem. Rev., 2008, 108, 3351 CrossRef CAS; (n) E. Jiménez-Núñez and A. M. Echavarren, Chem. Rev., 2008, 108, 3326 CrossRef; (o) A. Arcadi, Chem. Rev., 2008, 108, 3366 CrossRef; (p) A. S. K. Hashmi, Chem. Rev., 2007, 107, 3180 CrossRef CAS; (q) D. J. Gorin and F. D. Toste, Nature, 2007, 446, 395 CrossRef CAS; (r) A. S. K. Hashmi and G. J. Hutchings, Angew. Chem., Int. Ed., 2006, 45, 7896 CrossRef; (s) A. Fürstner and P. W. Davies, Angew. Chem., Int. Ed., 2007, 46, 3410 CrossRef.
- For comprehensive reviews, see: (a) L.-P. Liu and G. B. Hammond, Chem. Soc. Rev., 2012, 41, 3129 RSC; (b) A. S. K. Hashmi, Angew. Chem., Int. Ed., 2010, 49, 5232 CrossRef CAS.
- For a recent highlight on the potential for polynuclear activation of substrates by gold catalysts, see: A. Gómez-Suárez and S. P. Nolan, Angew. Chem., Int. Ed., 2012, 51, 8156 CrossRef.
- N. Mezailles, L. Ricard and F. Gagosz, Org. Lett., 2005, 7, 4133 CrossRef CAS.
- (a) D. Weber, M. A. Tarselli and M. R. Gagné, Angew. Chem., Int. Ed., 2009, 48, 5733 CrossRef CAS; (b) T. Brown, D. Weber, M. R. Gagné and R. A. Widenhoefer, J. Am. Chem. Soc., 2012, 134, 9134 CrossRef CAS.
- In the presence of excess Ag+ the reaction proceeded via a gold–silver complex, whose structure could not be verified, see: (a) D. Weber and M. R. Gagné, Org. Lett., 2009, 11, 4962 CrossRef CAS. For a recent publication detailing silver effects in gold catalysis, see: (b) D. Wang, R. Cai, S. Sharma, J. Jirak, S. K. Thummanapelli, N. G. Akhmedov, H. Zhang, X. Liu, J. L. Petersen and X. Shi, J. Am. Chem. Soc., 2012, 134, 9012 CrossRef CAS.
- (a) H. Schmidbaur and A. Schier, Chem. Soc. Rev., 2008, 37, 1931 RSC; (b) H. Schmidbaur, Gold Bull., 2000, 33, 1 CrossRef; (c) H. Schmidbaur, Chem. Soc. Rev., 1995, 24, 391 RSC; (d) A. N. Nesmeyanov, E. G. Perevalova, K. I. Grandberg and D. A. Lemenovskii, Izv. Akad. Nauk SSSR, Ser. Khim., 1974, 5, 1124 Search PubMed; (e) K. I. Grandberg and V. P. Dyadchenko, J. Organomet. Chem., 1994, 474, 1 CrossRef CAS; (f) K. I. Grandberg, Russ. Chem. Rev., 1982, 51, 249 CrossRef.
- G. Seidel, C. W. Lehmann and A. Fürstner, Angew. Chem., Int. Ed., 2010, 49, 8466 CrossRef CAS.
- (a) A. S. K. Hashmi, I. Braun, P. Nösel, J. Schädlich, M. Wieteck, M. Rudolph and F. Rominger, Angew. Chem., Int. Ed., 2012, 51, 4456 CrossRef CAS; (b) A. S. K. Hashmi, M. Wieteck, I. Braun, P. Nösel, L. Jongbloed, M. Rudolph and F. Rominger, Adv. Synth. Catal., 2012, 354, 555 CrossRef CAS; (c) A. S. K. Hashmi, I. Braun, M. Rudolph and F. Rominger, Organometallics, 2012, 31, 644 CrossRef CAS.
- π-complexated gold-vinyl species were synthesized from Fischer carbenes (with M = Cr, Mo, W) by electrophilic addition of Ph3PAu+, see: H. G. Raubenheimer, M. W. Esterhuysen, A. Timoshkin, Y. Chen and G. Frenking, Organometallics, 2002, 21, 3173 CrossRef CAS.
- (a) T. N. Hooper, M. Green and C. A. Russell, Chem. Commun., 2010, 46, 2313 RSC; (b) A. Himmelspach, M. Finze and S. Raub, Angew. Chem., Int. Ed., 2011, 50, 2628 CrossRef CAS; (c) T. J. Brown and R. A. Widenhoefer, Organometallics, 2011, 30, 6003 CrossRef CAS. One σ–π-diaurated alkyne complex was clearly in the range of an aurophilic interaction (Au–Au distance of 3.2880(5) Å), see ref. 11b.
- Complex 4b catalyzed the reaction but at a rate that is slower than 4a.
- Intermediate 6 could not be isolated in pure form. Only an enriched sample could be prepared. Impurities, such as DTBP, DTBP·HNTf2, or {(o-CH3–C6H4)3P}2AuNTf2, were not successfully removed.
- C. A. Tolman, Chem. Rev., 1977, 77, 313 CrossRef CAS.
- It is important to note that single crystals of 6 and of {(o-CH3–C6H4)3P}2AuNTf2 were obtained simultaneously. The third crystal picked gave a dataset by X-ray diffraction for compound 6. The formation of {(o-CH3–C6H4)3P}2AuNTf2 could imply decomposition via homo-coupling as observed by Fürstner for a geminally diaurated cyclopropyl-vinyl complex. However, analysis of the decomposed mixture was not conclusive to determine the mass balance.
- (a) T. J. Brown, M. G. Dickens and R. A. Widenhoefer, J. Am. Chem. Soc., 2009, 131, 6350 CrossRef CAS; (b) R. E. M. Brooner and R. A. Widenhoefer, Organometallics, 2011, 30, 3182 CrossRef CAS; (c) R. E. M. Brooner and R. A. Widenhoefer, Organometallics, 2012, 31, 768 CrossRef CAS.
- See the ESI† for the supplementary crystallographic data for 6.
- The potential energy surface for Au–Au interactions is rather flat for the approach of two gold atoms, see: ref. 7a.
- Couplings were determined through 31P and 1H NMR decoupling techniques.
- Relevant to these discussions are studies examining the structure and reactivity of digold-aryl and vinyl compounds. Grandberg and Nesmeyanov first characterized their reactivity through NMR and IR spectroscopy, see: (a) A. N. Nesmeyanov, E. G. Perevalova, A. B. Afanasova and K. I. Grandberg, Bull. Acad. Sci. USSR, Div. Chem. Sci. (Engl. Transl.), 1978, 27, 973 CrossRef; (b) A. N. Nesmeyanov, E. G. Perevalova, M. V. Ovchinnkov, Y. Y. Snakin and K. I. Grandberg, Bull. Acad. Sci. USSR, Div. Chem. Sci. (Engl. Transl.), 1978, 27, 1695 CrossRef. Proposed structures and thermodynamic properties of digold-aryl complexes have also been reported: (c) D. Weber, T. D. Jones, L. L. Adduci and M. R. Gagné, Angew. Chem., Int. Ed., 2012, 51, 2452 CrossRef CAS; (d) J. E. Heckler, M. Zeller, A. D. Hunter and T. G. Gray, Angew. Chem., Int. Ed., 2012, 51, 5924 CrossRef CAS.
- Gold-vinyl 3b was not isolated, but was strongly implicated based on characteristic chemical shifts and coupling constants of vinyl protons.
- K. E. Roth and S. A. Blum, Organometallics, 2010, 29, 1712 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental details and NMR data. CCDC 873041. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c2sc21281a |
| This journal is © The Royal Society of Chemistry 2013 |