Influence of adsorbed gas at liquid/solid interfaces on heterogeneous cavitation†
Valentina
Belova
*ab,
Marta
Krasowska
b,
Dayang
Wang
b,
John
Ralston
b,
Dmitry G.
Shchukin
ac and
Helmuth
Möhwald
*a
aMax Planck Institute of Colloids and Interfaces, Department of Interfaces, Research Campus, 14476 Potsdam, Germany. E-mail: valentina.belova@mpikg.mpg.de; Fax: +49 331 567 9202; Tel: +49 331 567 9235
bIan Wark Research Institute, University of South Australia, Mawson Lakes Campus, Mawson Lakes SA 5095, Adelaide, Australia
cStephenson Institute for Renewable Energy, Chemistry Department, University of Liverpool, Liverpool, L69 3BX, UK
First published on 16th October 2012
Abstract
The presence of dissolved gas at the solid/liquid interface can play a crucial role in heterogeneous cavitation. Here we focus our attention on the relationship between gas conditions and cavitation nucleation at planar solid surfaces with alternating hydrophobic/hydrophilic properties. Tapping mode atomic force microscopy and optical microscopy were used to monitor the gas adsorption on the patterns before sonication. Scanning electron microscopy revealed the effects of collapsing cavitation bubbles on the irradiated surfaces. High intensity ultrasonic irradiation (20 kHz) induces the formation of an interfacial gas layer at the solid surface immersed in different liquid media (water saturated with different gases, such as argon, nitrogen or carbon dioxide) by accelerating the adsorption of dissolved gas. Subsequently, the gas rearranges in diverse nano- or microstructures which take further part in the cavitation process. A solvent-exchange method was also applied to induce the formation of artificial gaseous domains accumulated at the solid surface in order to facilitate the cavitation process. By varying the gas adsorption time it is possible to accelerate or to slow down heterogeneous cavitation. The experimental findings on heterogeneous cavitation are discussed in terms of interfacial bubble nucleation and bubble attraction and growth on patterned solid surfaces in liquid media.
Introduction
Heterogeneous cavitation attracts a lot of attention as it involves the interaction of three interfaces (solid–liquid–gas) and leads to drastic modification of irradiated surfaces and changes in the liquid medium. Therefore the application of ultrasound, as a powerful and non-expensive tool to fabricate new composites with advanced physico-chemical properties or to modify the upper layers of irradiated materials is significantly increased.1–4Acoustic cavitation comprises several stages: nucleation, growth and collapse. Surface inhomogeneities, i.e., cracks, scratches and submicropores, generally provide pockets, where gas can be entrapped and the nucleation of cavitation bubbles can take place.5–8 The collapse of cavitation bubbles formed at the liquid/solid interface or very close to solid surfaces induces several physical and chemical effects, e.g. microjet impact, shockwave propagation in irradiated materials and the generation of free radicals, which can be involved in redox reactions of the irradiated materials.9–14 Additionally, strong shear forces and acoustic streaming can influence the surface of irradiated surfaces and contribute to morphological changes.15,16 It is worthwhile mentioning that bubble collapse is also accompanied by high temperatures and high pressures (5000 K and 1000 atm), which are favorable for physico-chemical reactions requiring extreme environmental conditions.1,2,17–21
Studies with chemically patterned surfaces provide the advantage that processes on surfaces can be studied by varying the surface but maintaining otherwise identical conditions. In addition bubbles can be localized and confined in a defined way providing further information on understanding their shapes. Several parameters, such as solution conditions (vapor pressure, composition, temperature, etc.), acoustic conditions (pressure and frequency) and reaction vessel geometry can influence cavitation.22–25 Furthermore, the surface energy was found to be strongly involved in the cavitation process.26,27 Recently, we showed that the control of surface energy via hydrophobic/hydrophilic balance permits cavitation events to exclusively occur on defined areas of irradiated materials.28–30 Microjets and shock waves resulting from collapsing bubbles preferably impact on the hydrophobic surface. We suggested that nanobubbles presented at the liquid–solid interface (mainly on the hydrophobic areas) could be involved in the cavitation process as they have a size close to critical radii (in a range of 100–200 nm). The evolution of cavitation bubbles in an acoustic field occurs in a short time interval, less than 1 μs; therefore, we expected to observe the surface damages immediately after ultrasound was applied. Contrary to our expectation, the first visual material response (pitting erosion) of the irradiated samples was observed after several minutes of ultrasonic treatment. Two unexpected observations have been made: (i) there is a delay in the response of the material to the applied acoustic field and (ii) the origin of cavitation bubble nuclei is on hydrophobic patterns with a negligible roughness (3 nm) and only chemical contrast. The knowledge on proper mechanisms of cavitation bubble formation at solid surfaces is rather limited at present, and this hinders both the control of the process and the development of new applications involving heterogeneous cavitation.
Here we aim to advance our understanding of the surface cavitation process by investigating the influence of gas conditions on the evolution of cavitation bubbles. It is known that the number and size of nuclei (in the case of homogeneous cavitation) are related to the concentration of the dissolved gas.31,32 Different gases influence the pressure to achieve bubble nucleation, the surface energies as well as the local pressure at collapse via their heat capacity, and in addition may enable different chemistry.26,27,33 Therefore, the investigation of the effect of dissolved gas on heterogeneous cavitation will be of crucial importance. The behavior of gas adsorbed at the solid surface from the liquid medium has been followed by tapping mode atomic force microscopy (TMAFM) and optical microscopy (OM) and further compared with scanning electron microscope (SEM) imaging of the samples after ultrasonic treatment. The effect of different dissolved gases (argon (Ar), nitrogen (N2) and carbon dioxide (CO2)) introduced into the liquid medium before and during ultrasonic treatment is also discussed.
Experimental section
Materials
Sylgard-184 poly(dimethylsiloxane) (PDMS) and curing agent were obtained from Dow Corning GmbH, Germany. A mixture with a ratio of 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :10 (curing agent/prepolymer) was used to prepare the stamps. Stamp replication was performed in contact with a silicon masters (size 10 × 10 mm2), which were purchased from GeSiM mbH, Germany (Dresden). The PDMS stamps were cured for about 2 h at 65 °C. After pealing off of the Si-Master, the PDMS stamps were oxidized by exposure of the surfaces to plasma Harrick Plasma PDS-32 G-2 (medium 60 W) for about 1 min. Optical quality Si wafers (111) (Silchem, Germany) with native silicon oxide layer were cut into pieces of 10 × 20 mm2, cleaned with piranha solution, washed with an excess of deionized water (Milli-Q water was purified in a three-stage Millipore Milli-Q Plus 185 purification system (>18.2 MΩ cm)), after 15 min of sonication in an ultrasonic bath and drying under nitrogen flow. An aluminium layer (40 nm) was deposited on the Si wafer by e-beam evaporation (Edwards Auto 306 FL400 e-beam evaporator) of pure Al under vacuum (10−5 to 10−7 mbar) at 80 °C. In order to improve the adhesion of the Al layer on the Si wafer, an intermediate layer of chromium (thickness less than 3 nm) as an adhesion promoter, was deposited. Soft material such as Al provides great opportunities to clearly visualize cavitation bubbles on the surface and to indirectly measure the cavitation impact of jets and shock waves formed upon bubble collapse.34n-Octadecylphosphonic acid (ODPA, 98% purity, Alfa Aesar), octadecanethiol (ODT, 96% purity, Alfa Aesar) and ethanol were used as received. All chemicals are reagent grade. Fresh ink solution was prepared before each set of experiments by dissolving ODPA and ODT in ethanol. Dissolution was achieved by immersion in an ultrasonic bath for a few minutes.
:10 (curing agent/prepolymer) was used to prepare the stamps. Stamp replication was performed in contact with a silicon masters (size 10 × 10 mm2), which were purchased from GeSiM mbH, Germany (Dresden). The PDMS stamps were cured for about 2 h at 65 °C. After pealing off of the Si-Master, the PDMS stamps were oxidized by exposure of the surfaces to plasma Harrick Plasma PDS-32 G-2 (medium 60 W) for about 1 min. Optical quality Si wafers (111) (Silchem, Germany) with native silicon oxide layer were cut into pieces of 10 × 20 mm2, cleaned with piranha solution, washed with an excess of deionized water (Milli-Q water was purified in a three-stage Millipore Milli-Q Plus 185 purification system (>18.2 MΩ cm)), after 15 min of sonication in an ultrasonic bath and drying under nitrogen flow. An aluminium layer (40 nm) was deposited on the Si wafer by e-beam evaporation (Edwards Auto 306 FL400 e-beam evaporator) of pure Al under vacuum (10−5 to 10−7 mbar) at 80 °C. In order to improve the adhesion of the Al layer on the Si wafer, an intermediate layer of chromium (thickness less than 3 nm) as an adhesion promoter, was deposited. Soft material such as Al provides great opportunities to clearly visualize cavitation bubbles on the surface and to indirectly measure the cavitation impact of jets and shock waves formed upon bubble collapse.34n-Octadecylphosphonic acid (ODPA, 98% purity, Alfa Aesar), octadecanethiol (ODT, 96% purity, Alfa Aesar) and ethanol were used as received. All chemicals are reagent grade. Fresh ink solution was prepared before each set of experiments by dissolving ODPA and ODT in ethanol. Dissolution was achieved by immersion in an ultrasonic bath for a few minutes.
Microcontact printing (μCP)
Microcontact printing (μCP) was applied to create hydrophobic–hydrophilic micropatterns. The printing procedure is thoroughly explained in our previous papers.28–30 The patterned substrates were designed with complex surface geometries, which exhibit different wetting properties and different widths of the hydrophobic and hydrophilic stripes. The pattern has six fields of regular hydrophobic and hydrophilic stripes of different length-to-width ratios.Liquid medium preparation
All aqueous solutions were made using deionized Milli-Q water with initial oxygen concentration about 7.51 mg l−1. Degassed water was prepared by stirring deionized water under vacuum for about 1 h. The oxygen concentration was reduced to 1.43 mg l−1. Degassed water was saturated with different gases, such as Ar, N2 and CO2 by purging water with desired gases for about 2 hours.Contact angle
Contact angle (CA) measurements were performed at room temperature with a contact angle meter (Software DSA 1, Krüss GmbH). Deionized Milli-Q water droplets (3 ml) were placed on the surfaces and the advancing CAs were measured at three different positions for each sample.Atomic force microscopy
Tapping-mode AFM was used to monitor the gas nucleation on the sample in a closed fluid cell using a MultiMode 8 with a Nanoscope V controller (Bruker, United States). For imaging the silicon nitride probes (NP-10, nominal resonance frequency 40–75 kHz, nominal spring constant 0.32 N m−1, nominal tip radius 10 nm, Bruker, United States) were used. All AFM measurements were conducted inside a clean room (Class 1000) at solution temperature of 22 °C.Optical microscopy
Optical microscopy (OM) was used to detect the formation of gas bubbles on patterns. Direct observations were made with a magnification factor of 20.Scanning electron microscopy
Scanning electron microscopy (SEM) was performed with a Gemini Leo 1550 instrument at an operating voltage of 3 keV. All samples were sputtered with a thin layer of gold (3 nm) before the measurements in order to increase the thermal conduction and to reduce sample charging. However, gold sputtering reduces the contrast between hydrophobic and hydrophilic domains that brings some difficulties in analyzing images.Gas content measurements
The dissolved oxygen content of the liquid medium was measured with an optical oxygen sensor based on the principle of fluorescence quenching (getOtwo, Sentronic GmbH, Germany). Detailed explanation of the gas content measurements are given in the ESI.†Software Image J
Software Image J was used to quantify the morphological changes of the samples after ultrasonic treatment.35The ultrasonic processor
The ultrasonic processor UIP 1000hd from Hielscher Ultrasonics, Germany with a cooling system inside the converter was used to sonicate the samples. The experiments were carried out at an excitation frequency of 20 kHz and ultrasonic power density of 51.3 W cm−2. During all experiments the samples were fixed parallel to the flat Ti sonotrode (3.14 cm2 area) at a distance of 15 mm in a water bath fitted with a thermostat. The temperature of the liquid medium was controlled and kept at about 45 °C.Results and discussion
Gas adsorption at patterns
The AFM image of a dry patterned sample shows a well-defined structure of homogeneous hydrophobic stripes separated by steps of 3 nm towards the hydrophilic regions (Fig. 1a). The observed height profile gives only a slight contrast difference between adsorbed amphiphilic materials and uncovered hydrophilic stripes (Al) due to their short chain length (less than 3 nm), hence we have basically a chemical and no geometrical pattern. However, the borderlines between hydrophobic and hydrophilic stripes are sufficiently sharp to allow us to distinguish the stripes' width. The roughness of the hydrophobic and hydrophilic areas is similar and equal to 3 nm. The advancing contact angle (CA) of the sputtered Al before patterning was about 70° and after patterning the CA was in a range of 105–112° depending on the designed patterns. The TMAFM image of the sample in degassed water exhibits a simple two phase liquid–solid interface without any nano- or micro-gas structures (Fig. 1b). No structural changes or gas presence on the patterns was observed with longer AFM measurements (up to 60 min).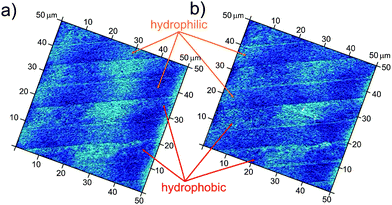 | ||
| Fig. 1 AFM images of the patterns: (a) dry samples (b) in degassed water. The width of the hydrophobic stripes is 7 μm and width of the hydrophilic stripes is 6.7 μm. | ||
Imaging of patterns in deionized water (standard conditions) demonstrates the appearance of a new interfacial gas state, preferentially located on the hydrophobic stripes. An optical microscopy observation projected perpendicular to the sample placed in a liquid cell for the period of the AFM measurements provides a better overview of the large surface areas where very interesting behavior of the gas adsorption to the patterned surfaces was observed (Fig. 2). Low-energy surfaces (hydrophobic) have been postulated from theory and experiment to exhibit a layer of reduced water density compared with high-energy surfaces (hydrophilic).36–38 However, the existence of a gas layer remains to be proved, and for this one should make use of the selectivity of gas adsorption depending on the wettability of the solids. Indeed Fig. 2a shows that a small amount of gas may adsorb on the hydrophobic stripes in a uniform way forming bubbles shaped as channels. When the amount of gas adsorbed onto the hydrophobic stripes is increased, they experience morphological changes including bulge formation (Fig. 2b). Furthermore, these bulges can form bridges between two or even more neighboring hydrophobic stripes, overlapping the underlying hydrophilic stripes. In a few minutes, small gas bulges move towards bigger ones and coalesce (Fig. 2c–e).
 | ||
| Fig. 2 Optical microscopy observation of selective gas adsorption onto patterned surfaces (the width of the hydrophobic stripes is 10 μm and the width of the hydrophilic stripes is 20 μm) in deionized water: (a) gas channel formation on the hydrophobic stripes and appearance of the first bulge formation; (b–d) the increased adsorbed gas volume promotes the formation of many bulges on the hydrophobic stripes, which may form bridges between two neighbourhood stripes; (e) the bulges increase their size by attracting the adsorbed gas from the hydrophobic stripes. | ||
A rearrangement of the adsorbed gas on the stripes, i.e., attraction of gas in the bulges due to the thinning of gas layers, was also observed in a series of AFM measurements in deionized water.
The behavior of the gas adsorbed at the hydrophobic areas in liquid medium is similar to that of liquid adsorbed at the hydrophilic areas in gas medium.
Gau et al. found that by patterning a substrate with hydrophobic and hydrophilic regions, it is possible to confine a liquid to micro-channels due to selective liquid adsorption along the hydrophilic stripes.39 Furthermore, it has also been found that in presence of a wettability gradient, a structured liquid film can be formed.40
A series of tapping mode AFM experiments presenting the time dependent gas rearrangement at the solid surface in liquid environment is presented in Fig. 3. In order to avoid artifacts in AFM imaging, the first image was taken after the laser signal was equilibrated. Hence, Fig. 3a demonstrates gas arrangement on the micro-structured surfaces at 20 min. A continuous gas film with a height in a range of 40–70 nm forms at the hydrophobic stripes. However, such a gas film was not stable and gradually evolved into separated bubbles (Fig. 3b–d) of different sizes and volumes (the height was in the range of 70–240 nm). It may indicate that the gas film is mobile and can move towards some energetically favorable points where the gas forms separate bubbles. Homogeneous distribution of gas bubbles on the hydrophobic areas was not observed even for much longer AFM experiment times.
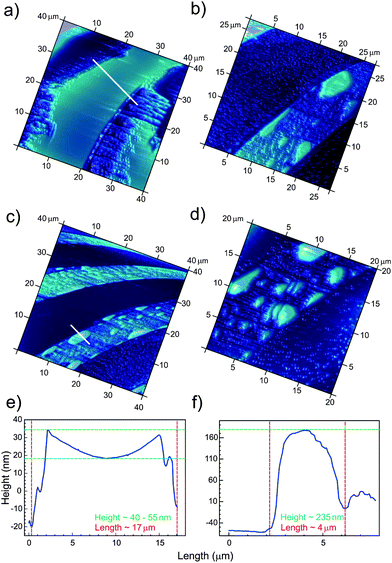 | ||
| Fig. 3 Formation of gas layers and nanobubbles (height profile) on the hydrophobic stripes (bright areas) of unirradiated patterns during AFM measurements at different times: (a) 20 min, (b) 30 min, (c) 40 min and (d) 50 min. The width of the hydrophobic stripes is 10 μm and the width of the hydrophilic stripes is 20 μm. Two cross-sections through the gas layer (e) and gas bubble (f) formed on the hydrophobic stripe. The unexpected shape in (e) may be due to a measurement artifact of the tapping mode or to a long-range attraction between the hydrocarbon/air and the air/liquid interfaces. Other cross-sections are given in the ESI (Fig. S1†). | ||
The biggest bubbles were mostly distributed along the borders of the hydrophobic domains, whereas the smaller ones were located on the central parts of the stripes. In most cases, the bubbles were elongated along the stripes, exhibiting different lengths of up to 7 μm.
A similar gas arrangement on hydrophobic surfaces was previously observed by Seddon et al.41,42 They obtained sufficiently flat gas nanostructures with height in the range of 10–15 nm and size of several μm, randomly formed on hydrophobized Si wafers. After 50 min of TMAFM measurements these gas structures (so-called micropancakes) increased their height by approximately 65% compared with the initial size and then remained unchanged for a long time. The interplay between nanobubbles and micropancakes, and micropancakes and an enriched gas layer was also investigated. Micropancakes were found to have at least one nanobubble sitting on top, possibly both having been nucleated from the same asperity.43 Moreover, it has been found that the nanobubble size increases mainly due to the influx of gas from the micropancakes at the contact line. In our case, the height of the gas layer on the hydrophobic areas is much larger (by a factor of ten) than for the gas structures obtained by Seddon's group, but the behavior is very similar. Hence, gas film rearrangements show the appearances of nanobubbles due to the balanced influx and outflux of gas molecules from layers to bubbles. Another remarkable example on adsorption of dissolved gases onto a substrate resulted from investigations by using a quartz crystal microbalance (QCM) in combination with TMAFM.44,45 It was demonstrated that CO2 gas molecules present in solution only adsorb on hydrophobic silica. At first gas adsorption/bubble growth undergoes a slow growth process due to the diffusion of gas molecules from an interfacial region to surface sites (Harvey nuclei). When the critical bubble/aggregate size is reached, further gas adsorption takes place by diffusion from the interfacial region into the surface nanobubbles.
Ultrasonic treatment of patterns in degassed and saturated water
The sonication of patterns in two different liquid media (degassed or standard conditions) does not show as great an extent of surface changes as was expected (Fig. 4). In both cases, cavitation damages the hydrophobic areas with a slight difference in pit distribution and shape. By reducing the concentration of dissolved gas in the liquid medium, the shear forces generated by pressure waves created by ultrasound are not changed; hence, transient cavitation events must contribute to the material loss of irradiated patterns under sonication. The degassing procedure simply removes nanobubbles from the solid surface, but sonication with high intensity ultrasound may very quickly lead to re-gassing of the liquid medium. The applied ultrasound creates strong vibrations as well as stirring and within the first few minutes, gas from the top of the reactor can be trapped into the liquid medium and therefore contribute to bubble formation. Gas content measurements showed that after about 5 min of sonication the gas concentration in the deionized water approached the initial equilibrium level of 7.2 mg l−1 (Fig. S2†). During the first minutes of sonication, the damaging cavitation events occur locally on the hydrophobic stripes; in fact, the nucleation sites were not homogeneously distributed on the patterns. The main difference between ultrasonically treated patterns under degassed and standard conditions was observed after 20 min of sonication (Fig. 4b and e). In the case of degassed water, the pits are predominantly located at the interfaces between hydrophobic and hydrophilic stripes. In the case of standard sonication conditions the pits are mainly located on the hydrophobic stripes. By increasing the sonication period, the pits grew fast and some of them coalesced (Fig. 4). Longer sonication does not illustrate any difference in surface modification. After 50 min of ultrasonic treatment the hydrophobic surfaces have completely reacted showing the formation of metal-foam corrugations. We have already discussed in a previous work the time-dependence of the pattern morphology changes under ultrasonic treatment.28–30 Initial surface defects, formed on solid surfaces under microjet impingement and shockwaves, entrap dissolved gas from the liquid medium and provide new nuclei for surface cavitation (secondary nucleation).6,46,47 Longer ultrasonic irradiation enhances the surface modification through the appearance of surface irregularities, which provide a large amount of gas pockets (metal-foams) favorable for surface cavitation. | ||
| Fig. 4 SEM of ultrasonically treated patterned surfaces at different sonication times: (a, d) at 10 min, (b, e) at 20 min, and (c, f) at 50 min. The left column is for degassed conditions and right column is for standard conditions. The hydrophobic stripes (bright regions) are impacted by collapsed cavitation bubbles. | ||
The TMAFM and SEM images (Fig. 3c and d and Fig. 4) demonstrate remarkable similarity between gas conditions on the patterns and pits formed due to the impact of the collapsing cavitation bubbles. Hence, the gaseous domains, formed on the patterns due to the adsorption of dissolved gas from the liquid medium, might be directly or indirectly involved in heterogeneous cavitation:
–A direct interplay would exist if these gaseous domains indeed served as nuclei for cavitation bubbles. The question then remains: why do we observe a time delay of the material response to the cavitation process?
–An indirect relationship to form cavitation bubbles would exist if gaseous domains simply served as gas reservoirs close to the liquid/solid interface which may further contribute to the cavitation process.
We exclude the idea that the gas film, formed at the patterned surfaces due to the adsorption of the dissolved gas from the bulk, screens the irradiated surfaces and prevents cavitation bubble formation at the liquid/solid interface at the first minutes of sonication. According to the Einstein diffusion equation d = (Dt)½, where D is the diffusion coefficient, typically 2 × 10−5 cm2 s−1 for air in water at room temperature, d is the size of a solid surface area (we assume it to be approximately 100 μm2), a time t for gas dissolution from the liquid/solid interface would be about 0.25 s.11
We do not exclude the idea that the gaseous domains, observed with TMAFM on the hydrophobic stripes of the unirradiated patterns, serve as nuclei for cavitation bubbles. However, we would like to refer here to independent findings of two groups on combined experiments of cavitation and AFM.48,49 They confirmed that interfacial nanobubbles are stable, immobile and therefore should not be involved in the cavitation process. Brotchie and Zhang found that nanobubbles formed on highly ordered pyrolytic graphite (HOPG) can grow moderately in an applied ultrasonic field (1.2 bar, duration 40 s), through an increase in height and consequently an increase in contact angle (by 2–5°) due to pinning of the contact line of the nanobubbles on the surface.48 Any lateral movement or displacement during sonication was not observed. However recently our own optical experiments revealed that micron sized bubbles form preferentially on hydrophobic stripes and can move laterally (to be published). The group of Lohse postulated the superstability of nanobubbles formed on flat polyamide and hydrophobized silicon wafers immersed in water under reduction of the liquid pressure down to −6 MPa (single shock pulse).49 It should also be mentioned, that using an AFM it is impossible to directly image changes of nanobubbles (which occurs in the order of minutes) during the shock wave passage (which is order of μs). Therefore, these studies are indirect and the stability of nanobubbles to applied ultrasonic irradiation remains an open question. The following assumptions, concerning the time delay of the material response during the first minutes of cavitation, can be made:
–Nucleation of cavitation bubbles on flat surfaces with a small roughness value (less than 3 nm) and without artificial gas pockets requires a sufficient amount of gas to be concentrated at the liquid–solid interface. Collapse of cavitation bubbles in the liquid medium forms new gas bubbles, which are further involved in bulk cavitation or forced to the solid surfaces by acoustic streaming, where they can adsorb. Therefore, during the first minutes of treatment, ultrasound assists in the enrichment of the irradiated surface with the gas required to start heterogeneous cavitation.
–The chemical robustness of Al substrates to the microjets and shockwaves formed under the collapse of cavitation bubbles can also be major contributors to material response delays. Therefore Al needs more energy input (or energy accumulation of cavitation collapse) to damage its upper layers to be visually detectable with SEM and AFM.
–Cavitation bubbles can be shielded by neighbouring ones which expand larger and collapse earlier. In this case, jetting flows produced by the collapsing bubbles have horizontal direction, and no damage of the irradiated materials can be observed.44
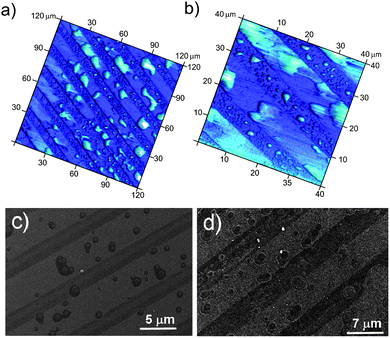
Additionally, we have found that the width of the stripes influences the distribution of the pits (Fig. 5). With increasing hydrophobic stripe width up to 100 μm (hydrophilic areas up to 80 μm) the impacts of cavitation on the surfaces are mainly located close to the boundary. With a relatively small width (about 3 μm) only a few large pits as wide as the stripes themselves can be observed. It can be seen that the design of solid surfaces strongly influences the cavitation process. Therefore, by controlling the hydrophobic/hydrophilic ratio it will be possible to direct the nucleation of gas at the liquid/solid interface and to achieve a better control over surface cavitation. The immersion time of patterns in water (standard conditions) prior to sonication can also influence the cavitation process. SEM imaging of samples that had been kept for about 30 min in water before applying ultrasonic irradiation, demonstrates the appearance of pits much earlier than in the case of the instant sonication procedure. After 5 min of sonication, the material damage was 30% higher. We also conducted experiments with samples that had been kept for about one hour before sonication, however no specific difference was observed. Hence, the maximum adsorption of dissolved gas at the solid surfaces occurred during the first 30 min, and the longer presence of the samples in water allowed equilibrium between the adsorbed gas at the solid surfaces and bulk to be reached.
 | ||
| Fig. 5 SEM imaging of patterned surfaces with different width of the hydrophobic and hydrophilic stripes after 30 min of sonication at standard conditions. | ||
In order to induce more gaseous domains on the patterned surfaces, an ethanol–water solvent exchange technique was applied.50,51 TMAFM images after the liquid exchange show the appearance of gas films on the hydrophobic stripes (average height of 50–70 nm) with gas bubbles of different size and shape sitting on top (Fig. 6). Moreover, the hydrophilic areas were partially covered with nanobubbles of much smaller size (an average height is 30 nm and the length is 100 nm) which remained stable for prolonged periods of time.
 | ||
| Fig. 6 Top: AFM images of the patterns after EtOH–water solvent exchange procedure: overview (a) and zoom (b) images. Bottom: SEM images of the patterns after EtOH–water solvent exchange procedure: after 3 min (c) and 10 min (d) of sonication. | ||
Ultrasonic treatment of the preconditioned samples was found to be more pronounced and already after 10 min of sonication the number of pits on the hydrophobic areas was increased by a factor of 2. However, at extended sonication times disordered surface changes appear, due to the high initial gas concentration at the solid surface.
Ultrasonic treatment of patterns in liquid medium saturated with different gases
Another set of experiments was conducted under flow of different gases (Ar, N2 and CO2). TMAFM imaging in water saturated with Ar (solubility of Ar in water is about 62 mg l−1 at 20 °C) also demonstrates that gas accumulation occurs only on the hydrophobic stripes similar to gas adsorption at standard conditions (Fig. 7a and b). Nevertheless, pitting of the materials under sonication in the liquid medium saturated with Ar occurs much faster than at standard conditions. This could be due to the high number of cavitation nuclei in the water saturated with Ar. Therefore, cavitation in the bulk can dominate the surface cavitation process. | ||
| Fig. 7 AFM images of the unirradiated patterns measured in water saturated with Ar (a and b). SEM images of the samples after different time of ultrasonic irradiation under argon flow: (c) 5 min, (d) 10 min and (e) 30 min. The hydrophobic stripes (bright regions) are impacted by collapsed cavitation bubbles. | ||
After 5 min of sample sonication in the solution saturated with Ar and under continuous Ar bubbling we observed the appearance of numerous small pits with an average size of 1 μm on the hydrophobic stripes (Fig. 7c). Extended sonication (up to 10 min) significantly increased the size of the pits located close to the hydrophobic/hydrophilic boundary. These pits are about 10 times larger than the initial pits and surrounded by asymmetrically distributed smaller pits (Fig. 7b). At longer treatment times, the impacts on the hydrophobic areas were found to be more homogeneously distributed (Fig. 7c). As a result, ultrasonic treatment of the samples under argon flow amplified the surface cavitation process, maintaining the selective material response.
The pit distribution on the hydrophobic areas was quantified with Image J software from the electron micrographs obtained at different sonication times (Fig. 8). A considerable contribution of the hydrophobic surface to the nucleation of cavitation bubbles starts in the time range of 10 to 50 min. It can be seen that the cavitation rate is slightly lower for the samples in degassed water. However, the nucleation rate increases under Ar flow and for the preconditioned samples after EtOH–water solvent exchange. Sonication of the samples in nitrogen saturated water did not show significant differences compared to sonication at standard conditions (Fig. 8). Solubility of N2 in water (20 mg l−1 at 20 °C) is much lower than the solubility of Ar; as a result the gas adsorption on the patterns is less pronounced, but it is slightly higher than in case of saturated water. Water saturated with CO2 does not enhance the formation of gaseous domains on the immersed substrates. On the contrary, it promotes surface damage, due to the low pH (4.7). Initial surface changes were observed after 10 min by TMAFM measurements (Fig. S3†).
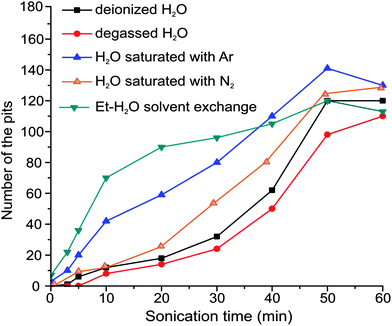 | ||
| Fig. 8 Plot of the pit density as a function of sonication time. The width of the hydrophobic stripes is 20 μm and the width of the hydrophilic stripes is 10 μm, reference area is 230 × 167 μm2. The density is derived only for the hydrophobic areas and it is negligible on the hydrophilic areas for a sonication period below 30 min. | ||
Longer AFM measurements showed that the surface corrugations grew exponentially and after 60 min the initially hydrophobic surface regions were completely eroded. Thus, investigations of heterogeneous cavitation in water saturated with CO2 do not provide sufficient data to quantify the cavitation impact at the solid surface. The self assembled monolayer of ODPA and ODT (hydrophobic areas) interacts very rapidly with dissolved CO2 and leads to surface modification of the hydrophobic stripes before ultrasonic irradiation can be applied. The surface damage occurs very fast due to the acidic character of the liquid medium and it is difficult to distinguish the difference between cavitation impact and influence of the dissolved CO2. ODPA/ODT SAMs can efficiently protect the Al surface against cavitation erosion for longer sonication times, but do not prevent the chemical etching of the Al in water saturated with CO2. This suggests that in the presence of CO2, the formation of cavitation bubbles is not efficient on the solid substrates during sonication; otherwise cavitation collapse could cause a huge increase of temperature and pressure that should damage the underlying irradiated material without any time delay.
Conclusions
Accumulation and assembly of adsorbed gas at the liquid/solid interface is demonstrated to lead to gas layers and bubbles that are involved in the heterogeneous cavitation process. Development of surface cavitation is time-dependent and can be accelerated when the surfaces have an initially high volume of gaseous domains. The gas adsorption was found to be strongly dependent on the surface-energy of the substrates, whereas the hydrophobic areas are more favourable for gas layer growth. As expected, the largest increase of heterogeneous cavitation is reported for surfaces after ethanol–water exchange. Surface cavitation on the preconditioned samples due to their high volume of adsorbed gas starts much earlier than in the case of sonication under standard conditions. Nevertheless, an excess of interfacial gas reduces the cavitation selectivity under extended ultrasonic treatment. The presence of argon and nitrogen in the liquid medium accelerates the surface cavitation, keeping the response selective on the patterned surfaces. However, water saturated with CO2 is not an appropriate liquid medium for our types of surfaces to study the relationship between interfacial gas and surface cavitation. Direct observation of cavitation bubble nucleation and growth on structured surfaces will yield further insights into the possible mechanism of surface selective cavitation behaviour.Acknowledgements
We thank Dr Hartmann, R. Pitschke and A. Heilig for helping with SEM and AFM measurements and acknowledge Max Planck Society for financial support. We are grateful to Dr Xue Hua Zhang for her contribution and discussions on nanobubble preparation at solid surfaces.Notes and references
- K. S. Suslick and L. A. Crum, Sonochemistry and Sonoluminiscence, Wiley-Interscience, New York, 1997, vol. 1 Search PubMed.
- T. J. Mason and J. P. Lorimer, Sonochemistry: Theory, Applications and Uses of Ultrasound in Chemistry, Harword, Chichester, UK, 1988 Search PubMed.
- K. S. Suslick and G. Price, Annu. Rev. Mater. Sci., 1999, 29, 295–326 CrossRef CAS.
- V. Sáez and T. J. Mason, Molecules, 2009, 14, 4284–4299 CrossRef.
- W. Lauterborn and H. Bolle, J. Fluid Mech., 1975, 72, 391–399 CrossRef.
- A. K. Khurana, H. Chen and C. G. Wall, Chem. Eng. Commun., 1998, 165, 199–215 CrossRef CAS.
- N. Bremond, M. Arora, C.-D. Ohl and D. Lohse, J. Phys.: Condens. Matter, 2005, 17, S3603–S3608 CrossRef CAS.
- D. G. Shchukin, E. Skorb, V. Belova and H. Möhwald, Adv. Mater., 2011, 23(17), 1922–1934 CrossRef CAS.
- M. Kornfeld and L. Suvorov, J. Appl. Phys., 1944, 15(6), 495 CrossRef CAS.
- K. S. Suslick, D. A. Hammerton and R. E. Cline, J. Am. Chem. Soc., 1986, 108(18), 5641 CrossRef CAS.
- C. E. Brennen, Cavitation and Bubble Dynamics, Oxford University Press, 1995, p. 282 Search PubMed.
- L. A. Crum, J. Acoust. Soc. Am., 1980, 68(1), 203 CrossRef.
- L. A. Crum and A. Eller, J. Acoust. Soc. Am., 1968, 44(1), 369 CrossRef.
- G. E. Reisman, Y.-C. Wang and C. E. Brennen, J. Fluid Mech., 1998, 355, 255–283 CrossRef.
- W. L. Nyborg, Acoustic Streaming, Academic Press, New York, 1965, vol. 2B Search PubMed.
- P. Tho, R. Manassen and A. Ooi, J. Fluid Mech., 2007, 576, 191–233 CrossRef.
- Y. T. Didenko, W. B. McNamara and K. S. Suslick, J. Am. Chem. Soc., 1999, 121, 5817 CrossRef CAS.
- M. Aschokkumar, R. Hall, P. Mulvaney and F. Grieser, J. Phys. Chem. B, 1997, 101, 10845 CrossRef.
- J. W. Westwater, Boiling of Liquids, New York, 1956, vol. 1 Search PubMed.
- C. R. Thomas, C. H. Farny, C. C. Coussios, R. A. Roy and R. G. Holt, Acoust. Res. Lett. Online, 2005, 6, 182 CrossRef.
- L. Rayleigh, Philos. Mag., Ser. 5, 1917, 34, 94 CrossRef.
- E. A. Neppiras, Phys. Rep., 1980, 61(3), 159–251 CrossRef.
- M. A. Margulis, Sonochemistry and Cavitation, 1993, p. 536 Search PubMed.
- M. Ashokkumar and F. Grieser, Phys. Chem. Chem. Phys., 2007, 9, 5631–5643 RSC.
- B. Nanzai, K. Okitsu, N. Takenaka, H. Bandow, N. Tajima and Y. Maeda, Ultrason. Sonochem., 2009, 16(1), 163–168 CrossRef CAS.
- J. S. Fisher, J. Appl. Phys., 1948, 19, 1062–1067 CrossRef.
- E. N. Harvey, Wm. D. McElroy and A. H. Whiteley, J. Appl. Phys., 1947, 18, 162–172 CrossRef CAS.
- V. Belova, D. A. Gorin, D. G. Shchukin and H. Möhwald, Angew. Chem., Int. Ed., 2010, 49, 7129–7133 CrossRef CAS.
- V. Belova, D. A. Gorin, D. G. Shchukin and H. Möhwald, ACS Appl. Mater. Interfaces, 2011, 3, 417–425 CAS.
- V. Belova, D. G. Shchukin, D. A. Gorin, A. Kopyshev and H. Möhwald, Phys. Chem. Chem. Phys., 2011, 13, 8015–8023 RSC.
- T. G. Leighton, The Acoustic Bubble, Academic Press, California, USA, 1994 Search PubMed.
- K. A. Mørch, J. Hydrodyn., Ser. B, 2009, 21, 176–189 CrossRef.
- S. D. Lubetkin, Langmuir, 2003, 19, 2575–2587 CrossRef CAS.
- A. Philipp and W. Lauternborn, J. Fluid Mech., 1998, 361, 75–116 CrossRef CAS.
- Free Software Foundation GNU General Public License, website: http://www.gnu.org/licenses/licenses.html#GPL Search PubMed.
- K. Lum, D. Chandler and J. D. Weeks, J. Phys. Chem. B, 1999, 103, 4570–4577 CrossRef CAS.
- F. H. Stillinger, J. Solution Chem., 1973, 2, 141–158 CrossRef CAS.
- U. K. Sur and V. Lakshminarayanan, J. Colloid Interface Sci., 2002, 254, 410–413 CrossRef CAS.
- H. Gau, S. Herminghaus, P. Lenz and R. Lipowsky, Science, 1999, 283, 46–49 CrossRef CAS , 1, 5398.
- H. G. Braun and E. Meyer, Thin Solid Films, 1999, 345, 222–228 CrossRef CAS.
- J. R. T. Seddon, O. Bliznyuk, E. S. Kooij, B. Poelsema, H. J. W. Zandvliet and D. Lohse, Langmuir, 2010, 26(12), 9640–9644 CrossRef CAS.
- J. R. T. Seddon and D. Lohse, J. Phys.: Condens. Matter, 2011, 23, 133001 CrossRef.
- X. H. Zhang, X. Zhang, J. Sun, Z. Zhang, G. Li, H. Fang, X. Xiao, X. Zeng and J. Hu, Langmuir, 2007, 23(4), 1778–1783 CrossRef CAS.
- J. Yang, J. Duan, D. Fornasiero and J. Ralston, Phys. Chem. Chem. Phys., 2007, 9, 6327–6332 RSC.
- J. Yang, J. Duan, D. Fornasiero and J. Ralston, J. Phys. Chem. B, 2003, 107(25), 6139–6147 CrossRef CAS.
- J. P. J. Nail, R. I. Vachon and J. Morehouse, J. Heat Transfer, 1974, 96, 132 CrossRef CAS.
- N. Bremond, M. Arora, C.-D. Ohl and D. Lohse, Phys. Rev. Lett., 2006, 96, 224501 CrossRef.
- A. Brotchie and X. H. Zhang, Soft Matter, 2011, 7, 265–269 RSC.
- B. M. Borkent, S. M. Dammer, H. Schönherr, G. J. Vancso and D. Lohse, Phys. Rev. Lett., 2007, 98, 204502 CrossRef.
- X. H. Zhang, N. Maeda and V. S. J. Craig, Langmuir, 2006, 22(11), 5025–5035 CrossRef CAS.
- X. H. Zhang, A. Quinn and W. A. Ducker, Langmuir, 2008, 24(9), 4756–4764 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Fig. S1–S2. See DOI: 10.1039/c2sc21321d |
| This journal is © The Royal Society of Chemistry 2013 |