Synthesis and properties of all-benzene carbon nanocages: a junction unit of branched carbon nanotubes†
Katsuma
Matsui
a,
Yasutomo
Segawa
*a,
Tomotaka
Namikawa
bc,
Kenji
Kamada
*bc and
Kenichiro
Itami
*a
aDepartment of Chemistry, Graduate School of Science, Nagoya University, Nagoya 464-8602, Japan. E-mail: ysegawa@nagoya-u.jp; itami.kenichiro@a.mbox.nagoya-u.ac.jp; Fax: +81 52-788-6098; Tel: +81 52-788-6098
bResearch Institute for Ubiquitous Energy Devices, National Institute of Advanced Industrial Science and Technology (AIST), Ikeda, Osaka 563-8577, Japan. E-mail: k.kamada@aist.go.jp; Fax: +81 72-751-9637; Tel: +81 72-751-9523
cDepartment of Chemistry, School of Science and Technology, Kwansei Gakuin University, Sanda, Hyogo 669-1337, Japan
First published on 29th August 2012
Abstract
The first synthesis of an all-benzene carbon nanocage 1, which represents a junction unit of branched carbon nanotubes, has been achieved. A stepwise assembly of six L-shaped units [cis-di(p-bromophenyl)cyclohexane derivative] and two three-way units (1,3,5-triborylbenzene) by cross-coupling and homocoupling provided the unstrained cyclic precursor. Acid-mediated aromatization of cyclohexane moieties in the precursor resulted in the formation of carbon nanocage 1. Photophysical measurements and DFT studies revealed the unique properties of 1, such as D3 symmetry with degenerate HOMO/HOMO − 1 and LUMO/LUMO + 1, high fluorescence quantum yield (ΦF = 0.87), and a relatively large two-photon absorption cross section (500 GM at 590 nm), which are attractive for various applications.
Introduction
Three-dimensional cage-shaped molecules are of great interest because of their sheer aesthetic appeal, host–guest chemistry, and catenane-like applications.1–3 In general, the synthesis of a cage molecule relies on the thermodynamic stability of products, where reversible bond formation such as supramolecular assembly2 and dynamic covalent bond formation3 are typically utilized. This strategy means that strained cages are still very rare. In particular, the bottom-up synthesis of strained hydrocarbon cages consisting of sp2- and/or sp-hybridized carbons is a formidable task.4–6 Herein we report the synthesis and photophysical properties of an all-benzene carbon nanocage as the first strained and conjugated hydrocarbon cage consisting solely of sp2-carbons and covalent bonds.We became interested in all-benzene carbon nanocages as part of our ongoing efforts to synthesize structurally uniform carbon nanotubes using carbon nanorings, such as cycloparaphenylene (a short sidewall segment of armchair carbon nanotubes), as templates (Fig. 1, top). Recently, the chemical synthesis of cycloparaphenylene (CPP)6 has been accomplished by the groups of Bertozzi,7 Itami,8–13 Yamago,14,15 and Jasti7,16,17 after many years.6,18 The modular,10 size-selective,8,10,13–17 and scalable11,12 synthesis of [n]CPPs is now possible, and [12]CPP has become commercially available.19 CPP is an interesting and unique molecular entity not only because it represents the shortest sidewall segment of armchair carbon nanotube structures but also because of its aesthetic appeal,6 unique cyclic conjugation, photophysical properties,7,14,15,20 and guest-encapsulating properties.11,12,21 More recently, short sidewall segments of chiral carbon nanotubes,22,23 pyridine-inserted CPP,24 and π-extended CPP (cyclo-1,4-naphthylene)25 have been also synthesized.
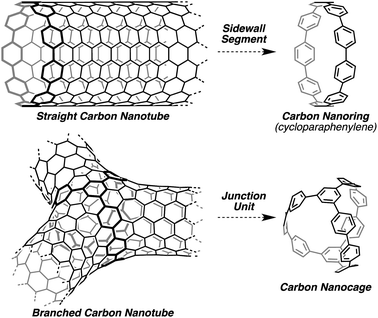 | ||
| Fig. 1 Carbon nanoring and carbon nanocage as segments of straight and branched carbon nanotubes. | ||
In addition to straight carbon nanotubes, branched or so-called Y-junction carbon nanotubes have also attracted attention as a small logic gates or small transistors (Fig. 1, bottom).26 Our simple retrosynthetic analysis of these structures led to the identification of a tri-bridged carbon nanocage consisting solely of benzene rings as a junction unit for branched carbon nanotubes. Moreover, the synthesis of such carbon nanocages is of great interest, as they would possess unique photophysical properties due to the hitherto unknown symmetric structures with a mix of linear and branched conjugations.27
For the synthesis of carbon nanocages, we used our synthesis platform for CPPs as a blueprint (Fig. 2). We previously reported that U-shaped units synthesized from L-shaped cis-diphenylcyclohexane units and a linear phenylene unit could be coupled by Suzuki–Miyaura coupling or nickel-mediated homocouplings to afford strain-free cyclic precursors (Fig. 2, left). The cyclohexane moieties in the precursors were aromatized to afford [14]–[16]CPP. We envisaged that the carbon nanocage 1, consisting of 20 benzene rings, could be synthesized by using 1,3,5-triborylbenzene as a “three-way” unit to assemble the L-shaped unit (Fig. 2, right).
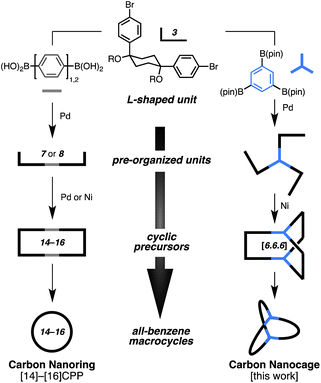 | ||
| Fig. 2 Our strategy for the synthesis of all-benzene macrocycles. | ||
Results and discussion
Synthesis and structure of the carbon nanocage
The outcome of our investigations based on this strategy is the first synthesis of carbon nanocage 1 shown in Scheme 1. Palladium-catalyzed borylation of 1,3,5-tribromobenzene with (pin)BB(pin) [B(pin) = (pinacolato)boryl] produced 1,3,5-triborylbenzene 2.28 The cis-di(p-bromophenyl)cyclohexane derivative 3 (L-shaped unit) was synthesized via twofold nucleophilic addition of 1-bromo-4-lithiobenzene to cyclohexane-1,4-dione followed by methoxymethylation of the hydroxy groups.10 The trifurcated unit 4, which has three L-shaped units on the benzene core with C3 symmetry, was obtained in 53% yield by a palladium-catalyzed Suzuki–Miyaura coupling reaction of 2 and 3, with 82% of unreacted 3 recovered after the reaction. Compound 4 was then subjected to a threefold homocoupling reaction, mediated by Ni(cod)2/2,2′-bipyridyl [cod = 1,5-cyclooctadiene], to produce bicyclic macrocycle 5 in 12% yield. High dilution conditions (2 mM of 4 in DMF) are necessary to prevent oligomerization of 4. Treatment of cage precursor 5 with NaHSO4·H2O and o-chloranil in m-xylene/DMSO under reflux conditions afforded 1,8(1,3,5),2,3,4,5,6,7,9,10,11,12,13,14,15,16,17,18,19,20(1,4)-icosabenzenabicyclo[6.6.6]icosaphane (1),29 the desired carbon nanocage, in 69% yield as a white solid.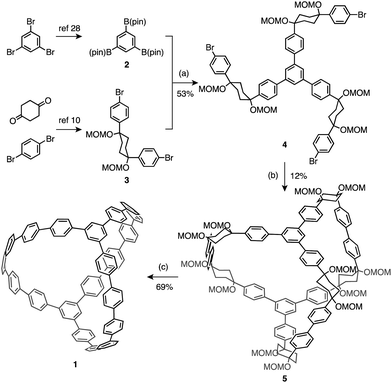 | ||
| Scheme 1 Synthesis of carbon nanocage 1. Reaction conditions: (a) PdCl2(dppf), K2CO3, DMF, 90 °C; (b) Ni(cod)2, 2,2′-bipyridyl, DMF, 90 °C; (c) NaHSO4·H2O, o-chloranil, m-xylene/DMSO, reflux. | ||
Characteristic signals of one singlet and three sets of doublets observed in the 1H NMR spectrum of 1 clearly support its D3h symmetric structure in solution state. The structure of 1 was also confirmed by the observation of 14 independent signals in the 13C NMR spectrum as well as HH COSY, HMQC, and high-resolution MALDI-TOF MS. Similarly to CPPs, carbon nanocage 1 has high solubility in most organic solvents such as CHCl3, CH2Cl2, EtOAc, acetone, THF, Et2O, benzene, and toluene.
To gain insight into the structural properties of 1, DFT calculations were performed (B3LYP/6-31G(d)). The optimized structure of 1 is shown in Fig. 3. The conformation with D3 symmetry was found to be the ground state of 1. As expected, 1 forms a beautiful cage-shape structure with a spherical void inside. The two tri-substituted benzene rings deviate from each other at an angle of 12.5°. The distance between these two benzene rings is 18.4 Å, a value which is between the diameters of [13]CPP (17.9 Å) and [14]CPP (19.4 Å).9 Other di-substituted benzene rings in 1 are slightly bent to form three arches. The strain energies of carbon nanocage 1 and a model of cyclic precursor 5 (5-H: all methoxymethoxy groups in 5 are removed) were determined to be 67.2 kcal mol−1 (1) and 2.1 kcal mol−1 (5-H) by using the hypothetical homodesmotic reaction (see the ESI for details†).9 It is clear from these values that the six cyclohexane moieties of 5 effectively help in attenuating the build-up of strain energy during the macrocyclization (4 → 5).
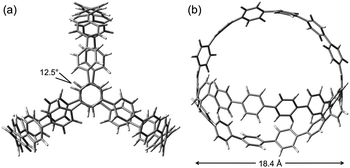 | ||
| Fig. 3 Optimized structure of 1. (a) Top view. (b) Side view. | ||
Photophysical properties of the carbon nanocage
We next measured the photophysical properties (UV-vis and fluorescence) of 1 in chloroform (Fig. 4). In the UV-vis absorption spectrum of 1, the absorption maximum (λabs) was observed at 325 nm with the molecular absorption coefficient (ε) of 1.9 × 105 cm−1 M−1. Nanocage 1 exhibits intense blue fluorescence (418, 431 nm) with a very high fluorescence quantum yield (ΦF = 0.87). The radiative (kr) and non-radiative (knr) decay rate constants of 1, based on the fluorescence lifetime (τs = 1.4 ns) and the equations (ΦF = kr × τs and kr + knr = τs−1), were determined to be 6.2 × 108 s−1 and 9.4 × 107 s−1, respectively. These values are comparable to those of [12]CPP (kr = 4.0 × 108 s−1, knr = 5.0 × 107 s−1).20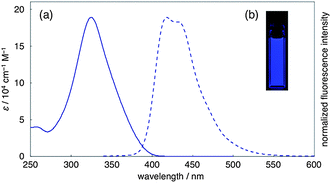 | ||
| Fig. 4 (a) UV-vis absorption (solid line) and fluorescence spectra (broken line) of 1. (b) Blue fluorescence emission of the chloroform solution of 1. | ||
We further performed time-dependent DFT (TD-DFT) studies of the optimized structure of 1 (B3LYP/6-31G(d)). The energy diagrams and pictorial representations of frontier MOs of 1 are shown in Fig. 5. Four orbitals, HOMO − 1, HOMO, LUMO, and LUMO + 1, have e-symmetry whereas HOMO – 2 and LUMO + 2 have a2- and a1-symmetries, respectively. Degeneracy in energy levels was found for the relevant pairs of frontier orbitals, HOMO − 1/HOMO (−5.39 eV) and LUMO/LUMO + 1 (−1.58 eV). The HOMO–LUMO gap of 3.81 eV is slightly higher than that of [18]CPP (3.71 eV (ref. 20)). The HOMO → LUMO and HOMO − 1 → LUMO + 1 transitions mainly contribute to the transition to the S3-state, which has a high oscillator strength (f = 3.04). This high oscillator strength value should be the reason for the high kr value of 1. On the other hand, the HOMO − 1 → LUMO and HOMO → LUMO + 1 transitions leading to the S4-state were found to be symmetrically forbidden (A1, f = 0.00).
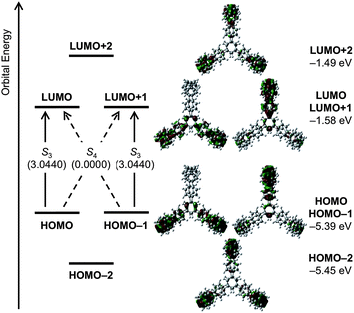 | ||
| Fig. 5 Energy diagrams and pictorial representations of the frontier MOs of 1 calculated at the B3LYP/6-31G(d) level of theory. Excitation energies were computed by TD-DFT at the same level. Values in parentheses represent oscillator strengths (f). | ||
To investigate more about the nature of the electronic excited states, thereby exploring new applications of 1, we measured the two-photon absorption (TPA) spectrum by the femtosecond open-aperture Z-scan method (Fig. 6). The TPA spectrum of 1, taken in CHCl3, is broad, spanning 640 nm to 480 nm, with a peak value of the TPA cross section of 500 ± 60 GM at 590 nm, beside the maximum value of 540 ± 80 GM at the shorter wavelength edge of the measurements (480 nm). These values are relatively large for a hydrocarbon molecule without any electron donor or acceptor substituents.30 The peak wavelength (590 nm) of 1 shows a large bathochromic shift compared to that of didecyl-substituted p-septiphenyl, which is a linear paraphenylene and can be a model compound of each arc of 1, measured under the same conditions (also in Fig. 6). This shows the exciton coupling among the arcs occurs in 1. Previously the exciton coupling among the arms was reported to enhance the TPA for a benzene-cored tribranched molecule.31 However, the peak cross section of 1 (500 GM) is close to or less than three times that of the p-septiphenyl (190 GM), suggesting the exciton coupling has little effect on the magnitude of TPA beside the drawback due to the curved shape of each arc. Moreover, the broad TPA spectrum of 1 is considered to consist of many TPA transitions in the wavelength range. This is supported by the TD-DFT calculations, which show that there are many TPA-allowed excited states [A1-states: S4 (3.57 eV), S7 (3.66 eV), and S13 (3.81 eV)], which can show strong TPA transitions beside E-states, which can show weak transitions in the wavelength range.
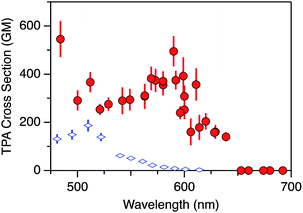 | ||
| Fig. 6 Two-photon absorption cross-section spectra of 1 (circles) and 2′′′,5′′′-didecyl-p-septiphenyl (rhombi) in CHCl3. 1 GM = 10−50 cm4 s per molecule per photon. | ||
Conclusions
In summary, we have synthesized a strained, conjugated carbon nanocage 1, consisting solely of benzene rings and covalent bonds, for the first time. A stepwise assembly of cyclohexane-based L-shaped units and benzene-based three-way units by cross-coupling and homocoupling provided an unstrained cyclic precursor, which was successfully converted into 1 by acid-mediated aromatization. Photophysical measurements and DFT studies revealed the unique properties of 1, such as D3 symmetry with degenerate HOMO/HOMO − 1 and LUMO/LUMO + 1, high fluorescence quantum yield (ΦF = 0.87), and relatively large two-photon absorption cross section (500 GM at 590 nm). In addition to exploring various applications of this newly developed carbon nanocage 1, such as optoelectronic application and host–guest chemistry, we are now in the position to initiate the bottom-up chemical synthesis of branched carbon nanotubes using 1 (junction unit) and CPP (sidewall unit) as key components.Acknowledgements
This work was supported by the Funding Program for Next Generation World-Leading Researchers (220GR049 to K.I.) and a Grant-in-Aid for Scientific Research (23550174 to K.K.) from JSPS. K.M. thanks the Nagoya University Leading Graduate School Program for fellowship. We thank Prof. Cathleen M. Crudden (Queen's Univ.) for discussion and comments. We thank Prof. Shigehiro Yamaguchi, Dr Aiko Fukazawa, and Dr Shohei Saito (Nagoya Univ.) for assistance in the photophysical measurements. We also thank Dr Teruhiro Shiono (Panasonic Corp.) for providing the p-septiphenyl derivative. Calculations were performed using the resources of the Research Center for Computational Science, Okazaki, Japan.Notes and references
- Reviews on cage molecules: (a) C. Seel and F. Vögtle, Angew. Chem., Int. Ed. Engl., 1992, 31, 528 CrossRef; (b) F. Hof, S. L. Craig, C. Nuckolls and J. Rebek, Angew. Chem., Int. Ed., 2002, 41, 1488 CrossRef CAS.
- Reviews on supramolecular cages: (a) M. Fujita, M. Tominaga, A. Hori and B. Therrien, Acc. Chem. Res., 2005, 38, 369 CrossRef CAS; (b) S. R. Seidel and P. J. Stang, Acc. Chem. Res., 2002, 35, 972 CrossRef CAS; (c) S. Leininger, B. Olenyuk and P. J. Stang, Chem. Rev., 2000, 100, 853 CrossRef CAS; (d) D. L. Caulder and K. N. Raymond, Acc. Chem. Res., 1999, 32, 975 CrossRef CAS.
- Reviews on dynamic covalent bond cages: (a) S. J. Rowan, S. J. Cantrill, G. R. L. Cousins, J. K. M. Sanders and J. F. Stoddart, Angew. Chem., Int. Ed., 2002, 41, 898 CrossRef; (b) N. Fujita, S. Shinkai and T. D. James, Chem.–Eur. J., 2008, 3, 1076 CAS; (c) K. Severin, Dalton Trans., 2009, 5254 RSC; (d) M. Mastalerz, Angew. Chem., Int. Ed., 2010, 49, 5042 CrossRef CAS.
- Examples of cage-shaped hydrocarbons: (a) Modern Cyclophane Chemistry, ed. R. Gleiter and H. Hopf, Wiley-VCH, Weinheim, Germany, 2004 Search PubMed; (b) V. Boekelheide and R. A. Hollins, J. Am. Chem. Soc., 1973, 95, 3201 CrossRef CAS; (c) J. Gross, G. Harder, A. Siepen, J. Harren, F. Vögtle, H. Stephan, K. Gloe, B. Ahlers, K. Cammann and K. Rissanen, Chem.–Eur. J., 1996, 2, 1585 CrossRef CAS.
- Carbon cages as precursors for fullerenes: (a) Y. Rubin, T. C. Parker, S. I. Kahn, C. L. Holliman and S. W. McElvany, J. Am. Chem. Soc., 1996, 118, 5308 CrossRef CAS; (b) Y. Rubin, T. C. Parker, S. J. Pastor, S. Jalisatgi, C. Boulle and C. L. Wilkins, Angew. Chem., Int. Ed., 1998, 37, 1226 CrossRef CAS; (c) Y. Tobe, N. Nakagawa, K. Naemura, T. Wakabayashi, T. Shida and Y. Achiba, J. Am. Chem. Soc., 1998, 120, 4544 CrossRef CAS; (d) Y. Tobe, N. Nakagawa, J. Kishi, M. Sonoda, K. Naemura, T. Wakabayashi, T. Shida and Y. Achiba, Tetrahedron, 2001, 57, 3629 CrossRef CAS; (e) Y. Tobe, R. Umeda, M. Sonoda and T. Wakabayashi, Chem.–Eur. J., 2005, 11, 1603 CrossRef CAS.
- Reviews: (a) T. Kawase and H. Kurata, Chem. Rev., 2006, 106, 5250 CrossRef CAS; (b) B. D. Steinberg and L. T. Scott, Angew. Chem., Int. Ed., 2009, 48, 5400 CrossRef CAS; (c) G. J. Bodwell, Nat. Nanotechnol., 2010, 5, 103 CrossRef CAS; (d) R. Jasti and C. R. Bertozzi, Chem. Phys. Lett., 2010, 494, 1 CrossRef CAS; (e) M. Iyoda, J. Yamakawa and M. J. Rahman, Angew. Chem., Int. Ed., 2011, 50, 10522 CrossRef CAS; (f) U. H. F. Bunz, S. Menning and N. Martín, Angew. Chem., Int. Ed., 2012, 51, 8094 CrossRef; (g) K. Itami, Pure Appl. Chem., 2012, 84, 907 CrossRef CAS; (h) H. Omachi, Y. Segawa and K. Itami, Acc. Chem. Res., 2012, 45, 1378 CrossRef CAS.
- R. Jasti, J. Bhattacharjee, J. B. Neaton and C. R. Bertozzi, J. Am. Chem. Soc., 2008, 130, 17646 CrossRef CAS.
- H. Takaba, H. Omachi, Y. Yamamoto, J. Bouffard and K. Itami, Angew. Chem., Int. Ed., 2009, 48, 6112 CrossRef CAS.
- Y. Segawa, H. Omachi and K. Itami, Org. Lett., 2010, 12, 2262 CrossRef CAS.
- H. Omachi, S. Matsuura, Y. Segawa and K. Itami, Angew. Chem., Int. Ed., 2010, 49, 10202 CrossRef CAS.
- Y. Segawa, S. Miyamoto, H. Omachi, S. Matsuura, P. Šenel, T. Sasamori, N. Tokitoh and K. Itami, Angew. Chem., Int. Ed., 2011, 50, 3244 CrossRef CAS.
- Y. Segawa, P. Šenel, S. Matsuura, H. Omachi and K. Itami, Chem. Lett., 2011, 40, 423 CrossRef CAS.
- Y. Ishii, Y. Nakanishi, H. Omachi, S. Matsuura, K. Matsui, H. Shinohara, Y. Segawa and K. Itami, Chem. Sci., 2012, 3, 2340 RSC.
- S. Yamago, Y. Watanabe and T. Iwamoto, Angew. Chem., Int. Ed., 2010, 49, 757 CrossRef CAS.
- T. Iwamoto, Y. Watanabe, Y.-I. Sakamoto, T. Suzuki and S. Yamago, J. Am. Chem. Soc., 2011, 133, 8354 CrossRef CAS.
- T. J. Sisto, M. R. Golder, E. S. Hirst and R. Jasti, J. Am. Chem. Soc., 2011, 133, 15800 CrossRef CAS.
- J. Xia and R. Jasti, Angew. Chem., Int. Ed., 2012, 51, 2474 CrossRef CAS.
- Representative attempts: (a) V. C. Parekh and P. C. Guha, J. Indian Chem. Soc., 1934, 11, 95 CAS; (b) R. Friederich, M. Nieger and F. Vögtle, Chem. Ber., 1993, 126, 1723 CrossRef CAS.
- [12]CPP is now commercially available from Tokyo Chemical Industry Co., Ltd (TCI), catalog no. C2449.
- Y. Segawa, A. Fukazawa, S. Matsuura, H. Omachi, S. Yamaguchi, S. Irle and K. Itami, Org. Biomol. Chem., 2012, 10, 5979 CAS.
- T. Iwamoto, Y. Watanabe, T. Sadahiro, T. Haino and S. Yamago, Angew. Chem., Int. Ed., 2011, 50, 8342 CrossRef CAS.
- H. Omachi, Y. Segawa and K. Itami, Org. Lett., 2011, 13, 2480 CrossRef CAS.
- S. Hitosugi, W. Nakanishi, T. Yamasaki and H. Isobe, Nat. Commun., 2011, 2, 492 CrossRef.
- K. Matsui, Y. Segawa and K. Itami, Org. Lett., 2012, 14, 1888 CrossRef CAS.
- A. Yagi, Y. Segawa and K. Itami, J. Am. Chem. Soc., 2012, 134, 2962 CrossRef CAS.
- D. Wei and Y. Liu, Adv. Mater., 2008, 20, 2815 CrossRef CAS , and references therein.
- K. Itami, K. Tonogaki, T. Nokami, Y. Ohashi and J. Yoshida, Angew. Chem., Int. Ed., 2006, 45, 2404 CrossRef CAS.
- Y. Liu, F. Niu, J. Lian, P. Zeng and H. Niu, Synth. Met., 2010, 160, 2055 CrossRef CAS.
- D. Hellwinkel, Systematic Nomenclature of Organic Chemistry: A Directory to Comprehension and Application of its Basic Principles, Springer-Verlag, Berlin, 2001, pp. 68–72 Search PubMed.
- K. Kamada, K. Ohta, T. Kubo, A. Shimizu, Y. Morita, K. Nakasuji, R. Kishi, S. Ohta, S. Furukawa, H. Takahashi and M. Nakano, Angew. Chem., Int. Ed., 2007, 46, 3544 CrossRef CAS.
- S. Kato, T. Matsumoto, M. Shigeiwa, H. Gorohmaru, S. Maeda, T. Ishi-i and S. Mataka, Chem.–Eur. J., 2006, 12, 2303 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental procedures, characterization of data for all new compounds, and details of photophysical and computational studies. See DOI: 10.1039/c2sc21322b |
| This journal is © The Royal Society of Chemistry 2013 |