On-surface cross-coupling methods for the construction of modified electrode assemblies with tailored morphologies†‡
Amber A. S. Gietter§, Rachel C. Pupillo§, Glenn P. A. Yap, Thomas P. Beebe Jr., Joel Rosenthal* and Donald A. Watson*
Department of Chemistry and Biochemistry, University of Delaware, Newark, DE, 19716 USA. E-mail: joelr@udel.edu; dawatson@udel.edu
First published on 23rd October 2012
Abstract
Controlling the molecular topology of electrode–catalyst interfaces is a critical factor in engineering devices with specific electron transport kinetics and catalytic efficiencies. As such, the development of rational methods for the modular construction of tailorable electrode surfaces with robust molecular wires (MWs) exhibiting well-defined molecular topologies, conductivities and morphologies is critical to the evolution and implementation of electrochemical arrays for sensing and catalysis. In response to this need, we have established modular on-surface Sonogashira and Glaser cross-coupling processes to synthetically install arrays of ferrocene-capped MWs onto electrochemically functionalized surfaces. These methods are of comparable convenience and efficiency to more commonly employed Huisgen methods. Furthermore, unlike the Huisgen reaction, this new surface functionalization chemistry generates modified electrodes that do not contain unwanted ancillary metal binding sites, while allowing the bridge between the ferrocenyl moiety and electrode surface to be synthetically tailored. Electrochemical and surface analytical characterization of these platforms demonstrate that the linker topology and connectivity influences the ferrocene redox potential and the kinetics of charge transport at the interface.
Introduction
The ability to control charge transport between conducting substrates and discrete redox sites is of critical importance to the development of electrochemical energy storing architectures such as lithium air batteries,1–3 solar water splitting assemblies4–7 and fuel cell devices.8,9 Furthermore, controlling the composition and molecular topology of electrode–catalyst interfaces is a critical factor in engineering devices with specific electron transport kinetics and catalytic efficiencies. An indication of the importance of optimized interfacial charge transfer to a system's energy conversion efficiency has been demonstrated by Kubiak and Mayer, who have shown that the current density for the photoelectrochemical reduction of CO2 at a styrene-modified p-Si electrode in the presence of a Re(I)bipyridine complex is enhanced roughly threefold compared to the same process at a hydrogen or hexane terminated p-Si electrode.10,11 Further indication that the properties of an electrode surface need to be addressed when developing artificial photosynthetic systems is illustrated by recent studies showing that DSC photocurrent is sensitive to topological variations at the semiconductor interface.12–15 Interfacial charge transport is also fundamentally important to the development of hybrid biomolecule-electrode assemblies designed for energy conversion16–20 and sensing applications.21,22Various strategies have been employed to tailor the molecular composition, topology and connectivity of electrode interfaces. A common method used to modify conducting substrates involves the formation of thiol-based self-assembled monolayers (SAMs) to which redox active moieties can be anchored.23 This approach is confronted by several practical limitations, however, as it is typically only applicable for modification of expensive gold substrates and produces modified electrodes with relatively low conductivities24 and poor thermal25 and oxidative stabilities.25–27
Another popular method for electrode modification is the arylation of conducting surfaces via the electrochemical reduction of aryl diazonium salts.28,29 Electrochemical generation of aryl radicals at a glassy carbon (GC) electrode leads to formation of strong C–C bonds between the aryl group and the carbon surface to furnish robust modified electrodes. There are, however, limitations associated with this general approach that compromise its utility in producing functionally modified electrodes. For example, preparation of the requisite diazonium appended synthons requires strongly oxidizing and/or acidic conditions, which are generally incompatible with complex arenes and catalysts for molecular energy conversion applications. Moreover, the highly reactive aryl radicals formed upon reduction of the diazonium-appended catalyst can participate in undesirable side reactions and contribute to catalyst decomposition. Equally problematic are side reactions between the aryl radical intermediates and arenes already grafted onto the conducting substrate, which often leads to formation of a disordered tangle of arene polymers with ill-defined molecular topologies and electronic properties.30 The longer and more complex the diazonium derivative used to functionalize the surface, the more side reactions are possible and the greater the likelihood of polylayer formation. Polylayers of this type are not ideal for controlling charge transfer at the modified electrode, since the molecular connectivities and electron-transfer pathways are ill-defined.30 As such, the development of new methods for the production of robust functionalised electrodes with uniform/well-defined pathways for electron transfer to surface appended redox sites are needed.
It has been demonstrated that aryl diazoniums with properly positioned steric bulk can be cleanly grafted onto carbon or copper electrodes to generate precise surface appended monolayers.31 For example, electrochemical reduction of 3,5-di-tert-butyl-phenyl diazonium at carbon does not generate polyarenes, as the bulky tert-butyl substituents protect the initially grafted layer from subsequent arylation.32 Similarly, Hapiot and coworkers have demonstrated the electrodeposition of a monolayer of a triisopropylsilyl (TIPS) protected ethynyl benzene diazonium onto carbon. In this case, the large footprint of the TIPS substituent precludes formation of the polyarene tangles observed for less sterically demanding systems. Moreover, it was shown that these monolayers could be synthetically modified via a 1,3-dipolar Huisgen cycloaddition reaction,33–35 following removal of the terminal TIPS groups. Although only Huisgen reactivity has been demonstrated for this system, we rationalized that this general method for presentation of a terminal alkyne monolayer on conducting surfaces could provide a versatile platform for several cross-coupling chemistries and allow for the modular construction of tailorable electrode surfaces with robust molecular wires (MWs) that exhibit well-defined structural topologies, conductivities and morphologies.36
In our efforts to develop modular and efficient methods for the preparation of such architectures we have sought to establish a modular on-surface metal-mediated cross-coupling chemistry to synthetically install arrays of MWs onto inexpensive conducting supports. To this end, we have developed on-surface variations of Sonogashira37,38 and Glaser39,40 cross-coupling reactions for modification of conducting substrates. Herein we describe the development and utility of these methods for fabrication of novel surface-modified assemblies, and demonstrate that these systems have notable electronic properties that are distinguished from more commonly employed surface-modification chemistries such as Huisgen cycloaddition reactions and non-specific small molecule adsorption.
Results and discussion
We selected fuel-cell grade carbon paper as our electrode–substrate for these studies. This material was chosen for diazonium grafting and surface modification since it is inexpensive (∼$0.1 cm−2), highly conducting, and of commercial utility for the construction of electrochemical energy conversion devices. As mentioned above, Hapiot and coworkers have demonstrated that a self-limiting monolayer of sterically bulky aryl diazonium units can be electrochemically grafted onto similar carbon electrodes.41 Building on this work we carried out cyclic voltammetry (CV) using a piece of carbon paper as a working electrode with an acetonitrile solution of TIPS-protected aryl diazonium (1) containing 0.1 M TBAPF6.42 Upon sweeping to reductive potentials, we observed a large, irreversible cathodic current. This response was suppressed upon subsequent CV passes, with stable CV traces emerging after approximately six cycles (Fig. 1a). This CV response is consistent with diazonium reduction and aryl grafting to the carbon paper substrate to generate CP1 (Fig. 1). The suppression of cathodic current as the number of CV sweeps increased indicated passivation of the carbon paper surface as the bulky TIPS groups formed an insulating monolayer that impeded subsequent diazonium reduction processes and hence, the formation of a self-limited monolayer. Integration of charge passed during the electrografting shows that the density of surface modification is 1.1 × 10−9 mol cm−2, which corresponds very well to the maximum theoretical surface coverage of a close packed monolayer (Γ = 1.35 × 10−9 mol cm−2).43 Treatment of CP1 with tetrabutylammonium fluoride (TBAF) in dry acetonitrile efficiently removed the silicon protecting groups from the modified carbon substrate, to reveal alkyne-terminated surface CP2 (Fig. 1). Survey and high-resolution XPS analyses were consistent with these proposed surface structures. The XPS data of the TIPS-modified surface of CP1 showed signals attributable to silicon (2p) at 100.4 eV. These features were almost completely absent in alkyne-terminated surface CP2 (Fig. 1b). The area reduction of the silicon 2p feature between CP1 and CP2 in Fig. 1b is 87%.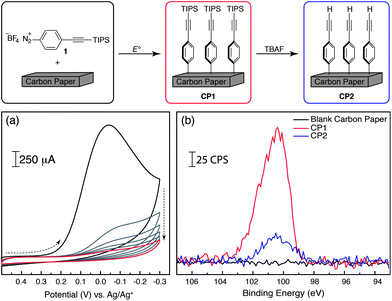 | ||
| Fig. 1 Electrochemical deposition and deprotection of an alkynyl TIPS-appended aryl diazonium. (a) Repeated CV scans using a carbon paper working electrode in a solution of 1.0 mM diazonium 1, containing 0.1 M TBAPF6 (scan rate = 50 mV s−1); (b) high-resolution XPS spectra for the silicon 2p region of unmodified carbon paper (black), CP1 (red) and CP2 (blue). | ||
On-surface cross-coupling chemistry
With an inexpensive phenyl acetylene-terminated electrode substrate in hand, we turned to the use of transition metal-catalysed cross-coupling reactions for surface modification. We began by examining the on-surface Sonogashira cross-coupling44–47 of 4-iodophenylferrocene (2) (Scheme 1). A ferrocene derivative was selected as a coupling partner, since it provides a convenient handle for both electrochemical and XPS analysis. To carry out the coupling and avoid surface fouling by precipitates, we first identified conditions under which all molecular reagents and catalysts were soluble. Model reactions between phenyl acetylene and 4-iodophenylferrocene (2) revealed that the use of catalytic Pd(PPh3)4 and CuI in DMF at 80 °C with Et3N as base resulted in homogenous reactions that afforded rapid C–C bond formation. These reaction conditions afforded Fc1 in 84% yield as a crystalline orange material (Scheme 1a). Crystals of Fc1 suitable for X-ray diffraction analysis were grown by slow evaporation of concentrated CH2Cl2 solutions. The solid-state structure of this compound is shown in Fig. 2 and relevant crystallographic data are summarized in Table S-1.‡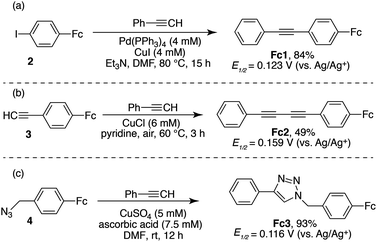 | ||
| Scheme 1 Optimized ferrocene cross-coupling methods including (a) Sonogashira coupling, (b) Glasier coupling and (c) Huisgen coupling. | ||
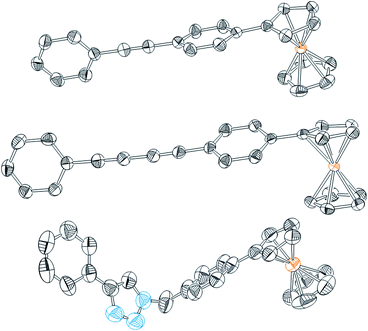 | ||
| Fig. 2 Thermal ellipsoid plots of Fc1–Fc3 (top–bottom) with ellipsoids drawn at the 50% probability level. All hydrogen atoms have been omitted for clarity. Blue = nitrogen; grey = carbon; orange = iron. | ||
Using the optimized methods for solution-phase preparation of Fc1, we found that the acetylene-terminated carbon support (CP2) could be easily modified to give the alkyne-bridged surface (CP3) as shown in Scheme 2. Several independent experiments were conducted to determine optimal reaction time and temperature for this surface modification. XPS analysis (vide infra) showed the greatest surface-bound iron content when the reactions were conducted at 80 °C for 15 min. Longer reaction times did not lead to enhanced coupling. XPS spectra recorded for the modified carbon substrate revealed the presence of surface-bound iron atoms (Fig. 3a). The peak shape and position for the iron 2p doublet at 708.2 and 720.9 eV are consistent with previously reported ferrocene XPS data.48 Significantly, only trace amounts of copper, palladium, and iodine were observed on the modified surfaces by high-resolution XPS (Fig. 3b–d). In all cases, these elements were detected at significantly lower levels than the signal for iron (Table 1), which strongly supports a successful on-surface cross-coupling reaction. Moreover, control reactions in which the Pd and Cu co-catalysts were omitted, or use of unfunctionalized carbon paper substrates showed no quantifiable grafting of iron by XPS.49
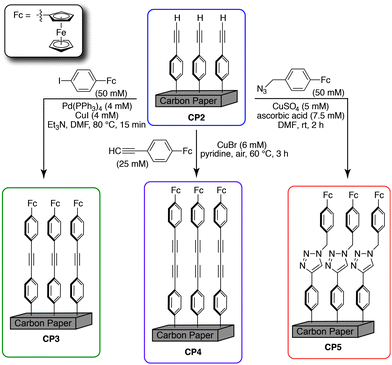 | ||
| Scheme 2 On-surface synthetic methodology developed for modular construction of ferrocene capped molecular wires on a conductive carbon paper support. | ||
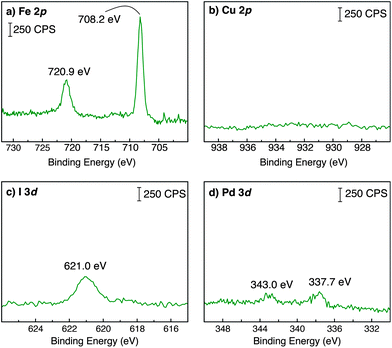 | ||
| Fig. 3 High-resolution XPS spectra recorded for a Sonogashira-modified carbon substrate (CP3) centered in the (a) Fe 2p; (b) Cu 2p; (c) I 3d and (d) Pd 3d regions. | ||
| Average atomic percent (%)b | Redox properties | |||||||
|---|---|---|---|---|---|---|---|---|
| C 1s | Fe 2p | Cu 2p | Br 2p | I 3d | Pd 3d | E1/2c | ETRCd | |
| a Dashed entries denote elements that were not introduced onto the surface and were not present as determined by XPS.b (n ≥ 40; 1σ).c Redox potentials are referenced to Ag/AgNO3.d Pair-wise t-tests at 90% confidence indicated that CP4 was significantly different than CP3 and CP5. | ||||||||
| CP3 | 94.3 ± 1.3 | 0.29 ± 0.08 | <0.1 | — | <0.1 | <0.1 | 0.197 V | 36 ± 6 s−1 |
| CP4 | 93.8 ± 0.9 | 0.12 ± 0.05 | <0.1 | <0.1 | — | — | 0.231 V | 44 ± 10 s−1 |
| CP5 | 92.3 ± 1.6 | 0.30 ± 0.05 | <0.1 | — | — | — | 0.189 V | 35 ± 3 s−1 |
Having demonstrated that Sonogashira reactions can be used to modify conducting carbon supports, we next determined if other types of transition metal-catalysed C–C bond forming reactions could also be used to similar effect. In particular, we envisioned that the Glaser reaction, which joins two terminal alkynes to form a new Csp–Csp bond would offer a powerful way to modify electrode surfaces by allowing moieties of interest to be tethered via a strongly conductive diyne linker.
As before, we began by identifying homogenous conditions for the model reaction of ferrocene-linked alkyne 2 and phenyl acetylene (Scheme 1b). We found that use of catalytic CuCl in pyridine at 60 °C under air led to the formation of Fc2 in 49% yield after 3 hours, along with homo-coupled byproducts. Crystals of Fc2 suitable for X-ray diffraction analysis were easily obtained, and the solid-state structure of this compound is reproduced in Fig. 2 and relevant crystallographic data are summarized in Table S-1.‡
The reaction conditions used for preparation of Fc2 translated well to on-surface cross-coupling by substituting CuBr for CuCl. Treatment of the acetylene-terminated carbon substrate (CP2) with alkynyl ferrocene derivative 3, resulted in the formation of the diacetylene modified surface CP4 (Scheme 2). Linkage of the ferrocene unit to the acetylene modified carbon paper was once again demonstrated by XPS, which showed the presence of significant surface-bound ferrocene (Fig. S-1‡) and only trace amounts of copper or bromine (Table 1). As with the case for the Sonogashira coupling, control experiments in which the copper catalyst was omitted, and independent runs employing non-functionalized carbon substrates did not result in surface-bound ferrocene, as judged by both XPS and electrochemical analysis (vide infra). These results are consistent with a successful on-surface Glaser cross-coupling.
Finally, in order to make a direct comparison with the triazole-bridged electrode assemblies that have been reported by Hapiot and others, we prepared ferrocene azide 4 (Scheme 1c). Treatment of this compound with phenyl acetylene, CuSO4 and ascorbic acid in DMF at room temperature, cleanly afforded Fc3, the structure of which is reproduced in Fig. 2. These Huisgen cycloaddition conditions, which are completely homogenous, were easily translated to the analogous on-surface coupling reaction allowing the acetylene-terminated electrode (CP2) to be transformed to the triazole-linked ferrocene-modified surface (CP5) as shown in Scheme 2.
Similar to the Sonogashira and Glasier reactions, XPS analysis of the Huisgen-modified carbon paper (CP5), showed the presence of bound ferrocene with a strong signal corresponding to iron at 708.0 and 720.8 eV (Fig. S-1‡). The presence of nitrogen on the modified surface was also evident from the high-resolution XPS spectrum, which showed a strong feature at ∼400 eV (Fig. S-2b‡). This signal corresponds to the nitrogen atoms that comprise the triazole moiety linking the ferrocene unit and carbon surface. This peak cannot simply be attributed to unreacted azide starting material adsorbed onto the carbon paper, which is characterized by a sharp XPS feature at 404.8 eV (Fig. S-2a‡). The Huisgen-modified substrate (CP5) also showed low levels of copper and sulfate deposition, although copper contamination was slightly more problematic than for the other on-surface couplings described above (Table 1).
Electrochemical characterization of modified electrodes
The success of the on-surface cross-coupling reactions described in the preceding section is also evident from electrochemical analysis. CV analysis carried out for each of the ferrocene-modified carbon substrates (CP3–CP5) showed a reversible redox wave attributable to electron transfer from the tethered ferrocene units (Fig. 4). This feature is absent in both the unmodified carbon paper (Fig. 4, black traces), as well as on both alkynyl-modified substrates lacking a ferrocene moiety (CP1 and CP2). The potential at which this redox wave appears (E1/2) is affected by varying the molecular topology of the spacer that links the carbon surface to the ferrocenyl adduct. These potentials were measured to be E1/2(CP3) = 0.197 V, E1/2(CP4) = 0.231 V and E1/2(CP5) = 0.189 V versus Ag/AgNO3 (Table 1). Further evidence that the ferrocenyl moieties of CP3–CP5 are chemically linked to the carbon surface is garnered by noting that these redox potentials are significantly perturbed from those recorded for the analogous small molecule analogues (i.e., Fc1–Fc3). For example, while the Sonogashira modified carbon substrate (CP3) displays a redox wave centred at 0.197 V, the homologous ferrocene monomer has an oxidation potential of E1/2(Fc1) = 0.123 V versus Ag/AgNO3. Similarly, while the Glaser (CP4) and Huisgen (CP5) modified surfaces display redox waves at 0.231 V and 0.189 V, respectively, the corresponding monomeric ferrocene derivatives show oxidation potentials of E1/2(Fc2) = 0.159 V and E1/2(Fc3) = 0.116 V. In all cases, anchoring of the ferrocenyl adduct to the carbon support shifted the associated E1/2 values to higher potentials by slightly more than 70 mV.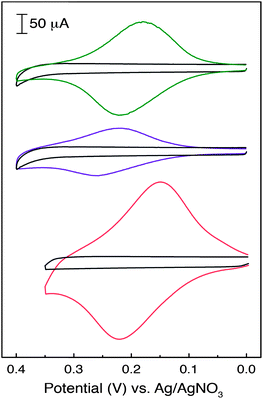 | ||
| Fig. 4 CV traces recorded for CP3 (green) CP4 (purple) and CP5 (orange) substrates suspended in a 0.1 M solution of TBAPF6 in MeCN (scan rate = 100 mV s−1). Each of the ferrocene-modified electrodes is superimposed onto a CV trace obtained for an unmodified carbon substrate (black) under identical conditions. | ||
Electrochemistry experiments have also revealed the contrasting surface properties of the triazole-containing modified surfaces (CP5) compared to the Sonogashira (CP3) and Glasier (CP4) modified electrodes. Initial XPS analysis of CP5 substrates showed slightly elevated levels of residual copper relative to CP3 or CP4. CV analysis of freshly prepared CP5 electrodes that had been rigorously washed to remove residual reagents and non-covalently linked species revealed an irreversible redox wave at ∼0.3 V versus Ag/AgNO3, which obscured the ferrocene couple. Notably, this irreversible wave only appears during the first few CV sweeps; in subsequent anodic cycles this feature does not appear, thereby allowing the ferrocene couple at 0.189 V to be resolved (Fig. S-3a‡).
XPS spectra recorded for CP5 after CV analysis show that the amount of residual copper on the surface was dramatically reduced, while the level of iron was virtually unchanged within experimental error (Fig. S-3b‡). Given that triazole-based complexes of copper are known, these results are consistent with enhanced binding of Cu(I) to the surface-anchored triazoles of CP5. Oxidation of the bound Cu(I) ions results in decreased affinity and loss of Cu(II) to the electrolyte solution. These results suggest that MWs formed via Huisgen couplings at a conducting surface may generate modified electrodes that display a coordination chemistry that is not innocent. This realization is important given the popularity of Huisgen methods for electrode modification chemistry.50–57 The on-surface Sonogashira and Glasier reactions we have developed are therefore distinguished, since they do not introduce ancillary metal ion binding sites onto the modified conducting substrates.
CV experiments have also allowed us to establish how the three surface-modification topologies descried above attenuate electronic communication between the ferrocene moiety and the conducting support. Unlike electron transfer at an electrode–solution interface under diffusion controlled conditions, the anodic and cathodic peak potentials (Ea and Ec, respectively) for redox events involving electro-active species anchored to an electrode surface diverge as scan rate (ν) is increased (Fig. 5a). By applying a formalism developed by Laviron, in which Ec–E1/2 and Ea–E1/2 are plotted as a function of log(ν), we have determined the electron-transfer rate constants (ETRCs) for CP3 through CP5. Laviron plots for each of these systems were constructed (Fig. 5) and ETRCs were extracted by fitting the portions of these plots in which (Ea–Ec) ≥ 200 mV. This analysis afforded ETRC values of 36 s−1, 44 s−1 and 35 s−1 for CP3, CP4 and CP5, respectively (Table 1).
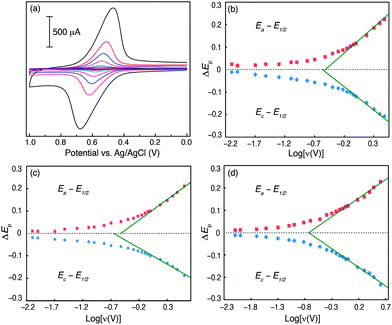 | ||
| Fig. 5 (a) Representative set of CV traces obtained for CP3 suspended in acetonitrile containing 0.1 M TBAPF6 at various scan rates; Laviron plots constructed for oxidation of surface bound ferrocene on (b) CP3; (c) CP4 and (d) CP5. | ||
At first glance, the fact that these values are so similar would appear to suggest that the three MW topologies we have probed do not significantly differ in their ability to modulate charge transport between the ferrocene moiety and carbon surface. A closer inspection of these modified electrodes, however, shows that the three MWs studied are not all of the same length. Based on the crystallographic structures of Fc1–Fc3, we can estimate the distal separation between the ferrocene centre and aryl carbon grafted to the electrode surface to be 12.59 Å for CP3, 15.01 Å for CP4 and 12.62 Å for CP5. Given that the rate of electron transport between two centres decreases as the distance between them is augmented, one would expect CP4 to display a smaller ETRC compared to CP3 and CP5 if each MW topology was equally proficient in modulating charge transport. The fact that the measured ETRC for CP4 is larger than for the other shorter systems studied suggests that the diyne linker may be a more conductive molecular architecture than the others we have probed.
Summary and conclusions
We have developed a modular method to prepare electrically conducting substrates with tailored morphologies at the electrode surface. In carrying out this work, we have adapted metal-mediated cross-coupling chemistry to synthetically modify carbon-paper substrates. These on-surface modification methods have been applied to build carbon-supported MWs with terminal ferrocene units. The current study has focused on the construction of MW-electrode assemblies with three distinct molecular compositions and topologies. Using the methodology described herein, we have prepared well-defined carbon-paper substrates appended with alkyne (CP3), diyne (CP4) and triazole (CP5) based units. We have also demonstrated that each different MW topology gives rise to distinct electronic properties, as evidenced by the disparate E1/2 values recorded for CP3–CP5. Notably, these systems also display variations in the kinetics of electron transfer between the ferrocene units and electrode surface, with the Glaser-based electrode (CP4) displaying a larger ETRC than the other systems we have studied.The observation that the diyne MWs of CP4 afford a modified electrode with enhanced electron transport kinetics, despite the fact that this linker is nearly 2.5 Å longer than the alkyne (CP3) or triazole (CP5) containing MWs indicates that the diyne linker is a more efficient bridge for electron transport. This result is consistent with prior work that has demonstrated polyacetylenic linkers to be superior mediators of electronic communication in donor–acceptor assemblies.58–62 Similarly, polyacetylenic linked porphyrin-fullerene dyads have been shown to undergo extremely efficient and rapid photoinduced electron transfer, with associated attenuation factors that are exceptionally small (β ∼ 0.06 Å−1).63 When taken together, these prior studies reinforce our observation that electron transport between small molecules anchored at an electrode surface and the bulk material is facilitated by a diyne bridge. Building on this work, we would expect electrode substrates grafted with longer polyacetylenic-based MWs to also display relatively large ETRCs and efficient charge transport kinetics. Future work from our laboratories will take advantage of the on-surface coupling methods we have developed to enable the construction and physical interrogation of such systems.
Acknowledgements
We thank Dr. Holt P. Bui (UD) and Jillian Jastrzembski for helpful discussion and assistance with XPS and electrochemical analysis, respectively. The instrumentation in UD's Surface Analysis Facility was purchased with funding from the NSF (CHE-9814477 and DMR-9724307). Financial support for this work was provided to JR and DAW in part through COBRE seed funding (NIH P20 RR017716) and a grant from the US Army Power Division (GTS-S-11-1-243). JR also thanks the University of Delaware Research Foundation for support of this project and RCP thanks the University of Delaware for a University Graduate Scholars Award. NMR and other data were acquired at UD using instrumentation obtained with assistance from the NSF and NIH (NSF-CRIF MU CHE-0541775, CHE-0840401 and CHE-1048367, NIH P20 RR017716 and S10 RR02692).Notes and references
- M. S. Whittingham, Chem. Rev., 2004, 104, 4271–4302 CrossRef CAS.
- G. Girishkumar, B. McCloskey, A. C. Luntz, S. Swanson and W. Wilcke, J. Phys. Chem. Lett., 2010, 1, 2193–2203 Search PubMed.
- M. Armand and J. M. Tarascon, Nature, 2008, 451, 652–657 CrossRef CAS.
- J. J. H. Pijpers, M. T. Winkler, Y. Surendranath, T. Buonassisi and D. G. Nocera, Proc. Natl. Acad. Sci. U. S. A., 2011, 108, 10056–10061 CrossRef CAS.
- S. Y. Reece, J. A. Hamel, K. Sung, T. D. Jarvi, A. J. Esswein, J. J. H. Pijpers and D. G. Nocera, Science, 2011, 334, 645–648 CrossRef CAS.
- D. G. Nocera, Acc. Chem. Res., 2012, 45, 767–776 CrossRef CAS.
- Y. Zhao, J. R. Swierk, J. D. Megiatto, B. Sherman, W. J. Youngblood, D. Qin, D. M. Lentz, A. L. Moore, T. A. Moore, D. Gust and T. E. Mallouk, Proc. Natl. Acad. Sci. U. S. A., 2012, 109, 15612–15616 Search PubMed.
- C.-Y. Wang, Chem. Rev., 2004, 104, 4727–4766 CrossRef CAS.
- M. Winter and R. J. Brodd, Chem. Rev., 2004, 104, 4245–4270 CrossRef CAS.
- J. M. Smieja, E. E. Benson, B. Kumar, K. A. Grice, C. S. Seu, A. J. M. Miller, J. M. Mayer and C. P. Kubiak, Proc. Natl. Acad. Sci. U. S. A., 2012, 109, 15646–15650 Search PubMed.
- B. Kumar, J. M. Smieja, A. F. Sasayama and C. P. Kubiak, Chem. Commun., 2012, 48, 272–274 RSC.
- M. Law, L. E. Greene, J. C. Johnson, R. Saykally and P. Yang, Nat. Mater., 2005, 4, 455–459 CrossRef CAS.
- J. Wang and Z. Lin, Chem. Mater., 2010, 22, 579–584 CrossRef CAS.
- G. K. Mor, K. Shankar, M. Paulose, O. K. Varghese and C. A. Grimes, Nano Lett., 2006, 6, 215–218 CrossRef CAS.
- K. Zhu, N. R. Neale, A. Miedaner and A. J. Frank, Nano Lett., 2007, 7, 69–74 CrossRef CAS.
- T. W. Woolerton, S. Sheard, E. Pierce, S. W. Ragsdale and F. A. Armstrong, Energy Environ. Sci., 2011, 4, 2393–2399 RSC.
- T. W. Woolerton, S. Sheard, E. Reisner, E. Pierce, S. W. Ragsdale and F. A. Armstrong, J. Am. Chem. Soc., 2010, 132, 2132–2133 CrossRef CAS.
- K. A. Vincent, J. A. Cracknell, O. Lenz, I. Zebger, B. Friedrich and F. A. Armstrong, Proc. Natl. Acad. Sci. U. S. A., 2005, 102, 16951–16954 CrossRef CAS.
- M. Kato, T. Cardona, A. W. Rutherford and E. Reisner, J. Am. Chem. Soc., 2012, 134, 8332–8335 CrossRef CAS.
- J. A. Cracknell, K. A. Vincent and F. A. Armstrong, Chem. Rev., 2008, 108, 2439–2461 CrossRef CAS.
- J. Wang, Chem. Rev., 2008, 108, 814–825 CrossRef CAS.
- I. Willner and E. Katz, Angew. Chem., Int. Ed., 2000, 39, 1180–1218 CrossRef.
- A. Ulman, Chem. Rev., 1996, 96, 1533–1554 CrossRef CAS.
- B. Liu, A. Bard, M. Mirkin and S. Creager, J. Am. Chem. Soc., 2004, 126, 1485–1492 CrossRef CAS.
- E. Delamarche, B. Michel, H. Kang and C. Gerber, Langmuir, 1994, 10, 4103–4108 CrossRef.
- M. Schoenfisch and J. Pemberton, J. Am. Chem. Soc., 1998, 120, 4502–4513 CrossRef CAS.
- N. Flynn, T. Tran, M. Cima and R. Langer, Langmuir, 2003, 19, 10909–10915 CrossRef CAS.
- M. Delamar, R. Hitmi, J. Pinson and J. M. Saveant, J. Am. Chem. Soc., 1992, 114, 5883–5884 CrossRef CAS.
- P. Allongue, M. Delamar, B. Desbat, O. Fagebaume, R. Hitmi, J. Pinson and J.-M. Savéant, J. Am. Chem. Soc., 1997, 119, 201–207 CrossRef CAS.
- A. Devadoss and C. E. D. Chidsey, J. Am. Chem. Soc., 2007, 129, 5370–5371 CrossRef CAS.
- C. Saby, B. Ortiz, G. Y. Champagne and D. Bélanger, Langmuir, 1997, 13, 6805–6813 CrossRef CAS.
- C. Combellas, D.-e. Jiang, F. d. r. Kanoufi, J. Pinson and F. I. Podvorica, Langmuir, 2009, 25, 286–293 CrossRef CAS.
- H. C. Kolb, M. G. Finn and K. B. Sharpless, Angew. Chem., Int. Ed., 2001, 40, 2004–2021 CrossRef CAS.
- E. Lallana, R. Riguera and E. Fernandez-Megia, Angew. Chem., Int. Ed., 2011, 50, 8794–8804 CrossRef CAS.
- L. Liang and D. Astruc, Coord. Chem. Rev., 2011, 10, 1791–1815 Search PubMed.
- S. Mahouche-Chergui, S. Gam-Derouich, C. Mangeney and M. M. Chehimi, Chem. Soc. Rev., 2011, 40, 4143–4166 RSC.
- R. Chinchilla and C. Nájera, Chem. Rev., 2007, 107, 874–922 CrossRef CAS.
- R. Chinchilla and C. Nájera, Chem. Soc. Rev., 2011, 40, 5084–5121 RSC.
- G. W. Gribble, in Name Reactions for Homologations, Part 1, ed. J. J. Li, John Wiley and Sons, Hoboken, New Jersey, 2009, pp. 236–257 Search PubMed.
- P. Siemsen, R. C. Livingston and F. Diederich, Angew. Chem., Int. Ed., 2000, 39, 2632–2657 CrossRef CAS.
- Y. R. Leroux, H. Fei, J.-M. Noël, C. Roux and P. Hapiot, J. Am. Chem. Soc., 2010, 132, 14039–14041 CrossRef CAS.
- In our hands, the electrographing step was best preformed under nitrogen atmosphere in order to avoid surface oxidation, as determined by XPS analysis.
- J. Pinson and F. Podvorica, Chem. Soc. Rev., 2005, 34, 429–439 RSC.
- Y. Kashiwagi, S. Chiba, H. Ikezoe and J.-i. Anzai, Synlett, 2004, 2513–2516 Search PubMed.
- J.-C. Lin, J.-H. Kim, J. A. Kellar, M. C. Hersam, S. T. Nguyen and M. J. Bedzyk, Langmuir, 2010, 26, 3771–3773 CrossRef CAS.
- M. Qu, Y. Zhang, J. He, X. Cao and J. Zhang, Appl. Surf. Sci., 2008, 255, 2608–2612 CrossRef CAS.
- S. A. Testero and E. G. Mata, J. Comb. Chem., 2008, 10, 487–497 CrossRef CAS.
- M. Umaña, D. R. Rolison, R. Nowak, P. Daum and R. W. Murray, Surf. Sci., 1980, 101, 295–309 Search PubMed.
- Without CuI, the on-surface Sonogashira reaction did occur, but the amount of surface-bound ferrocene was decreased approximately 10-fold.
- D. Evrard, F. Lambert, C. Policar, V. Balland and B. Limoges, Chem.–Eur. J., 2008, 14, 9286–9291 CrossRef CAS.
- H. Li, F. Cheng, A. M. Duft and A. Adronov, J. Am. Chem. Soc., 2005, 127, 14518–14524 CrossRef CAS.
- S. Mahouche-Chergui, A. Ledebt, F. Mammeri, F. Herbst, B. Carbonnier, H. Ben Romdhane, M. Delamar and M. M. Chehimi, Langmuir, 2010, 26, 16115–16121 CrossRef CAS.
- J. P. Collman, N. K. Devaraj and C. E. D. Chidsey, Langmuir, 2004, 20, 1051–1053 CrossRef CAS.
- R. E. Ruther, M. L. Rigsby, J. B. Gerken, S. R. Hogendoorn, E. C. Landis, S. S. Stahl and R. J. Hamers, J. Am. Chem. Soc., 2011, 133, 5692–5694 CrossRef CAS.
- L. M. Bishop, J. C. Yeager, X. Chen, J. N. Wheeler, M. D. Torelli, M. C. Benson, S. D. Burke, J. A. Pedersen and R. J. Hamers, Langmuir, 2012, 28, 1322–1329 CrossRef CAS.
- A. C. Cardiel, M. C. Benson, L. M. Bishop, K. M. Louis, J. C. Yeager, Y. Tan and R. J. Hamers, ACS Nano, 2012, 6, 310–318 CrossRef CAS.
- S. Shah, M. C. Benson, L. M. Bishop, A. M. Huhn, R. E. Ruther, J. C. Yeager, Y. Tan, K. M. Louis and R. J. Hamers, J. Mater. Chem., 2012, 22, 11561–11567 RSC.
- A. Osuka, N. Tanabe, S. Kawabata, I. Yamazaki and Y. Nishimura, J. Org. Chem., 1995, 60, 7177–7185 CrossRef CAS.
- S. A. Vail, D. I. Schuster, D. M. Guldi, M. Isosomppi, N. Tkachenko, H. Lemmetyinen, A. Palkar, L. Echegoyen, X. Chen and J. Z. H. Zhang, J. Phys. Chem. B, 2006, 110, 14155–14166 CrossRef CAS.
- V. Grosshenny, A. Harriman and R. Ziessel, Angew. Chem., Int. Ed. Engl., 1995, 34, 1100–1102 CrossRef CAS.
- V. Grosshenny, A. Harriman and R. Ziessel, Angew. Chem., Int. Ed. Engl., 1995, 34, 2705–2708 CrossRef.
- G. Sedghi, V. M. Garcia-Suarez, L. J. Esdaile, H. L. Anderson, C. J. Lambert, S. Martin, D. Bethell, S. J. Higgins, M. Elliott, N. Bennett, J. E. Macdonald and R. J. Nichols, Nat. Nanotechnol., 2011, 6, 517–523 CrossRef CAS.
- S. A. Vail, P. J. Krawczuk, D. M. Guldi, A. Palkar, L. Echegoyen, J. P. C. Tomé, M. A. Fazio and D. I. Schuster, Chem.–Eur. J., 2005, 11, 3375–3388 CrossRef CAS.
- M. Müri, B. Gotsmann, Y. R. Leroux, M. Trouwborst, E. Lörtscher, H. Riel and M. Mayor, Adv. Funct. Mater., 2011, 21, 3706–3714 Search PubMed.
Footnotes |
| † For a recent related example in which Sonogashira chemistry was used to modify surface polyarenes bound to carbon surfaces see ref. 64. |
| ‡ Electronic supplementary information (ESI) available: Fig. S-1–S-4, Table S-1 and NMR spectra of new compounds. CCDC 898334–898336. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c2sc21413j |
| § These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2013 |