 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
The curious case of (caffeine)·(benzoic acid): how heteronuclear seeding allowed the formation of an elusive cocrystal†
Dejan-Krešimir Bučar*a, Graeme M. Dayb, Ivan Halaszcd, Geoff G. Z. Zhange, John R. G. Sanderf, David G. Reida, Leonard R. MacGillivrayf, Melinda J. Duera and William Jonesa
aDepartment of Chemistry, University of Cambridge, Cambridge CB2 1EW, UK. E-mail: dkb29@cam.ac.uk
bSchool of Chemistry, University of Southampton, Southampton, SO17 1BJ, UK
cDepartment of Chemistry, Faculty of Science, University of Zagreb, 10002 Zagreb, Croatia
dDivision of Materials Chemistry, Ruđer Bošković Institute, 10002 Zagreb, Croatia
eResearch and Development, AbbVie Inc., North Chicago, IL 60064, USA
fDepartment of Chemistry, University of Iowa, Iowa City, IA 52242, USA
First published on 20th August 2013
Abstract
Cocrystals are modular multicomponent solids with exceptional utility in synthetic chemistry and materials science. A variety of methods exist for the preparation of cocrystals, yet, some promising cocrystal phases have proven to be intractable synthetic targets. We describe a strategy for the synthesis of the pharmaceutically relevant (caffeine)·(benzoic acid) cocrystal (1), which persistently failed to form using a broad range of established techniques. State-of-the-art crystal structure prediction methods were employed to assess the possible existence of a thermodynamically stable form of 1, hence to identify appropriate heteronuclear seeds for cocrystallization. Once introduced, the designed heteronuclear seeds facilitated the formation of 1 and, significantly they (or seeds of the product cocrystal) continued to act as long-lasting laboratory “contaminants”, which encouraged cocrystal formation even when present at such low levels as to evade detection. The seeding technique described thus enables the synthesis of cocrystals regarded as unobtainable under desired conditions, and potentially signifies a new direction in the field of materials research.
Introduction
In many areas of applied solid-state research, including the pharmaceutical field, it is crucial that a compound in development be prepared and characterized in the largest possible number of solid forms (e.g. polymorphs, salts, cocrystals).1 The identification of a wide range of solid forms improves the likelihood of identifying solids with optimal physicochemical properties,2,3 and simultaneously maximizes patent protection opportunities.4,5 For those industries focused on crystalline solids, this approach may alleviate the risk of encountering new solid forms of a compound after substantial investments in product development and marketing.6 Consequently, extensive screening for alternative solid forms is seen as a vital step in any solid materials development program.The risks inherent in the development and marketing of organic solids in the pharmaceutical arena are well illustrated by the widely-known cases of the HIV protease inhibitor Ritonavir7 (Norvir), and the dopamine agonist Rotigotine8 (Neupro). A second polymorph of Ritonavir was found two years after Norvir was placed on the market. It transpired that this unexpected second polymorph was thermodynamically more stable than the marketed one,9 rendering the initial polymorph unobtainable for a substantial period and thus necessitating its temporary removal from the market.7 Rotigotine, which was used for the treatment of Parkinson's disease, was administered through a skin patch to minimize the unpleasant side effects of the drug.8 Soon after Neupro was released on the market in 2006, a previously unknown and thermodynamically more stable polymorph started to emerge in the Neupro skin patches.10 The unexpected appearance of this new polymorph drastically reduced the efficacy of the patches, and led to a temporary withdrawal of the drug from the market.
The appearance of a previously unknown polymorph, accompanied by the complete disappearance of the initially observed form, has been reported before.11 Such disappearing polymorphs may be recovered by the tedious determination of the precise crystallisation conditions that led to their formation in the first place.12 In addition, computational crystal structure predictions,13 as well as knowledge-based hydrogen-bond prediction,14 can be utilized to assess the likelihood that additional polymorphs may exist, and to evaluate relative polymorph stability. Despite these possible solutions, the prospect of encountering new and unanticipated solid forms that might lead to the withdrawal of a product from the market – even for a short period of time – still remains daunting.5,7
While the inability to constrain the formation of new polymorphs can have disastrous consequences for a marketed drug, unsuccessful attempts to prepare a multi-component solid (e.g. salt or cocrystal15) can raise the question of whether the solid is actually obtainable in the first place. In particular, since the crystallization of such a solid competes with the crystallization of the pure components or their solvates/hydrates, it can be quite difficult to estimate whether the formation of the crystallized solid is even thermodynamically favorable. Unobtainable (or at least apparently “hidden”16) solid forms with potentially desirable properties are problematic for multiple reasons: for example, an initial failure to obtain a cocrystal might give competing interests a market entry if the synthesis of this “hidden” solid was subsequently accomplished.6
Crystal engineering and cocrystallization (i.e. the formation of multi-component crystalline solids) have recently emerged as important tools for the development of functional organic materials17–29 and for maximum patent protection.4,5 A burgeoning area in this field is the development of efficient and rapid cocrystal screening techniques. Techniques are sought to assure cocrystal formation, assuming that a cocrystal phase exists under desired conditions (e.g. standard ambient conditions). Numerous screening methods (mechanochemical,30,31 thermal,32,33 and solution-based34) and crystallization techniques (such as heteronuclear seeding35,36) have been applied to cocrystal screening. These methods are generally used in an integrated manner to prepare the maximum number of crystal forms of a given molecule. The field, however, still lacks a general methodology that would allow and ensure the crystallization of all designed solid forms predicted to be attainable.
Here, we present an approach that uses pre-designed heteronuclear seeds to facilitate the synthesis of the elusive cocrystal 1 based on caffeine (caf) and benzoic acid (BA) in a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 ratio. Despite the components' apparent propensity for self-assembly,37,381 has failed to form in the past,39,40 nor was it prepared by our persistent cocrystallization attempts in four laboratories that involved the use of various established cocrystallization methods. Following a computational study using crystal structure prediction methods, which showed that formation of 1 is thermodynamically favorable, the predicted cocrystal was ultimately obtained by heteronuclear seeding, using structurally related cocrystals, consisting of caf and fluorobenzoic acids (FBA), as seeds (Fig. 1). The adoption of such a seeding strategy41 was motivated by the successful use of crystal-structure prediction in the synthesis of a previously unobserved polymorph of a pharmaceutical compound.42
:1 ratio. Despite the components' apparent propensity for self-assembly,37,381 has failed to form in the past,39,40 nor was it prepared by our persistent cocrystallization attempts in four laboratories that involved the use of various established cocrystallization methods. Following a computational study using crystal structure prediction methods, which showed that formation of 1 is thermodynamically favorable, the predicted cocrystal was ultimately obtained by heteronuclear seeding, using structurally related cocrystals, consisting of caf and fluorobenzoic acids (FBA), as seeds (Fig. 1). The adoption of such a seeding strategy41 was motivated by the successful use of crystal-structure prediction in the synthesis of a previously unobserved polymorph of a pharmaceutical compound.42
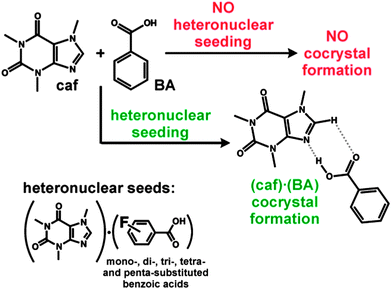 | ||
| Fig. 1 While cocrystallization attempts of caf and BA regularly fail, cocrystal 1 is readily accessible when (caf)·(FBA) cocrystals are used as heteronuclear seeds. | ||
It is particularly noteworthy that, after our initial success with seeding, 1 then remained attainable for several months in all four laboratories where these experiments were conducted, without the need for the deliberate introduction of the seeds.
The findings presented here have significant implications for solid-state and materials science, as well as for pharmaceutical and biomedical researchers. Specifically, this work suggests that elusive cocrystals, sometimes rashly regarded as nearly unobtainable (or even nonexistent under desired conditions) because of their presumed unfavorable lattice energies, might actually be readily available using designed heteronuclear seeds. Our findings also highlight the failings in our current understanding of the nucleation process of cocrystals, and the consequences of such deficient knowledge for progress in materials development.
The work presented here describes the efforts of four laboratories to prepare 1, a solid of pharmaceutical interest whose synthesis has been previously unsuccessfully attempted by others during the last sixty years.39,40 Our interest in 1 is threefold. First, it arises from the sunscreen effect of physical mixtures of caf and sodium benzoate (SB).43 Second, equimolar solutions of caf and SB were found to facilitate electroconvulsive therapy,44 and used to treat postdural puncture headaches and migraine attacks.45 In such solutions, SB (or alternatively, citric acid) is added to enhance the solubility of caf and to maintain the solution as sterile. Notably, the presence of undissociated BA in the caf solution might be more desirable (as BA is a more effective antimicrobial agent for preservation purposes than the dissociated SB46), further driving efforts to prepare 1. Third, our interest in 1 was also triggered by our previous findings in related studies that caf invariably cocrystallizes with a broad range of carboxylic acids, and that cocrystal 1 was the only cocrystal that remained elusive.37,38,47,48
Experimental section
Crystal structure prediction
The crystal structure prediction study involved four steps. First, trial crystal structures were generated in 15 space groups (i.e. P1, P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) , P21, P21/c, P21212, P212121, Pna21, Pca21, Pbca, Pbcn, C2/c, Cc, C2, Pc and P2/c) using the CrystalPredictor program,49 which generates structures using a low-discrepancy sequence to sample the degrees of freedom that define the crystal structure (unit cell parameters, molecular positions and orientations). All trial structures were built from one caf and one BA molecule in the asymmetric unit. Molecular structures were kept rigid at this stage, at geometries obtained from isolated molecule optimization at the B3LYP/6-311G** level of theory, using Gaussian03.50 Intermolecular interactions were modeled using an exp-6 repulsion–dispersion model and an atomic partial charge electrostatic model. The FIT parameter51 set was used for repulsion–dispersion parameters and atomic partial charges were calculated by fitting to the B3LYP/6-311G** molecular electrostatic potential, using the CHelpG scheme of fitting points. A total of 1.6 million trial structures were generated and energy minimized during the initial search. In the second step, rigid-molecule lattice energy minimizations were performed on approximately 13000 of the lowest energy structures with an improved intermolecular model potential, using the same exp-6 repulsion–dispersion parameters as in the structure generation step, now coupled with an atomic multipole model for electrostatic interactions, deriving multipoles up to hexadecapole on each atom from a distributed multipole analysis (DMA)52 of the B3LYP/6-311G** charge densities. These crystal structure calculations were performed using DMACRYS.51 All intermolecular interactions were summed to a 30 Å cutoff, apart from charge–charge, charge–dipole and dipole–dipole interactions, for which Ewald summation was applied. The resulting structures were then clustered using the COMPACK algorithm to remove duplicate structures, leaving 83 distinct crystal structures in a 10 kJ mol−1 energy range from the global energy minimum. In the third step, the 83 crystal structures were re-optimized allowing the optimization of the caf methyl group orientations, and of the dihedral angle between the phenyl ring and the COOH plane in BA. These were performed using the Crystal Optimizer53 code, which finds a minimum in the total energy, calculated as the sum of intermolecular (FIT + DMA) and intramolecular (B3LYP/6-311G**) energies, calling DMACRYS51 for crystal structure calculations and Gaussian03 (ref. 50) for molecular calculations. Finally, all structures obtained in the third step were subjected to a final rigid-molecule lattice energy minimization using the same exp-6 repulsion–dispersion parameters, but with the DMA of each molecule calculated from a charge density calculated with B3LYP/6-311G** within a PCM54 representation of its solid state environment, which has been shown to effectively model the influence of charge density polarization on relative lattice energies.55 The PCM dielectric was chosen as ε = 3.0, which is a typical value for the organic solid state. The same lattice energy minimization steps described in the third and fourth step were applied to ordered models of caf and BA, as described in the main text.
, P21, P21/c, P21212, P212121, Pna21, Pca21, Pbca, Pbcn, C2/c, Cc, C2, Pc and P2/c) using the CrystalPredictor program,49 which generates structures using a low-discrepancy sequence to sample the degrees of freedom that define the crystal structure (unit cell parameters, molecular positions and orientations). All trial structures were built from one caf and one BA molecule in the asymmetric unit. Molecular structures were kept rigid at this stage, at geometries obtained from isolated molecule optimization at the B3LYP/6-311G** level of theory, using Gaussian03.50 Intermolecular interactions were modeled using an exp-6 repulsion–dispersion model and an atomic partial charge electrostatic model. The FIT parameter51 set was used for repulsion–dispersion parameters and atomic partial charges were calculated by fitting to the B3LYP/6-311G** molecular electrostatic potential, using the CHelpG scheme of fitting points. A total of 1.6 million trial structures were generated and energy minimized during the initial search. In the second step, rigid-molecule lattice energy minimizations were performed on approximately 13000 of the lowest energy structures with an improved intermolecular model potential, using the same exp-6 repulsion–dispersion parameters as in the structure generation step, now coupled with an atomic multipole model for electrostatic interactions, deriving multipoles up to hexadecapole on each atom from a distributed multipole analysis (DMA)52 of the B3LYP/6-311G** charge densities. These crystal structure calculations were performed using DMACRYS.51 All intermolecular interactions were summed to a 30 Å cutoff, apart from charge–charge, charge–dipole and dipole–dipole interactions, for which Ewald summation was applied. The resulting structures were then clustered using the COMPACK algorithm to remove duplicate structures, leaving 83 distinct crystal structures in a 10 kJ mol−1 energy range from the global energy minimum. In the third step, the 83 crystal structures were re-optimized allowing the optimization of the caf methyl group orientations, and of the dihedral angle between the phenyl ring and the COOH plane in BA. These were performed using the Crystal Optimizer53 code, which finds a minimum in the total energy, calculated as the sum of intermolecular (FIT + DMA) and intramolecular (B3LYP/6-311G**) energies, calling DMACRYS51 for crystal structure calculations and Gaussian03 (ref. 50) for molecular calculations. Finally, all structures obtained in the third step were subjected to a final rigid-molecule lattice energy minimization using the same exp-6 repulsion–dispersion parameters, but with the DMA of each molecule calculated from a charge density calculated with B3LYP/6-311G** within a PCM54 representation of its solid state environment, which has been shown to effectively model the influence of charge density polarization on relative lattice energies.55 The PCM dielectric was chosen as ε = 3.0, which is a typical value for the organic solid state. The same lattice energy minimization steps described in the third and fourth step were applied to ordered models of caf and BA, as described in the main text.Cocrystal screens
The cocrystal screens were conducted in four laboratories under various conditions and using several screening methods, including neat grinding31 (NG), liquid-assisted grinding31 (LAG), sonic slurry30 (SS) and solution-mediated phase transformation34 (SMPT). The obtained solids were characterized using powder X-ray diffraction and/or Raman spectroscopy. Experimental details and summaries of results obtained from each laboratory are provided in the ESI.†Cocrystal characterisation
The prepared cocrystals were characterised using powder X-ray diffraction (PXRD), infrared spectroscopy, thermogravimetric analysis, differential-scanning calorimetry, Raman spectroscopy and, in some instances, 15N cross-polarisation magic-angle spinning nuclear magnetic resonance spectroscopy (CP-MAS NMR). Experimental details are given in the ESI.† The crystal structures of the cocrystals were determined by ab initio crystal structure solution by simulated annealing and Rietveld refinement using powder X-ray data obtained from a laboratory powder diffractometer. Experimental details and Rietveld plots for all cocrystal structures are also available in the ESI.†Results and discussion
The preparation of cocrystal 1 was attempted in independent trials by groups from the University of Cambridge, AbbVie Inc., the University of Zagreb and the University of Iowa, using several highly reliable cocrystal screening methods viz., LAG,31 NG,31 SS30 and SMPT.34 These well-established screening methods are trusted to facilitate cocrystal formation, provided that such a cocrystal phase exists. In the SMPT and SS methods, for example, the potential for cocrystal formation is maximized once the activities of both components are kept at values higher than their respective critical values, which is certainly accomplished in suspensions of physical mixtures of the cocrystal components.34 The use of ultrasound in the SS method is also known to facilitate cocrystal nucleation. The mechanochemical methods (i.e. LAG and NG), on the other hand, enable the formation of amorphous phases or eutectic mixtures through the local heating induced by colliding mill balls.31Despite the use of reliable screening methods, however, all cocrystallization attempts failed in all four laboratories over several years, and the negative outcome was largely attributed to “crystal packing effects”, i.e. the inability of the two components to pack together in a sufficiently stable crystal lattice.56 Considering that caf and BA form complexes in solution,57 as well as the fact that caf is known to readily form cocrystals with a broad variety of benzoic acid derivatives,58 it was intriguing that such complex formation did not occur in the crystalline solid state.
If it were able to form a solid-state complex, the complex between caf and BA was felt more likely to be a cocrystal, rather than a salt. This assumption was based primarily on the ΔpKa value of the caf:BA pair (i.e., ΔpKa < −3.5, see Table S2 in ESI†), which clearly suggests the formation of a cocrystal.59 To assess the probability that such a cocrystal is stable, global lattice energy minimization calculations were performed to generate the possible low energy structures of 1. A 1:1 stoichiometry was assumed and crystal structures were generated using quasi-random structure generation in 15 commonly observed space groups, each with Z′ = 1 (i.e. one caf and one BA per asymmetric unit). The most promising trial cocrystal structures generated from an initial search were further optimized in several stages, allowing for molecular flexibility within each structure, applying anisotropic models for interatomic interactions and treating polarization of the molecular electron density by the solid state environment. Energies and volumes of the resulting predicted cocrystal structures are summarized in Fig. 2a.
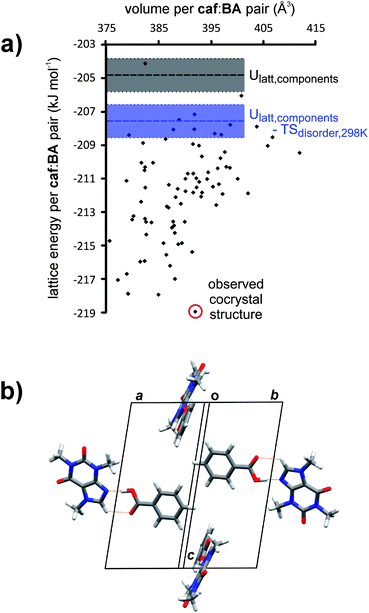 | ||
| Fig. 2 (a) Calculated lattice energies of the predicted cocrystal structures of 1, compared to the sum of the pure component lattice energies (horizontal black dashed line) and the sum of lattice energies and entropy resulting from orientational disorder in pure caf (blue dashed line) and pure BA (the shaded areas represent uncertainties in calculated energies due to the disorder); (b) crystal packing in the P21/c lowest energy predicted cocrystal structure, viewed approximately down the ab diagonal. | ||
To a first approximation, the thermodynamic driving force for cocrystallization can be assessed by comparing the calculated energies of the putative cocrystal structures with those of the pure crystalline components; this approach has been previously applied to predicting cocrystal formation, stoichiometry and structure.60,61 In the present study, the known crystal structure of BA, and an ordered version of the thermodynamically stable form of caf at room temperature (Lehmann and Stowasser's Z′ = 5 ordered structure of β-caf62), were lattice energy minimized using the same methods as used in the cocrystal predictions. The calculated energy of this ordered model of β-caf is an approximation of the lattice energy of the orientationally disordered crystal structure, and we estimated an uncertainty in the lattice energy of ±1 kJ mol−1 from the spread in calculated lattice energies of the 512 unique possible orientational configurations reported by Habgood.63 Disorder in the crystal structures of pure caf and BA provides an entropic contribution to the stability of the pure forms, which, for β-caf, were taken from Habgood63 as –TS ≈ −1 kJ mol−1 at room temperature and were estimated as Sdisorder ≈ Rln2 for BA, due to the two occupied configurations of the carboxylic acid group in the hydrogen-bonded dimers in crystalline BA.
Even after accounting for uncertainties in the calculated lattice energies, and for the entropic contributions to the stability of the pure forms, we found about 70 computer-generated cocrystal structures that were predicted to be energetically preferable to the pure single-component crystal structures (Fig. 2a). The lowest energy predicted cocrystal structure was shown to be more than 10 kJ mol−1 more stable than the pure components. We concluded from these computational studies that caf and BA are able to form a stable cocrystal that, on thermodynamic grounds, should be realizable. Furthermore, we were able to predict the likely primary interaction: all but one of the 70 lowest-energy predicted cocrystal structures exhibit carboxylic-acid:imidazole O–H⋯N hydrogen bonding (the lowest-energy predicted cocrystal structures are available as ESI†). The hydrogen-bonded molecules are nearly co-planar in the lowest-energy cocrystal structure, which is sustained by C–H⋯O and π⋯π interactions (Fig. 2b).
Parallel to the computational study, the Cambridge group commenced a structural study of related cocrystals, in order to identify heteronuclear seeds that could potentially facilitate the growth of 1. This study attempted the cocrystallization of caf with structural isomers of mono-, di-, tri-, tetra- and penta-fluorobenzoic acids (FBAs). Heteronuclear seeding has previously been used to control polymorphism in metal–organic and organic compounds, as well as in cocrystals.35,36,42,64 Our work, however, applied this technique to facilitate the synthesis of multicomponent solids that could not be obtained otherwise.
The working hypothesis for this study was that the formation of 1 is presumably hindered by a high kinetic barrier65 and that such a high barrier may be overcome by introducing a heteronuclear seed that matches the target cocrystal structurally or epitaxially.66,67 Specifically, it was proposed that fluorinated benzoic acids (FBAs) would likely form cocrystals based on molecular assemblies that are similar in size and shape to those present in 1. Such an analogous assembly could be rationalized by the relatively small size difference between hydrogen and fluorine atoms (van der Waals radii: 120 pm vs. 147 pm,68 respectively).69
Indeed, it was found that 17 (out of 19) isomers of FBAs form cocrystals with caf.70 All the experimentally obtained cocrystal structures were solved using powder diffraction data collected on a laboratory diffractometer. The structural studies showed that all (caf)·(FBA) cocrystals are based on caf:FBA complexes being sustained by the same hydrogen-bonding motifs that are found in the predicted structure of 1, namely imidazole–carboxylic-acid heterosynthons utilizing O–H⋯N hydrogen bonds (Fig. 3). Out of these cocrystals, four (i.e. cocrystals involving 2-fluorobenzoic (2FBA), 2,3-difluorobenzoic (23diFBA), 2,5-difluorobenzoic (25diFBA) and 2,6-difluorobenzoic (26diFBA)) were found to be isomorphous with the lowest-energy predicted crystal structure of 1 (i.e. the structures display the same space group and unit-cell dimensions, as well as the same atom types and the positions, except for an interchange of hydrogen and fluorine within the different cocrystals).
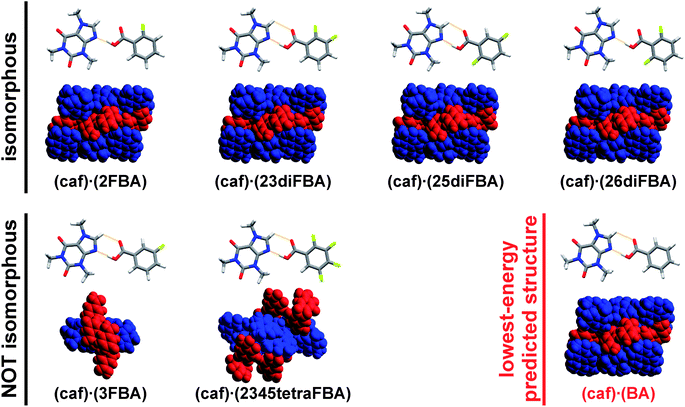 | ||
| Fig. 3 X-ray crystal structures of (caf)·(FBA) cocrystals used as heteronuclear seeds for the crystallization of 1 (black labels). Structures shown in the first row are isomorphous with the lowest-energy predicted crystal structure of 1 (red label), unlike the structures shown in the second row. Each cocrystal system is represented with a depiction of the caf:FBA (or caf:BA) assembly (top) and their crystal packing diagrams (bottom). Colour scheme for the crystal packing diagrams: caf – blue, BA – red. | ||
Interestingly, once the (caf)·(FBA) cocrystals were synthesized in the Cambridge laboratory, and before the actual seeding experiments began there, the Cambridge group became unable to replicate the previously negative results concerning the cocrystallization of caf and BA. Specifically, a new crystal phase unexpectedly became achievable in every crystallization attempt, as evidenced by powder diffraction studies and thermal analyses (see Fig. S1, ESI†). This new crystal phase was subsequently identified as the lowest energy predicted form of 1 (Fig. 4a–c) using crystal structure solution from powder X-ray diffraction, natural abundance 15N CP-MAS NMR, and thermogravimetric analysis.
![(a) X-ray crystal structure of a caf : BA assembly in 1; (b) X-ray crystal structure of a molecular sheet in 1 sustained by π⋯π and C–H⋯π interactions (inset depicts two sheets stacked in an offset manner); (c) overlay of the predicted (shown in red) and observed (blue) crystal structure of 1; (d) PXRD traces of solids obtained in cocrystal screens with and without seeds at AbbVie Inc. [black: β-caf; grey: BA; red – from bottom to top: physical mixtures of caf and BA obtained in cocrystallization attempt via LAG using nitromethane as liquid, LAG using ethanol as liquid, LAG using acetonitrile as liquid, NG, SMPT using nitromethane as solvent, SMPT using ethanol as solvent, SMPT using acetonitrile as solvent; green – from bottom to top: 1 obtained via LAG using nitromethane as liquid and (caf)·(2FBA) as seed, LAG using nitromethane as liquid and (caf)·(23diFBA) as seed, LAG using nitromethane as liquid and (caf)·(26diFBA) as seed, LAG using nitromethane as liquid and (caf)·(3FBA) as seed, LAG using nitromethane as liquid and (caf)·(2345tetraFBA) as seed; blue: 1 obtained via LAG without the deliberate use of a seed in the (caf)·(FBA) contaminated laboratory (utilising nitromethane as liquid)]; (e) Raman spectra of a caf : BA physical mixture before and after being exposed to a seed-contaminated atmosphere [from bottom to top: β-caf (black), BA (black), physical mixtures of caf and BA sonicated for 5 min (red), physical mixtures of caf and BA sonicated for 5 min and subsequently slurried for 19 hours (red), caf : BA physical mixture from a sealed screening vial that converted to 1 upon exposure to the seed-contaminated atmosphere (green), 1 (black)].](/image/article/2013/SC/c3sc51419f/c3sc51419f-f4.gif) | ||
| Fig. 4 (a) X-ray crystal structure of a caf:BA assembly in 1; (b) X-ray crystal structure of a molecular sheet in 1 sustained by π⋯π and C–H⋯π interactions (inset depicts two sheets stacked in an offset manner); (c) overlay of the predicted (shown in red) and observed (blue) crystal structure of 1; (d) PXRD traces of solids obtained in cocrystal screens with and without seeds at AbbVie Inc. [black: β-caf; grey: BA; red – from bottom to top: physical mixtures of caf and BA obtained in cocrystallization attempt via LAG using nitromethane as liquid, LAG using ethanol as liquid, LAG using acetonitrile as liquid, NG, SMPT using nitromethane as solvent, SMPT using ethanol as solvent, SMPT using acetonitrile as solvent; green – from bottom to top: 1 obtained via LAG using nitromethane as liquid and (caf)·(2FBA) as seed, LAG using nitromethane as liquid and (caf)·(23diFBA) as seed, LAG using nitromethane as liquid and (caf)·(26diFBA) as seed, LAG using nitromethane as liquid and (caf)·(3FBA) as seed, LAG using nitromethane as liquid and (caf)·(2345tetraFBA) as seed; blue: 1 obtained via LAG without the deliberate use of a seed in the (caf)·(FBA) contaminated laboratory (utilising nitromethane as liquid)]; (e) Raman spectra of a caf:BA physical mixture before and after being exposed to a seed-contaminated atmosphere [from bottom to top: β-caf (black), BA (black), physical mixtures of caf and BA sonicated for 5 min (red), physical mixtures of caf and BA sonicated for 5 min and subsequently slurried for 19 hours (red), caf:BA physical mixture from a sealed screening vial that converted to 1 upon exposure to the seed-contaminated atmosphere (green), 1 (black)]. | ||
The cocrystal was found to crystallize in the monoclinic P21/c space group with one molecule of caf and BA in the asymmetric unit interacting via the imidazole-carboxylic-acid heterosynthon [d(O⋯N) = 2.663(7) Å] (Fig. 4a). Assemblies of caf:BA form head-to-tail-oriented pairs that further stack in an offset manner (offset: ∼7 Å) held together by π⋯π interactions. The stacks interact via weak C–H⋯π forces to form molecular sheets exhibiting a herringbone motif (Fig. 4b). The sheets form a 3D lattice being held together by C–H⋯π interactions.
Furthermore, 15N CP-MAS NMR was utilized to determine whether the basic N(imidazole) atom of caf was protonated by BA. Specifically, it was shown that the solid-state 15N NMR spectrum exhibits a signal at a high frequency (i.e. 194 ppm; Fig. S16 in ESI†) that is consistent with the presence of an unprotonated N(imidazole) atom in the studied solid.71 Considering that protonated N(imidazolate) atoms exhibit peaks at lower-frequency chemical shifts (i.e. 120–140 ppm),48 it is was concluded that caf and BA crystallize as a cocrystal (rather than a salt), as expected. Thermogravimetric analysis was utilized to determine that the investigated caf:BA phase consists solely of caf and BA (Fig. S18, ESI†).
One possible explanation for the unexpected formation of 1 was that the crystallization vessels or either batch of caf or BA (or the environment in general) were contaminated with the synthesized (caf)·(FBA) cocrystals, thus seeding the growth of the target cocrystal. Surprisingly, 1 was found to form even after numerous thorough laboratory and equipment cleanups, and the use of newly purchased glassware and batches of caf and BA. More specifically, the laboratory benches were thoroughly scrubbed using bleach and ethanol, whereas the milling jars were cleaned using concentrated solutions of strong inorganic acids and bases (i.e. HCl and NaOH, respectively). New crystallization vials and milling balls were used in each experiment, whereas all used solvents were heated and subsequently filtered through syringe filters with polyvinylidene fluoride membranes displaying 0.2 μm pores. The very strong seeding effect of either (caf)·(FBA) crystals or crystals of 1, however, persisted even one year after the seeding experiments were conducted, thus preventing comprehensive studies of the nucleation process and the seeding mechanisms in the Cambridge laboratories.
Seeding experiments were also performed at AbbVie Inc. after earlier attempts to prepare 1 without the use of heteronuclear seeds failed. The initial “seedless” cocrystal screens were based on the NG, LAG and SMPT techniques whereby ethanol, acetonitrile and nitromethane were used as liquids/solvents. After attempts to prepare the target cocrystal failed, four cocrystals that were isomorphous with the lowest-energy predicted target cocrystal (i.e. (caf)·(2FBA), (caf)·(23diFBA), (caf)·(25diFBA) and (caf)·(26diFBA)) were used as heteronuclear seeds, to successfully accomplish the synthesis of 1 (Fig. 4d).
The AbbVie group subsequently performed another series of screening experiments, this time without the use of seeds in order to determine the occurrence of possible laboratory contamination and unintentional seeding, as seen in the Cambridge laboratory. Indeed, a LAG experiment confirmed that 1 could be now obtained without the deliberate use of seeds. Another SMPT experiment was conducted using a physical mixture of caf and BA that was stored in a rubber-septa-sealed vial before any heteronuclear cocrystal seeds were introduced to the AbbVie laboratory. The seedless SMPT cocrystal screen was initialized by injecting acetonitrile through the rubber septum into the solid mixture. In situ Raman spectroscopy measurements showed the presence of a physical mixture in the sealed vial even after an appreciable amount of time (i.e. 19 h). During the isolation of the physical mixture by ultracentrifugation, however, it was observed that the physical mixture converted almost immediately into 1 (as evident from a visible change in the particle morphology, Raman spectroscopy data (Fig. 4e) and PXRD data (Fig. S4†)), thus demonstrating that seemingly minuscule amounts of contaminants of either 1 or a (caf)·(FBA) in the laboratory are capable of inducing a phase transformation in extremely short periods of time. It is to be further noted that additional seeding experiments involving two cocrystal seeds that are not isomorphous with the target cocrystal, namely (caf)·(3FBA) and (caf)·(2345tetraFBA), have also successfully led to the formation of the (caf)·(BA) cocrystal (Fig. 4d). Interestingly, the (caf)·(BA) cocrystal was obtainable without the use of seeds even after more than one year after the initial seeding experiments were conducted.
Original efforts to prepare 1 at the University of Zagreb also failed. Both FBA and (caf)·(FBA) seeds were thus utilised to facilitate nucleation and growth of the target cocrystal (see Fig. S7, ESI†). The use of crystals of pure FBAs as seeds was attempted to determine whether a heteronuclear seed could be formed in situ through addition to a caf:BA physical mixture, while the non-isomorphous (caf)·(FBA) cocrystals were selected as seeds to explore the role of epitaxy in the formation of 1.67 Specifically, 3FBA, 2345tetraFBA, (caf)·(2345tetraFBA) and (caf)·(3FBA) were initially used to attempt the formation of the target cocrystal, using LAG as the crystallization method. PXRD studies showed that all four experiments yielded the target cocrystal. These findings indicate that: (a) that the cocrystal seed and the cocrystal target do not have to be isomorphous, again pointing to the possible role of epitaxy in the nucleation of 1, and (b) that the heteronuclear seed could be generated in situ by addition of the cocrystal former (i.e.FBAs) to the reaction vessel. Crystallization experiments wherein 2FBA, (caf)·(2FBA), 23diFBA, (caf)·(23diFBA), 25diFBA, (caf)·(25diFBA), 26diFBA and (caf)·(26diFBA) were utilized as seeds also resulted in the formation of the target cocrystal, as expected. Notably, the target cocrystal was obtainable without the use of seeds for up to six weeks after the earlier seeding experiments were performed; thereafter, cocrystallization attempts involving caf and BA alone began to fail again.
All results obtained by the Iowa group were consistent with the ones gathered in Cambridge, Zagreb and at AbbVie – cocrystal 1 could only be obtained after the crystallization experiments were aided by the use of (caf)·(FBA) seeds (see Fig. S8, ESI†). Specifically, (caf)·(2FBA) was found to facilitate the quantitative formation of the target cocrystal in a SMPT experiment using acetonitrile as solvent. While 2FBA, 23diFBA, 25diFBA and 26diFBA led to cocrystal formation during LAG experiments performed by the Zagreb group, the same FBAs were not found to seed cocrystal formation during SMPT-based screens at Iowa. This was attributed to the inability of the FBA to form (caf)·(FBA) seeds under the screening conditions owing to the complete solubilisation of the FBA in the solvent.
Finally, given the remarkable sensitivity of the formation of 1 to the presence of miniscule amounts of contaminants in the laboratory environment, it was unclear whether the testing laboratories were suitable for the evaluation of the seeding ability of any (caf)·(FBA) cocrystal seed. To determine whether each of the studied (caf)·(FBA) cocrystals indeed acts as a seed for the target cocrystal, the AbbVie group has preformed a series of control experiments.
Physical mixtures of caf and BA (being stored in rubber-septa-sealed vials before the initial heteronuclear cocrystal seeds were conducted) were subjected to an SMPT cocrystal screen by injecting a small amount of acetonitrile through the rubber septum into the vial. In situ Raman spectroscopy measurements showed that no cocrystal formation occurred even after 112 h of slurrying. Suspensions containing small amounts of one of the cocrystals that are isomorphous with 1, namely (caf)·(2FBA), (caf)·(23diFBA), (caf)·(25diFBA), and (caf)·(26diFBA), were then injected to the suspension of the physical mixture. In situ Raman spectroscopy measurements revealed that the physical mixture converted to the cocrystal within a few minutes post-injection, thus proving that each of the utilised (caf)·(FBA) cocrystals can seed the formation of the target cocrystal. The outcomes of control experiments involving the non-isomorphous (caf)·(3FBA) and (caf)·(2345tetraFBA) were not conclusive and could not be used to determine whether these cocrystals can or cannot seed the formation of the target cocrystal.
To verify that FBA cocrystal formers can also be utilised to facilitate the formation of 1, a suspension of 25diFBA crystals was injected into the vial with the physical mixture of caf and BA. The formation of 1 was observed only after 45 minutes. The longer period required to achieve the crystallisation of the target compound is consistent with our hypothesis that the addition of the FBA cocrystal former first leads to the in situ formation of the (caf)·(FBA) cocrystal seeds, which then facilitates the formation of 1.
Conclusion and outlook
That the cocrystal 1 failed to form, even though a stable cocrystal form was predicted to exist, is possibly attributable to a kinetic barrier that hinders the nucleation and growth of the thermodynamically stable target cocrystal. The results presented herein illustrate that elusive multi-component crystal forms can be obtained using cocrystals based on structurally similar cocrystal formers. The results also demonstrate the utility of crystal structure prediction calculations in assessing the likelihood of cocrystal formation. In addition, the results clearly demonstrate that current cocrystal-screening methods need to be improved in order to eliminate the occurrence of false negative results in cocrystal screens, which can seriously impede the development of medicines and functional materials (e.g. cocrystals and coordination compounds with relevant electronic or catalytic properties). Further studies will focus on elucidating the cause of the problematic nucleation of 1, as well as on the development of a thorough understanding of the presumed epitaxial growth of 1 on the surfaces of the (caf)·(FBA) cocrystals.Acknowledgements
DKB acknowledges the Royal Society for a Newton International Fellowship and the Isaac Newton Trust (Trinity College, University of Cambridge) for funding. GMD acknowledges the Royal Society for a University Research Fellowship. IH thanks the Ministry of Science, Education and Sport of the Republic of Croatia (Grant no. 098-0982904-2953 and 119-1191342-1334). DGR acknowledges support from the BBSRC. JRGS thanks the Center for Biocatalysis and Bioprocessing at the University of Iowa for a Fellowship.Notes and references
- S. L. Morissette, Ö. Almarsson, M. L. Peterson, J. F. Remenar, M. J. Read, A. V. Lemmo, S. Ellis, M. J. Cima and C. R. Gardner, Adv. Drug Delivery Rev., 2004, 56, 275–300 CrossRef CAS PubMed.
- Polymorphism: In the Pharmaceutical Industry, ed. R. Hilfiker, Wiley VCH, Weinheim, Germany Search PubMed.
- Developing Solid Oral Dosage Forms: Pharmaceutical Theory & Practice: Pharmaceutical Theory and Practice, ed. Y. Qiu, Y. Chen, G. G. Z. Zhang, L.Liu and W. Porter, Academic Press Inc., London, 2009 Search PubMed.
- A. V. Trask, Mol. Pharmaceutics, 2007, 4, 301–309 CrossRef CAS PubMed.
- Ö. Almarsson, M. L. Peterson and M. Zaworotko, Pharm. Pat. Anal., 2012, 1, 313–327 CrossRef.
- W. A. Rakoczy and D. M. Mazzochi, Journal of Generic Medicines, 2006, 3, 131–139 CrossRef.
- S. R. Chemburkar, J. Bauer, K. Deming, H. Spiwek, K. Patel, J. Morris, R. Henry, S. Spanton, W. Dziki, W. Porter, J. Quick, P. Bauer, J. Donaubauer, B. A. Narayanan, M. Soldani, D. Riley and K. McFarland, Org. Process Res. Dev., 2000, 4, 413–417 CrossRef CAS.
- US Pat. US 2005175678 A1, 2005.
- J. Bauer, S. Spanton, R. Henry, J. Quick, W. Dziki, W. Porter and J. Morris, Pharm. Res., 2001, 18, 859–866 CrossRef CAS.
- US Pat. US 8232414 B2, 2012.
- J. D. Dunitz and J. Bernstein, Acc. Chem. Res., 1995, 28, 193–200 CrossRef CAS.
- J.-O. Henck, J. Bernstein, A. Ellern and R. Boese, J. Am. Chem. Soc., 2001, 123, 1834–1841 CrossRef CAS PubMed.
- S. L. Price, Acc. Chem. Res., 2008, 42, 117–126 CrossRef PubMed.
- P. T. A. Galek, F. H. Allen, L. Fabian and N. Feeder, CrystEngComm, 2009, 11, 2634–2639 RSC.
- Guidance for Industry: Regulatory Classification of Pharmaceutical Co-Crystals, Food and Drug Administration, Silver Spring, MD, USA, 2013 Search PubMed.
- D.-K. Bučar, R. F. Henry, X. Lou, R. W. Duerst, T. B. Borchardt and G. G. Z. Zhang, Chem. Commun., 2007, 525–527 Search PubMed.
- J. Ferraris, D. O. Cowan, V. Walatka and J. H. Perlstein, J. Am. Chem. Soc., 1973, 95, 948–949 CrossRef CAS.
- A. N. Sokolov, T. Friščić and L. R. MacGillivray, J. Am. Chem. Soc., 2006, 128, 2806–2807 CrossRef CAS PubMed.
- L. R. MacGillivray, G. S. Papaefstathiou, T. Friščić, T. D. Hamilton, D.-K. Bučar, Q. Chu, D. B. Varshney and I. G. Georgiev, Acc. Chem. Res., 2008, 41, 280–291 CrossRef CAS PubMed.
- M. Morimoto, S. Kobatake and M. Irie, Chem. Commun., 2008, 335–337 RSC.
- N. Schultheiss and A. Newman, Cryst. Growth Des., 2009, 9, 2950–2967 CAS.
- M. Morimoto and M. Irie, J. Am. Chem. Soc., 2010, 132, 14172–14178 CrossRef CAS PubMed.
- O. Bolton and A. J. Matzger, Angew. Chem., Int. Ed., 2011, 50, 8960–8963 CrossRef CAS PubMed.
- D. Yan, A. Delori, G. O. Lloyd, T. Friščić, G. M. Day, W. Jones, J. Lu, M. Wei, D. G. Evans and X. Duan, Angew. Chem., Int. Ed., 2011, 50, 12483–12486 CrossRef CAS PubMed.
- S. Ghosh and C. M. Reddy, Angew. Chem., Int. Ed., 2012, 51, 10319–10323 CrossRef CAS PubMed.
- D.-K. Bučar, S. Filip, M. Arhangelskis, G. O. Lloyd and W. Jones, CrystEngComm, 2013, 15, 6289–6291 RSC.
- K. B. Landenberger, O. Bolton and A. J. Matzger, Angew. Chem., Int. Ed., 2013, 52, 6468–6471 CrossRef CAS PubMed.
- S. K. Park, S. Varghese, J. H. Kim, S.-J. Yoon, O. K. Kwon, B.-K. An, J. Gierschner and S. Y. Park, J. Am. Chem. Soc., 2013, 135, 4757–4764 CrossRef CAS PubMed.
- J. W. Steed, Trends Pharmacol. Sci., 2013, 34, 185–193 CrossRef CAS PubMed.
- T. Friščić, S. L. Childs, S. A. A. Rizvi and W. Jones, CrystEngComm, 2009, 11, 418–426 RSC.
- T. Friščić and W. Jones, Cryst. Growth Des., 2009, 9, 1621–1637 Search PubMed.
- E. Lu, N. Rodriguez-Hornedo and R. Suryanarayanan, CrystEngComm, 2008, 10, 665–668 RSC.
- E. Lekšić, G. Pavlović and E. Meštrović, Cryst. Growth Des., 2012, 12, 1847–1858 Search PubMed.
- G. G. Z. Zhang, R. F. Henry, T. B. Borchardt and X. Lou, J. Pharm. Sci., 2007, 96, 990–995 CrossRef CAS PubMed.
- D. Braga, G. Cojazzi, D. Paolucci and F. Grepioni, CrystEngComm, 2001, 3, 159–161 RSC.
- M. Lang, A. L. Grzesiak and A. J. Matzger, J. Am. Chem. Soc., 2002, 124, 14834–14835 CrossRef CAS PubMed.
- A. V. Trask, W. D. S. Motherwell and W. Jones, Cryst. Growth Des., 2005, 5, 1013–1021 CAS.
- D.-K. Bučar, R. F. Henry, X. Lou, R. W. Duerst, L. R. MacGillivray and G. G. Z. Zhang, Cryst. Growth Des., 2009, 9, 1932–1943 Search PubMed.
- K. Sekiguchi, Yakugaku Zasshi, 1961, 81, 669–674 CAS.
- S. Heiden, L. Trobs, K.-J. Wenzel and F. Emmerling, CrystEngComm, 2012, 14, 5128–5129 RSC.
- M. A. Neumann and M.-A. Perrin, CrystEngComm, 2009, 11, 2475–2479 RSC.
- J.-B. Arlin, L. S. Price, S. L. Price and A. J. Florence, Chem. Commun., 2011, 47, 7074–7076 RSC.
- Y.-P. Lu, Y.-R. Lou, J.-G. Xie, Q.-Y. Peng, S. Zhou, Y. Lin, W. J. Shih and A. H. Conney, Carcinogenesis, 2006, 28, 199–206 CrossRef PubMed.
- B. Shapira, B. Lerer, D. Gilboa, H. Drexler, S. Kugelmass and A. Calev, Am. J. Psychiatry, 1987, 144, 1199–1202 CAS.
- A. Yücel, S. Özyalçin, G. K. Talu, E. C. Yücel and S. Erdine, Reg. Anesth. Pain Med., 1999, 24, 51–54 Search PubMed.
- A. Wibbertmann, J. Kielhorn, G. Koennecker, I. Mangelsdorf and C. Melber, in CICAD, United Nations Environment Programme, International Labour Organization, World Health Organization, Geneva, 2000, pp. 1–48 Search PubMed.
- D.-K. Bučar, R. F. Henry, X. Lou, R. W. Duerst, T. B. Borchardt, L. R. MacGillivray and G. G. Z. Zhang, Mol. Pharmaceutics, 2007, 4, 339–346 CrossRef PubMed.
- T. Friščić, D. G. Reid, G. M. Day, M. J. Duer and W. Jones, Cryst. Growth Des., 2011, 11, 972–981 Search PubMed.
- P. G. Karamertzanis and C. C. Pantelides, J. Comput. Chem., 2005, 26, 304–324 CrossRef CAS PubMed.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, J. J. A. Montgomery, T. Vreven, K. N. Kudin, J. C. Burant, J. M. Millam, S. S. Iyengar, J. Tomasi, V. Barone, B. Mennucci, M. Cossi, G. Scalmani, N. Rega, G. A. Petersson, H. Nakatsuji, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, M. Klene, X. Li, J. E. Knox, H. P. Hratchian, J. B. Cross, V. Bakken, J. J. C. Adamo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, P. Y. Ayala, K. Morokuma, G. A. Voth, P. Salvador, J. J. Dannenberg, V. G. Zakrzewski, S. Dapprich, A. D. Daniels, M. C. Strain, O. Farkas, D. K. Malick, A. D. Rabuck, K. Raghavachari, J. B. Foresman, J. V. Ortiz, Q. Cui, A. G. Baboul, S. Clifford, J. Cioslowski, B. B. Stefanov, G. Liu, A. Liashenko, P. Piskorz, I. Komaromi, R. L. Martin, D. J. Fox, T. Keith, M. A. Al-Laham, C. Y. Peng, A. Nanayakkara, M. Challacombe, P. M. W. Gill, B. Johnson, W. Chen, M. W. Wong, C. Gonzalez and J. A. Pople, Gaussian 03 Revision D.01, Gaussian, Inc., Wallingford, CT, USA, 2004 Search PubMed.
- S. L. Price, M. Leslie, G. W. A. Welch, M. Habgood, L. S. Price, P. G. Karamertzanis and G. M. Day, Phys. Chem. Chem. Phys., 2010, 12, 8478–8490 RSC.
- A. J. Stone and M. Alderton, Mol. Phys., 1985, 56, 1047–1064 CrossRef CAS.
- A. V. Kazantsev, P. G. Karamertzanis, C. S. Adjiman and C. C. Pantelides, J. Chem. Theory Comput., 2011, 7, 1998–2016 CrossRef CAS.
- B. Mennucci and J. Tomasi, J. Chem. Phys., 1997, 106, 5151–5158 CrossRef CAS.
- T. G. Cooper, K. E. Hejczyk, W. Jones and G. M. Day, J. Chem. Theory Comput., 2008, 4, 1795–1805 CrossRef CAS.
- J. J. Wolff, Angew. Chem., Int. Ed. Engl., 1996, 35, 2195–2197 CrossRef CAS.
- T. Higuchi and D. A. Zuck, J. Am. Pharm. Assoc., 1952, 41, 10–13 CAS.
- A survey of the Cambridge Structural Database (version 5.33, update of May 2012) was carried out using ConQuest (version 1.14) and limited to organic and nonionic compounds with determined 3D-coordinates. The survey revealed 21 entries that corresponded to a structure containing caffeine and an aromatic carboxylic acid.
- A. J. Cruz-Cabeza, CrystEngComm, 2012, 14, 6362–6365 RSC.
- A. J. Cruz-Cabeza, G. M. Day and W. Jones, Chem.–Eur. J., 2008, 14, 8830–8836 CrossRef CAS PubMed.
- N. Issa, P. G. Karamertzanis, G. W. A. Welch and S. L. Price, Cryst. Growth Des., 2008, 9, 442–453 Search PubMed.
- C. W. Lehmann and F. Stowasser, Chem.–Eur. J., 2007, 13, 2908–2911 CrossRef CAS PubMed.
- M. Habgood, Cryst. Growth Des., 2011, 11, 3600–3608 CAS.
- T. Friščić and L. R. MacGillivray, Chem. Commun., 2009, 773–775 RSC.
- G. A. Stephenson, J. Kendrick, C. Wolfangel and F. J. J. Leusen, Cryst. Growth Des., 2012, 12, 3964–3976 CAS.
- While cocrystal seeds isomorphous to 1 are expected to provide a good epitaxial match, non-isomorphous cocrystals might be suited as seed for 1 owing to a sufficient epitaxial match (see ref. 67).
- C. A. Mitchell, L. Yu and M. D. Ward, J. Am. Chem. Soc., 2001, 123, 10830–10839 CrossRef CAS PubMed.
- A. Bondi, J. Phys. Chem., 1964, 68, 441–451 CrossRef CAS.
- R. Dubey, M. S. Pavan and G. R. Desiraju, Chem. Commun., 2012, 48, 9020–9022 RSC.
- Details related to this series of cocrystals will be reported at a later date.
- J. Sitkowski, L. Stefaniak, L. Nicol, M. L. Martin, G. J. Martin and G. A. Webb, Spectrochim. Acta, Part A, 1995, 51, 839–841 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available: Details regarding computational studies, cocrystal-screening experiments, as well as crystallographic, thermal and spectroscopic analyses. CCDC 898406–898412. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c3sc51419f |
| This journal is © The Royal Society of Chemistry 2013 |