DOI:
10.1039/C2SM26945G
(Paper)
Soft Matter, 2013,
9, 138-145
Effect of regeneration of liquid silk fibroin on its structure and characterization
Received
22nd August 2012
, Accepted 25th September 2012
First published on 15th October 2012
Abstract
Silk derived from cocoons of the female silkworm (Bombyx mori) was made into three kinds of test material, degummed fiber, liquid fibroin and regenerated silk fibroin membrane (RSFM). The effect of the silk degumming and fiber dissolution system (SD–FDS) on the molecular structure and properties of silk fibroin were studied in detail. Degumming with Na2CO3 solution had the greatest impact on silk fibroin fiber, resulting in significant reduction of the thermal stability and crystallinity, and the fiber tensile property was only half of that degummed with urea buffer. The moderate degumming solution was urea buffer, followed by strongly alkaline electrolyzed water (SAEW). SDS-PAGE showed that the silk fibroin degummed with 8.0 M urea at 80 °C and dissolved in 9.3 M LiBr at 25 °C was similar to the natural silk fibroin in vivo, but boiling in Na2CO3 solution leads to serious breakage of the silk fibroin peptide chain. The degree of breakage of the peptide chain is positively correlated with the storage time of liquid silk fibroin. DSC showed the more nearly intact the silk fibroin peptide chain, the higher the thermal decomposition temperature. The regenerated condition of the silk protein is often overlooked during the research process, while our study showed that different silk degumming and fiber dissolution systems will have different influences. The results of this test help us gain a deeper understanding of the effect of silk degumming and fiber dissolution on the molecular structure and properties of silk fibroin and provide important experimental data for screening and using silk protein as advanced functional biomaterials, such as medical tissue engineering materials.
Silk, a well-known natural fiber produced by the silkworm (Bombyx mori), is composed of two kinds of protein; fibroin forms the thread core and the glue-like sericin surrounds the fibroin fibers and cements them together. Fibroin comprises both heavy and light chains and has an amorphous region of about one-third and a crystalline portion of about two-thirds,1 in which there are two crystalline forms, silk I and silk II, reported as the dimorphs of silk fibroin, on the basis of extensive investigations by a variety of analytical methods. Silk II, the structure of the fiber after spinning, is mainly an anti-parallel β-sheet.2–5 Silk I is the conformation of fibroin in the solid state before spinning; conformations similar to Silk I are obtained as a film from the liquid silk in the silk gland or from an aqueous solution of regenerated fibroin.6 The physical and mechanical properties of regenerated silk fibroin depend on the conformation of the molecular chain and the crystal structure.7 In the traditional process of making textile fibers or medical biomaterials, sericin is removed partially or completely to make silk textiles or further processed into regenerated silk fibroin biomaterials,8–10 such as medical tissue engineering materials.11 Silkworm silk fibers are influenced by processing conditions. Many environmental factors such as pH and salts and the concentration of silk protein are quantitatively correlated to the properties and stabilization of silk fibroin solution.12,13 It is very valuable to gain a window of protection against premature crystallization of the fibroin during processing, assuring safe storage, transport, and successful fiber spinning. Silk degumming and fiber dissolution induce breakage of the peptide chain of regenerated silk fibroin to various degrees, and they then directly affect the structure and properties of various silk biomaterials. Sericin, a water-soluble, protein-based polymer coated around the silk fibroin fiber, has three main components of different solubilities and β-sheet content in the cocoon shell sericin of the silkworm.14 According to its solubility in water at different temperatures and under different pressures, sericin is divided into the outer, middle and inner sericin layers. Furthermore, the differences in amino acid composition among the three layers are very significant.15 Boiling in water removes only sericin in the outermost layer of the silk fiber. Reported degumming methods, such as the use of Na2CO3 solution, neutral soap-alkali and highly concentrated urea buffer,16 focus on degumming efficiency and the features of sericin peptide and hydrolyzates. Degumming with acid, alkali, protease and microwave might impact the surface characteristics and mechanical properties of silk fibroin,17–20 and our latest research indicated that strong alkaline electrolyzed water (SAEW) was a good swelling and degumming solution for silk fiber.21 Silk is a protein-based fiber with a crystalline structure, which can be dissolved only in some highly concentrated or mixed salts, such as LiSCN,22 LiBr, LiBr–MeOH–H2O23 and the CaCl2–EtOH–H2O ternary system (CaCl2 TS);24 9.3 M LiBr and CaCl2 TS are the most common dissolution systems. Ordinarily, the degummed silk fiber is dissolved in 9.3 M LiBr (≥50 °C) or CaCl2 TS (≥70 °C). The length of peptide chains and the stability of the resulting liquid silk fibroin are very different, which has been studied from the perspective of fluidics and infrared spectroscopy.25 Post-processing methods affect the secondary structure of the regenerated silk fibroin nanocomposites.26 Therefore, degumming and dissolving techniques are particularly important with regard to molecular mass, structure and features of regenerated liquid silk and even to the resulting 3D fibroin scaffold as tissue engineering materials. However, there are few reports in the literature of systematic studies. This work is focused on the impact of silk degumming and fiber dissolving on the structure and characteristics of silk fibroin fiber, regenerated liquid silk and the resulting fibroin membrane.
1. Materials and methods
1.1. Materials
B. mori cocoons were provided by the sericulture Institute of Soochow University, Suzhou, P. R. China. The male cocoon shows yellow fluorescence and the female cocoon shows purple fluorescence under UV irradiation.27 Female cocoons chosen for this work to reduce error of fiber size between female and male cocoons were air-dried at 80 °C for 5 h.
1.2. Silk degumming
Three degumming methods were tested. Degumming by urea buffer was done by heating in 30 times (v/w) of 8 M aqueous urea containing 0.04 M Tris–SO4 (pH 7) and 0.5 M mercaptoethanol for 2 h at 80 °C.28 (1) Degumming with Na2CO3 solution, the B. mori cocoon shell was degummed twice in boiling 0.2% (w/v) Na2CO3 for 0.5 h.29 (2) The SAEW degumming method was carried out in 25 times (v/w) of SAEW (pH 11.50) for 30 min at 100 °C.21 (3) After these degumming treatments, the resulting silk materials were washed repeatedly in deionized water and then air-dried.
1.3. Dissolution of silk fibroin fiber
Method 1 (LiBr): the degummed silk fibers were added to 20 times (v/w) of 9.3 M LiBr and incubated with shaking at 25 °C until dissolved. Method 2 (Ajisawa's method): the degummed silk fibers were added to 20 times (v/w) of Ajisawa's reagent (CaCl2/ethanol/water, 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2:8 molar ratio) and stirred at 70 °C to form a clear solution.
:2:8 molar ratio) and stirred at 70 °C to form a clear solution.
Three silk degumming and two fibroin fiber dissolution methods were used. These systems of silk degumming and fiber dissolution are given in Table 1.
Table 1 The degumming–dissolution system (DDS) of silk fiber
| Treated solution |
Silk degumming–fibroin dissolution system (SD–FDS) |
| 1 |
2 |
3 |
4 |
| Degumming solution |
8.0 M urea |
pH 11.5 SAEW |
0.2% Na2CO3 |
0.2% Na2CO3 |
| Dissolving solution |
9.3 M LiBr |
9.3 M LiBr |
9.3 M LiBr |
CaCl2 TS |
| Abbreviated SD–FDS system |
Urea–LiBr |
SAEW–LiBr |
Na2CO3–LiBr |
Na2CO3–CaCl2 TS |
After the silk fibroin/salt solution was filtered through deionized water or centrifuged at 8000 rpm for 10 min, the filtered solution or supernatant was dialyzed continuously for 48 h with running water to remove CaCl2, smaller molecules and some impurities using a cellulose semi-permeable membrane (cutoff 5 kDa). Four aqueous solutions of silk fibroin were used for preparation of the regenerated silk fibroin membrane (RSFM).
1.4. Preparation of RSFM
The silk fibroin solution was dropped onto a PVC plate with a pipette and then air-dried at 25 °C. According to the experimental requirements, part of the silk membrane was soaked in 80% (v/v) methanol for 30 s and then drained on paper towels.
1.5. SDS-PAGE
The molecular mass range of regenerated silk fibroin was determined by sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE).30,31
1.6. Structure analysis of silk fibroin fiber and RSFM
Thermal analysis of the silk fibroin fiber and RSFM was carried out with a differential scanning calorimeter (DSC) instrument (DSC 2010, TA Instruments) at a heating rate of 20 °C min−1 in the temperature range 50–350 °C. The mechanical properties of degummed silk fibroin fiber were measured with a universal testing machine (3365 VNCLSYJ-001, INSTRON). X-ray diffraction analysis of the silk fibroin fiber and SFM was carried out with an X-ray diffractometer (X'Pert-Pro MPD, PANalytical) recording spectra in the angle range 5–40° (2θ). FTIR spectra of the silk films were obtained using an FTIR spectrometer (ProStar LC240; Jasco UK) in the spectral region 4000–400 cm−1 to determine the molecular conformation of silk fibroin. Samples were cut into 3 × 3 cm2 squares and the thickness of the samples was about 10 μm. Samples were peeled from the PVC plate using tweezers and were fixed to the iron window with circinate magnets. For each measurement, scanning was repeated 32 times with a resolution of 4 cm−1. Five-point smoothing was used for the IR spectroscopy. A Microscopic Confocal Raman Spectrometer (LabRAM HR800, Horiba Jobin Yvon), equipped with a LMPLFL optical microscope (50× objective with an aperture of 400 μm) was used to analyze the surface texture of silk films in the wavelength range 400–4000 cm−1 at a resolution of 4 cm−1. The wavelength of the laser was 633 nm and the samples have been irradiated for 20 s during spectra recording. Adjacent Averaging ten-point smoothing was used for Raman spectroscopy.
2. Results and discussion
2.1. Thermal behavior of degummed silk fibroin fiber
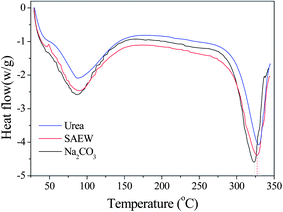
Fig. 1 shows the DSC curves of silk fibroin for three degumming methods. There is no obvious glass transition temperature in DSC curves, which means that the different silk degumming treatments had no significant impact on the structure of the fiber. However, these degumming methods had a marked impact on the thermal decomposition temperature. The DSC curve for 8.0 M urea showed an endothermic peak at 329.30 °C, which was attributed to thermal degradation of the β sheet material, and the peak for SAEW was at 326.75 °C, whereas the peak for Na2CO3 was shifted to the lower temperature of 322.96 °C. These results indicate that the degumming methods appear to be related to the decrease of thermal stability of silk fibroin. Urea had the smallest impact on fiber thermal stability and Na2CO3 had the greatest.
 |
| | Fig. 1 The impact of three degumming methods on the thermal properties of silk fibroin fiber: (blue) urea buffer, (red) SAEW, (black) Na2CO3. | |
2.2. Effect of degumming solution on the mechanical properties of silk fibroin fiber
Table 2 shows the effect of degumming solutions on the tensile performance of silk fibroin fiber. The mechanical properties of silk fibroin degummed with 8 M urea were slightly better than those after degumming with SAEW and much better than those after degumming with 0.2% Na2CO3. The maximum load of silk fibroin degummed with 8 M urea buffer was 5.83 cN, which was slightly higher than the value for fibers degummed with SAEW. However, the maximum load of silk fibroin degummed with Na2CO3 was only 63% of that after degumming with 8 M urea and the fracture stress was only 3.70 cN, similar to the displacement at the maximum load. The fracture energy of the silk fibroin fiber differed greatly among the different degumming agents in the order:
Table 2 The impact of the degumming methods on the mechanical properties of silk fibera
| SD–FDSs |
Maximum load (cN) |
Displacement at the max. load (mm) |
Energy at the max. load (kJ) |
Tensile strain at the max. load (%) |
|
Values are mean (±SD).
|
| Urea |
5.83 |
(0.76) |
4.30 |
(0.17) |
173.92 |
(29.96) |
45.11 |
(1.74) |
| SAEW |
5.40 |
(1.12) |
3.96 |
(0.37) |
152.67 |
(38.48) |
41.42 |
(4.22) |
| Na2CO3 |
3.70 |
(0.74) |
2.42 |
(0.47) |
66.66 |
(24.83) |
24.88 |
(4.95) |
Similar to the previous two indices, the tensile strain at the maximum load of fiber degummed with 8 M urea buffer was 45%, with SAEW was 41%, and it was reduced by almost half with Na2CO3 at <25%. These results suggest strongly that degumming with 8 M urea buffer at 80 °C for 2 h yields a silk fibroin fiber with good mechanical properties. Degumming with SAEW for 30 min also produced a very good silk fiber. Na2CO3, however, seriously damaged silk fibroin and markedly reduced the fracture stress, fracture energy and tensile strain of the fibers by ∼37%, ∼62% and ∼20%, respectively. Therefore, we conclude that the fiber mechanical properties decrease in the order:
2.3. Effect of degumming solvent on the conformation and crystalline structure of silk fibroin fiber
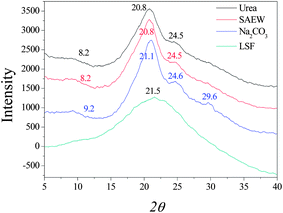
Naturally spun silk fibers are semi-crystalline, with β-sheet crystals embedded in a less ordered matrix. Dissolution and reconstitution of silk fibroin yields materials with a variety of structures, which are dependent upon the solvents employed and the processing procedure. X-ray diffraction peaks appear at 12.2°, 19.7°, 24.7° and 28.2° for silk I and at 9.1°, 18.9° and 20.7° for silk II.32 The three curves of X-ray diffraction (XRD) were similar to each other and it can be seen in Fig. 2 that the degumming methods have little influence on the crystalline structure of silk fibroin fiber. Silk fibroin fiber exists mainly in the form of silk I and silk II, which is very different from the lyophilized liquid silk fibroin regenerated with 8 M urea buffer. The XRD curve of it had a very broad peak that was a scatter peak at the top, 2θ = 21.5°. This is a characteristically typical diffraction pattern of amorphous silk fibroin. However, there are still fine distinctions among the degumming methods. The fibroin fiber degummed with Na2CO3 showed little swerve at 9.2° and 21.1°, which correspond to silk II, but had an obvious peak at 29.6°, which was likely owing to the severe damage caused by degumming with Na2CO3. The results of XRD analysis are similar to the conclusion given above, in showing that degumming with Na2CO3 caused the most severe damage to the silk fibroin structure, and the content of silk II was less than that of silk I.
 |
| | Fig. 2 X-ray diffraction curves of silk fibroin. LSF: lyophilized liquid silk fibroin. | |
2.4. Effect of SD–FDSs on the peptide chains of regenerated silk fibroin
It is not difficult to see that the four kinds of SD–FDSs have an influence on the distribution and molecular mass of regenerated liquid silk fibroin (Fig. 3). As the procedure of preparing solubilized fibroin from cocoons requires many steps, we asked which step(s) damage the fibroin molecule. First, the solvent for silk fiber dissolution was the same in the three systems urea–LiBr, SAEW–LiBr and Na2CO3–LiBr, thereby allowing us to compare the results of the different degumming solutions. Evidently, the molecular mass range of these regenerated liquid fibroins with the same degumming solution (9.3 M LiBr) was elongated in the order:
| SD–FDS 1 < SD–FDS 2 < SD–FDS 3 |
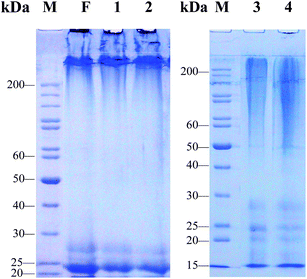 |
| | Fig. 3 SDS-PAGE analysis of regenerated liquid silk fibroins prepared with different SD–FDSs. F: intact silk fibroin in vivo, (1) urea–LiBr, (2) SAEW–LiBr, (3) Na2CO3–LiBr, (4) Na2CO3–CaCl2 TS, M: standard gradient marker. | |
The urea–LiBr system had the narrowest molecular mass distribution and clear bands at 25 kDa (L-chain) and 27 kDa (P25) (Fig. 3, lane 1). The H-chain of the regenerated liquid silk was degraded only slightly, and its distribution range was >200 kDa. These protein bands are very similar to those of the natural silk fibroin in the silk gland of the fifth instar silkworm (Fig. 3, lane F). Like the urea–LiBr system, the SAEW–LiBr system (SD–FDS 2) had a clear band at 25 kDa and another at 27 kDa and a continuous band of H-chain >200 kDa, but its silk peptide H-chain was damaged only slightly (Fig. 3, lane 2), showing that SAEW is also a good degumming solution. The Na2CO3–LiB system (SD–FDS 3) had a broad smear at 50–200 kDa, which indicated that degumming with Na2CO3 causes serious damage to the peptide chains of silk fibroin (Fig. 3, lane 3). Similarly, the regenerated silk fibroin peptide strand was seriously degraded in SD–FDS 4 (Na2CO3–CaCl2 TS; Fig. 3, lane 4) and the distribution range of molecular mass was >45 kDa, indicating that serious breakage of the silk fibroin peptide was caused by boiling in 0.2% (w/v) Na2CO3 solution, and dissolution in CaCl2 TS at 70 °C resulted in slight degradation of the degummed fiber peptide chain. These results showed that intact silk fibroin could be obtained by degumming with 8.0 M urea at 80 °C and dissolving in 9.3 M LiBr at 25 °C. Dissolving in CaCl2 TS at 70 °C did not cause serious damage to the protein. Boiling in SAEW had little effect on silk fibroin, whereas Na2CO3 caused serious degradation of the fibroin heavy chain.
2.5. The storage time of liquid silk fibroin
Table 3 shows images and related values that illustrate the relative situations for four silk fibroin salt solutions and regenerated liquid silk. The mixed solution derived from the urea–LiBr system was separated into an upper and a lower section. The upper layer was transparent and the lower layer was ivory. The other three solutions were very similar and retained a homogeneous turbidity. After dialysis, the fibroin salt solution became transparent liquid silk owing to the removal of salt from the mixed solution. There was no marked difference during the SD–FDSs of urea–LiBr, SAEW–LiBr and Na2CO3–LiBr, which were all nearly transparent, while it was distinctly ivory in Na2CO3–CaCl2 TS, indicating that liquid silk fibroin regenerated from the four different SD–FDSs had significant differences in structure. Yamada et al. reported that the intact fibroin solution was unstable which showed a rapid increase in viscosity and subsequently converted to a gel. The deionized fibroin solution gelled within 24 h and it can be dialyzed against water just before use, if necessary.28 The storage time of these regenerated silk fibroin solutions given in Table 3 showed that storage time increased from SD–FDS 1 to SD–FDS 4 at 4 °C and at 25 °C. The intact liquid silk generated from the urea–LiBr system is stable (and still existed in liquid form) for 3 days at 25 °C and for 13 days at 4 °C. The other three silk fibroin solutions can be stored for a longer time, indicating that the degree of integrity is closely related to the stability of the silk solution. As the degree of hydrolysis of the silk fibroin peptide chain is increased, the storage time increased gradually, which we suggest is due to hydrolytic cleavage of hydrophobic segments of the backbone of the protein known to form β-sheets.
Table 3 Liquid state before and after dialysis of silk fibroin and salt solution and stabilization of regenerated fibroin solution at 4 °C and 25 °Ca
| |
SD–FDSs |
| 1 |
2 |
3 |
4 |
|
(1) Urea–LiBr system; (2) SAEW–LiBr system; (3) Na2CO3–LiBr; (4) Na2CO3–TS system; ST: semi-transparent.
|
| Dialysis of silk fibroin–salt mixed solution |
Before |

|

|

|

|
| Layering |
Turbid |
Turbid |
Turbid |
| |

|

|

|

|
| Liquid silk |
20 mg mL−1 |
| Transparent |
ST |
| Storage time (days) |
4 °C |
13 |
≥45 |
≥45 |
≥45 |
| 25 °C |
3 |
12 |
16 |
≥45 |
2.6. Spectral characteristics of the RSFMs
Earlier studies on the structure of silk fibroin revealed two different structural models; silk I, known to be rich in helical structures, and silk II, known to be rich in β-sheets.31 The infrared absorption peaks of β-sheets in amides I, II and III are observed at 1625–1640, 1515–1525 and 1265 cm−1, respectively; those of α-helices occur at 1650–1658, 1545, and 1240 cm−1, respectively; and those of random coils occur at 1640–1648, 1535–1545 and 1235 cm−1, respectively.32,33 We found that the films as cast from aqueous solution had similar spectra in amides I, II and III, with bands at 1654 cm−1, 1540 cm−1 and 1242 cm−1, respectively, indicating that the four kinds of RSFM are predominantly a mixture of random coils, as shown in Fig. 4a. However nuances could be seen among the four samples, and the infrared absorption peak of random coils in amides I occur at 1651, 1654, 1655 and 1657 cm−1, respectively. The single peaks of the amide I band in the membrane cast from SD–FDS 1–4 increased gradually. Chen et al.25 also found that the secondary structure of the silk fibroin in the membrane cast from LiBr–H2O was slightly different from those prepared using the other solvents, such as LiBr–EtOH and Ca(NO3)2–MeOH–H2O. However, our results showed that the secondary structure did not have an evident bearing on the size of the molecular mass or the different regenerated methods. Raman spectroscopic studies supported this, and we observed a strong peak centered at 1660 cm−1, which is characteristic of silk in a random coil conformation,34 and interestingly we observed a shoulder at 1663 cm−1, which is evidence of a certain amount of β-sheet structure (see Fig. 4b), which is not completely consistent with the IR results mentioned above. This may be due to the fact that the surface structure of silk fibroin films is easier to influence by environmental conditions during the drying process. After we immersed the films in methanol, the characteristic infrared absorption bands of amides I, II, III and IV of all samples show band shifts indicative of the adoption of a silk II structure due to the formation of β-sheets (see Fig. 4c), as characterized by the amide I and amide II bands at 1627.9 cm−1 and 1531.5 cm−1. There is no significant difference in the characteristic absorption bands among different RSFMs. Therefore, it can be said that the degree of degradation has little difference on the FT-IR and Raman spectroscopy of silk fibroin. It seems that the large molecular weight proteins were easier to form into β-sheets and that the content of the silk fibroin β-sheet is increased following immersion in methanol.
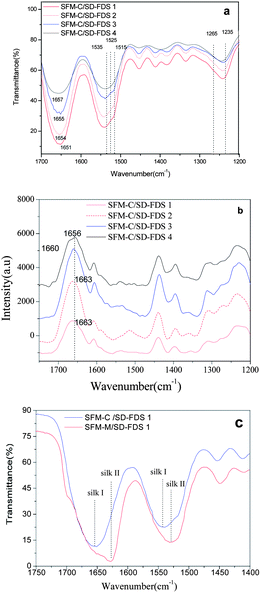 |
| | Fig. 4 (a) FTIR spectra for the films, as cast from different aqueous solutions; (b) Raman spectra for the films, as cast from different aqueous solutions; (c) FTIR spectra for the films before and after MOH immersion. | |
2.7. DSC analysis of the RSFMs
Section 2.1 (above) shows that the thermal decomposition temperature of silk fibroin degummed with 8 M urea at 80 °C for 2 h was 329.30 °C. Fig. 5 shows DSC curves of RSFM derived from the urea–LiBr system. Before immersion in methanol (SFM-C), the dehydration peak was at 75 °C, which was raised to 110 °C after treatment with methanol (SFM-M). Treatment with methanol also changed the glass transition temperature. SFM-C had a wide transition peak at 190 °C and SFM-M had a markedly increased temperature. The exothermic peak at 235.82 °C (SFM-C) indicated that α-helix molecules had strenuous exercise and the random coil was changed to a β-sheet structure35 but, because the SFM-M had already changed into the β-sheet conformation, there was no exothermic peak. SFM-M showed an endothermic peak at 292.4 °C and SFM-C showed an endothermic peak at 293.4 °C, indicating that the change in β-sheet structure improved the thermal decomposition temperature. Compared to the silk fiber degummed with 8 M urea, the RSFM, whether or not it was impregnated with methanol, created a drop in temperature of 23–24 °C. Thermal stability decreased remarkably after the degumming, dissolution and regeneration processes.
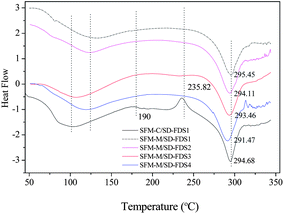 |
| | Fig. 5 DSC curves of RSFMs derived from SD–FDSs. SFM-C, control; SFM-M, treatment with methanol. | |
Fig. 5 shows the DSC curves of RSFMs immersed in methanol; the RSFMs had significantly different thermal decomposition temperatures. The temperature decreased in the order:
| SD–FDS 1 > SD–FDS 2 > SD–FDS 3 > SD–FDS 4 |
indicating that the thermal decomposition temperature and the degree of fracture were closely linked. The
thermal decomposition temperature decreased as the degree of completeness decreased, which is consistent with the thermal behavior of silk fibroin discussed above.
2.8. XRD analysis of the RSFMs
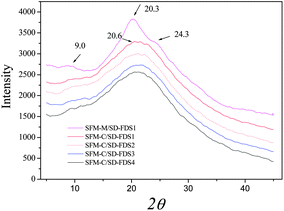
The XRD diffraction peaks for silk II appear at 9.1°, 18.9° and 20.7°, among which 20.7° is the most important. The RSFM had a broad peak at 20.5° (Fig. 6), which was attributed to the randomly coiled structure. After treatment with methanol, it had a strong peak at 20.3° and minor peaks at 9.0° and 24.3°, known characteristic peaks of a β-sheet crystalline structure, illustrating that when the RSFM is dipped in methanol, the characteristic diffraction peaks of a β-sheet crystalline structure occur and the crystallinity is improved significantly. Fig. 6 shows the XRD curves of RSFM obtained from SD–FDSs. The results show that the XRD diffraction peaks of different kinds of RSFM were very similar and existed mainly in the form of a random coil structure, indicating that the degree of degradation had little influence on the XRD diffraction peaks of silk fibroin.
3. Conclusion
This study is the first report to study the comprehensive effect of a silk degumming and fiber dissolution system on the molecular structure and properties of silk fibroin. The thermal behavior, mechanical properties and XRD analysis of silk fibroin showed that degumming in 8.0 M urea had the smallest effect on silk fibroin, then SAEW, while Na2CO3 had the greatest impact on the thermal properties of silk fibroin; the thermal decomposition temperature decreased by 6.34 °C. The mechanical properties and XRD analysis were basically the same with DSC analysis; boiling in Na2CO3 caused a serious fracture of the silk fibroin peptide chains, leading to a sharp decline in the mechanical properties of the fiber. Degumming for 2 h with 8 M urea at 80 °C and dissolving in LiBr at 25 °C could yield intact silk fibroin. SC caused significant degradation of the heavy chain of silk fibroin, and the regenerated silk fibroin peptide strand was seriously degraded. The degree of fracture of the silk peptide chains was positively correlated with the storage time of the liquid silk fibroin. Storage was better at 4 °C than it was at 25 °C. The degree of degradation has little influence on the FT-IR and Raman spectroscopy of silk fibroin, and the content of the silk fibroin β-sheet is increased following immersion in methanol. The thermal decomposition temperature was closely linked to the degree of fracture and decreased as the degree of completeness decreased. XRD diffraction peaks of different RSFMs were very similar, existing mainly as a random coil structure. These results suggest that different SD–FDSs will bring different influences to silk protein. It is very important to choose or unify the silk degumming and fiber dissolution systems during silk experiments.
Acknowledgements
The authors gratefully acknowledge the earmarked fund for China Agriculture Research System (CARS) and a project funded by the Priority Academic Program Development of Jiangsu Higher Education Institutions, P. R. China.
References
- K. Mita, S. Ichimura and T. C. James, Highly repetitive structure and its organization of the silk fibroin gene, J. Mol. Evol., 1994, 38, 583–592 CAS.
- R. E. Marsh, R. B. Corey and L. Pauling, An investigation of the structure of silk fibroin, Biochim. Biophys. Acta, 1955, 16, 1–34 CrossRef CAS.
-
R. D. B. Fraser, T. P. MacRae, Conformations of Fibrous Proteins and Related Synthetic Polypeptides, Academic Press, New York, 1973 Search PubMed.
- Y. Takahashi, M. Gehoh and K. Yuzuriha, Structure refinement and diffuse streak scattering of silk (Bombyx mori), Int. J. Biol. Macromol., 1999, 24, 127–138 CrossRef CAS.
- B. Lotz and F. C. Cesari, The chemical structure and the crystalline structures of Bombyx mori, Biochimie, 1979, 61, 205–214 CrossRef CAS.
- T. Asakura, K. Suita, T. Kameda, S. Afonin and A. S. Ulrich, Structural role of tyrosine in Bombyx mori silk fibroin, studied by solid-state NMR and molecular mechanics on a model peptide prepared as silk I and II, Magn. Reson. Chem., 2004, 42, 258–266 Search PubMed.
- D. J. Wen, H. Wang, X. S. Zhu and J. P. Sun, Conformation and crystallinity of silk fibroin, Acta Sericol. Sin., 2005, 26(1), 110–112 Search PubMed.
- Y. Q. Zhang, Natural silk fibroin as a support for enzyme immobilization, Biotechnol. Adv., 1998, 16, 961–971 CrossRef CAS.
- C. Vepari and D. L. Kaplan, Silk as a biomaterial, Prog. Polym. Sci., 2007, 32, 991–1007 CrossRef CAS.
- Y. Wang, H.-j. Kim, G. Vunjak-novakovic and D. L. Kaplan, Stem cell-based tissue engineering with silk biomaterials, Biomaterials, 2006, 27, 6064–6082 CrossRef.
- R. Nazarov, H.-j. Jin and D. L. Kaplan, Porous 3-D scaffolds from regenerated silk fibroin, Biomacromolecules, 2004, 5, 718–726 CrossRef CAS.
- A. Matsumoto, A. Lindsay, B. Abedian and D. L. Kaplan, Silk fibroin solution properties related to assembly and structure, Macromol. Biosci., 2008, 8, 1006–1018 Search PubMed.
- F. G. Omenetto and D. L. Kaplan, New opportunities for an ancient material, Science, 2010, 329(5991), 528–531 CrossRef CAS.
- Y. Takasu, H. Yamada and K. Tsubouchi, The silk sericin component with low crystallinity, Sanshi-Konchu Biotec, 2006, 75(2), 133–139 Search PubMed.
- Y. J. Wang and Y. Q. Zhang, Three-layered sericins around the silk fibroin fiber from Bombyx mori cocoon and their amino acid composition, Adv. Mater. Res., 2011, 175–176, 158–163 Search PubMed.
- Y. Q. Zhang, Comparison analysis of degumming procedures for silkworm silk, Acta Sericol. Sin., 1997, 28(1), 75–79 Search PubMed.
- M. Majibur, R. Khana, M. Tsukada, Y. Gotohb and H. Morikawab,
et al. Physical properties and dyeability of silk fibers degummed with citric acid, Bioresour. Technol., 2010, 101, 8439–8445 Search PubMed.
- R. H. Walters and O. A. Hougen, Silk degumming I. Degradation of silk sericin by alkalies, Text. Res. J., 1934, 5, 92 Search PubMed.
- G. Freddi, R. Mossotti and R. Innocenti, Degumming of silk fabric with several proteases, J. Biotechnol., 2003, 106, 101–112 Search PubMed.
- K. Haggag, H. E. Sayed and O. G. Allam, Degumming of silk using microwave-assisted treatments, Journal of Natural Fibers, 2007, 4(3) Search PubMed.
-
Y. Q. Zhang, Y. J. Wang, M. H. Wu, H. Y. Wang, L. Zhu, A silk reeling method in strong alkaline electrolyzed water for low temperature, China Patent. 201210082082.X, 2012-03-26.
- Y. Shimizu, The swelling and dissolution of silk with organic solvents, J. Seric. Sci. Jpn., 1978, 47, 417–420 Search PubMed.
-
K. Matsumoto and H. Uejimaet al., New solvent for silk fibroin, Japan Patent, JP7173192, 1995.
- A. Ajisawa, Studies on the dissolution of silk fibroin by CaCl2–H2O–R(OH)n, ternary system solutions, Sen'i Gakkaishi, 1968, 24, 61–64 Search PubMed.
- X. Chen, P. K. David and Z. Z. Shao,
et al., Regenerated Bombyx silk solutions studied with rheometry and FTIR, Polymer, 2001, 42(25), 9969–9974 CrossRef CAS.
- B. D. Lawrence, F. Omenetto, K. Chui and D. L. Kaplan, Processing methods to control silk fibroin film biomaterial features, J. Mater. Sci., 2008, 43, 6967–6985 CrossRef CAS.
- Y. Q. Zhang, X. H. Yu, W. D. Shen, Y. L. Ma and L. X. Zhou,
et a1. Mechanism of fluorescent cocoon sex identification for silkworms Bombyx mori, Sci. China, Ser. C: Life Sci., 2010, 53(11), 1330–1339 Search PubMed.
- H. Yamada, H. Nakao and Y. Takasu,
et al. Preparation of undegraded native molecular fibroin solution from silkworm cocoons, Mater. Sci. Eng., C, 2001, 14, 41–46 CrossRef.
- Y. Q. Zhang, M. L. Tao, W. D. Shen, Y. Z. Zhou and Y. Ding,
et a1. Immobilization of L-asparaginase on the microparticles of the natural silk sericin protein and its characters, Biomaterials, 2004, 25(17), 3751–3759 CrossRef.
- U. K. Laemmli, Cleavage of structural proteins during the assembly of the head of bacteriophage T4, Nature, 1970, 227, 680–685 CAS.
- Y. Q. Zhang, W. L. Zhou, W. D. Shen, Y. H. Chen and X. M. Zha,
et al. Synthesis, characterization and immunogenicity of silk fibroin-L-asparaginase bioconjugates, J. Biotechnol., 2005, 120(3), 315–326 CrossRef CAS.
- H. Y. Wu, Z. M. Jing and L. Q. Xu, Studies on the changes of crystallinity and structure of silk fibroin, Acta Sericol. Sin., 1993, 19(2), 105–110 Search PubMed.
- X. Chen, Z. Z. Shao, N. S. Marinkovic, L. M. Millerd and P. Zhou,
et al. Conformation transition kinetics of regenerated Bombyx mori silk fibroin membrane monitored by time-resolved FTIR spectroscopy, Biophys. Chem., 2001, 89, 25–34 CrossRef CAS.
- K. A. Karve, E. S. Gil, S. P. McCarthy and D. L. Kaplan, Effect of β-sheet crystalline content on mass transfer in silk film, J. Membr. Sci., 2011, 383, 44–49 CrossRef.
- Y. Q. Zhang, W. D. Shen, R. L. Xiang, L. J. Zhuge, W. J. Gao and W. B. Wang, Formation of silk fibroin nanoparticles in water-miscible organic solvent and their characterization, J. Nanopart. Res., 2007, 9, 885–900 CrossRef CAS.
|
| This journal is © The Royal Society of Chemistry 2013 |
Click here to see how this site uses Cookies. View our privacy policy here.