Multifunctionality in metal@microgel colloidal nanocomposites
Jorge
Pérez-Juste
*a,
Isabel
Pastoriza-Santos
a and
Luis M.
Liz-Marzán
abc
aDepartamento de Química Física, Universidade de Vigo, 36310 Vigo, Spain. E-mail: juste@uvigo.es
bCentre for Cooperative Research in Biomaterials (CIC biomaGUNE), Paseo de Miramón 182, 20009 San Sebastián, Spain
cIkerbasque, Basque Foundation for Science, 48011 Bilbao, Spain
First published on 29th August 2012
Abstract
The integration of metal nanoparticles within crosslinked polymeric microgels, in a well-defined core–shell structure, offers unique possibilities in various fields due to the potential to introduce multiple functionalities by tailoring the properties of the inorganic and the organic components. This Highlight describes recent developments related to hybrid nanocomposites comprising a metal core and a smart microgel shell. Different synthesis approaches for the preparation of such hybrid systems are described, including not only surface-initiated polymerization, but also in situ chemical reactions for core optimization and functionality addition. The main properties derived from the multifunctionality of these nanocomposites are also addressed and potential applications in the fields of catalysis and sensing are presented.
 Jorge Pérez-Juste | Jorge Pérez-Juste obtained his Chemistry degree from the Universidade de Santiago de Compostela in 1995 and his PhD degree in Chemistry from the Universidade de Vigo in 1999. He worked as a postdoctoral fellow at UCSC in 2000 and at University of Melbourne (Australia) in 2002 and 2003. Since 2009, he has been an Associate Professor at the University of Vigo. His current interests include the synthesis and assembly of nanoparticles and nanostructured materials with tailored optical and sensing properties. |
 Isabel Pastoriza-Santos | Isabel Pastoriza-Santos obtained her PhD degree from the Universidade de Vigo in 2001. She worked as a postdoctoral fellow at University of Melbourne (Australia) in 2002 and 2003. Since 2009, she has been an Associate Professor at the University of Vigo. She has co-authored over 80 articles and holds 2 patents. Her current interests include the synthesis of plasmonic nanoparticles with size and shape control, as well as their assembly. |
 Luis M. Liz-Marzán | Luis M. Liz-Marzán is a PhD from the University of Santiago de Compostela (1992) and has been a postdoc at Utrecht University and (more recently) a visiting professor at Tohoku University, Michigan, Melbourne, Hamburg and Max-Planck Institute Golm. He has been a Professor in Physical Chemistry at the University of Vigo (1995–2012), and he is currently an Ikerbasque Research Professor and Scientific Director of CIC biomaGUNE in San Sebastián. He is co-author of over 250 publications and 5 patents, and has received several research awards. His current interests include nanoparticle synthesis and assembly, nanoplasmonics, and development of nanoparticle-based sensing and diagnostic tools. |
Introduction
Novel materials are essential for technological progress. While in the past organic and inorganic materials attracted interest in separate disciplines, today the combination of different components leads to hybrid materials displaying interesting properties. More precisely, a variety of core–shell nanocomposites comprising inorganic cores and organic shells or vice versa have been reported.1–3 Within the inorganic part, metal nanoparticles are a popular choice, since a large number of available synthetic protocols allow not only a suitable control over particle size, shape and composition,4,5 but also a wide range of fascinating properties and applications in mature fields such as catalysis or electronics,6,7 and in emerging areas such as photonics, sensing, imaging and medicine.8,9 Regarding the organic component, the possibility of using a wide variety of polymers with different functionalities renders them ideal candidates, in particular for the case of crosslinked polymers. Water-based polymer matrices are called hydrogels when they form macroscopic networks, but when they are confined to smaller dimensions, typically in the submicron size range, they are called microgels or nanogels. Several methods have been explored for the preparation of microgels, including the free radical polymerization of water-soluble or hydrophilic monomers in the presence of either bifunctional or multifunctional cross-linkers.10 Interestingly, the composition of different components in the crosslinked polymer strongly influences its responsiveness toward external stimuli such as temperature, pH, and ionic strength, which ultimately determines the final applications.11,12 Therefore, the combination of both types of materials comprising the core and the shell will lead to hybrid nanocomposites that ideally should retain the properties of the individual components while allowing us to use one of these components as a trigger that tunes the function of the other part.In this Highlight we focus particularly on hybrid nanocomposites consisting of a single metal nanoparticle core and a thermoresponsive polymer shell. We restrict ourselves to shells made of poly-N-isopropylacrylamide (pNIPAM), since this is the standard microgel model system and it has been used in most of the work related to this particular type of nanocomposites. We provide an overview of the different synthetic approaches, with an emphasis on the effect of the obtained structure on the main properties that are characteristic of these materials. Finally, different applications will be addressed, as well as future perspectives that we trust will be accomplished soon.
Synthesis and properties
During the past five years, the encapsulation of gold nanoparticles within pNIPAM microgels has been reported for different particle sizes, morphologies (spheres, decahedrons, octahedrons, stars) and capping agents (cetyltrimethylammonium bromide (CTAB), citrate, poly(vinylpyrrolidone) (PVP)). Although different strategies have been used, they all comprise the initial modification of the metallic surface with molecules containing vinyl groups, which serve as starting points for the polymerization and thus facilitate the encapsulation (Scheme 1). Once the particles are surface grafted with vinyl groups, they can be dispersed in an aqueous mixture of N-isopropylacrylamide (NIPAM, monomer) and N,N-methylenebisacrylamide (BIS, cross-linker), where the polymerization is initiated at 60 °C by adding a water soluble initiator.10 The differences between different methods stem from the way in which the vinyl functionality is placed at the particle surface, which is in turn determined by the nature of the capping agents.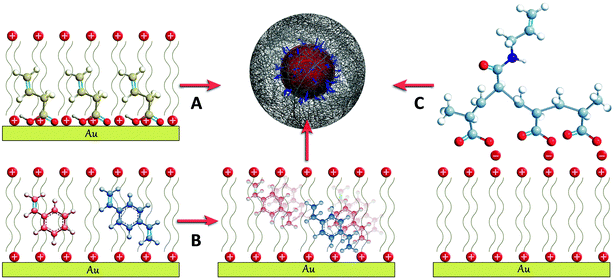 | ||
| Scheme 1 Schematic representation of the three different approaches developed for the surface functionalization with vinyl groups of CTAB stabilized gold nanoparticles: (A) butenoic acid, (B) styrene and divinylbenzene and (C) polyacrylic-N-allyl acid polyelectrolyte (see the text for details). | ||
An initial protocol was developed that involved the polymerization of styrene and divinylbenzene at the surface of the metal particles prior to introducing them in the pNIPAM polymerization medium.13 This method can be applied to nanoparticles stabilized with CTAB, since this cationic surfactant is known to form a bilayer on the gold surface,14 in which hydrophobic molecules such as styrene and divinylbenzene can be dissolved15 and subsequently polymerized into a thin polystyrene layer containing free vinyl functionalities that are available for further polymerization (Scheme 1B). An alternative approach relies on the surface modification of Au nanoparticles with butenoic acid or butenylamine. These molecules display a carboxylic acid or an amine group that can adsorb onto the nanoparticles, as well as the vinyl functionality from which polymerization and thus encapsulation can start (Scheme 1A).16,17 This method has been successfully applied for both citrate and surfactant stabilized metal nanoparticles, but it was found to fail for particles capped with another common stabilizer, PVP (see Scheme 1A). A more general approach was recently reported, which allows the encapsulation of particles stabilized with either CTAB or PVP. In this case, the particles were first functionalized with a polyelectrolyte, such as polyacrylic acid, by means of the layer by layer technique.18 However, it was required that the polyelectrolyte had been modified with allylamine through the well-known carbodiimide chemistry,19 to provide it with the necessary vinyl functionality (Scheme 1C).20–22
In all cases, once the particles have been surface grafted with vinyl groups, they can be dispersed in the aqueous mixture of NIPAM and BIS. The polymerization is usually initiated by adding a water soluble initiator at a moderate temperature (60 °C). This process typically yields a mixture of coated particles with free microgels and unreacted monomers, which can be easily washed by repeated centrifugation and redispersion in water. As examples of typical final products, Fig. 1 shows representative TEM images of gold nanoparticles of different shapes coated with pNIPAM, which were obtained through any of the three synthetic approaches described above.
 | ||
| Fig. 1 Representative TEM images of gold nanoparticles coated with pNIPAM: spheres (A), octahedra (B), nanorods (C) and nanostars (D). | ||
These colloidal nanocomposites based on a metallic core and a pNIPAM shell preserve the main properties of both components, i.e. the optical response from the plasmonic nanoparticle and the thermosensitive behavior of the polymer shell.13Fig. 2A shows a typical plot of the variation of the hydrodynamic diameter of Au@pNIPAM composites as a function of temperature, determined by photon-correlation spectroscopy (PCS). Regardless of the nature, size or shape of the metal core, Au@pNIPAM colloids show a volume phase-transition temperature (also known as lower critical solution temperature, or LCST) around 32 °C, which is also the characteristic value for pure pNIPAM microgels.10 Interestingly, in these colloids the localized surface plasmon resonance (LSPR) frequency of the metal cores becomes sensitive (in a reversible way) to the thermal-induced collapse–swelling transition, because the changes in shell volume also lead to variations in the local refractive index around the metal.23Fig. 2B shows a representative example of the heating–cooling effect on the LSPR band for 67 nm gold nanostars coated with pNIPAM. As predicted by theory, the magnitude of the LSPR shift during the heating–cooling cycles is determined by the sensitivity of the specific type of nanoparticles toward changes in the local refractive index. It has been reported that particles with sharp tips and edges, such as nanorods and nanostars, show a higher sensitivity toward refractive index changes because of the high confinement of the surface plasmon modes localized at these sites.24 In Fig. 2A we plotted a typical example of the LSPR band shift as a function of temperature, relative to the completely swollen state of the pNIPAM shell, for metal cores of different shapes. Gold nanostars and nanorods display significantly larger LSPR shifts as compared to shapes with lower anisotropy, such as decahedra and spheres.25,26
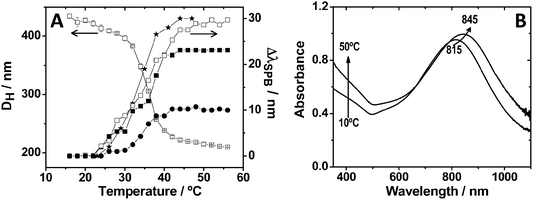 | ||
| Fig. 2 (A) Influence of temperature on the hydrodynamic diameter (left Y axis) of Au@pNIPAM composite particles containing 67 nm gold nanoparticle spherical cores, and the surface plasmon band shift (right Y axis) of different Au@pNIPAM composites, with respect to the fully swollen state at 15 °C: (★) nanostars with 115 nm overall size; (□) nanorods with dimensions 70.0 × 17.6 nm; (■) nanorods with dimensions 82.1 × 21.6 nm; and (●) 67 nm spheres. (B) UV-Vis-NIR spectra at 10 °C and 50 °C of an aqueous dispersion of 67 nm star-shaped Au@pNIPAM nanoparticles. | ||
However, not only the shell may be able to influence the optical features of the core, but these can also be exploited to act on the shell. For example, the large absorption cross-section of metal nanoparticles at their plasmon resonance makes them efficient and fast optothermal converters when irradiated with an external laser source operating at the LSPR wavelength.27,28 This is shown in Fig. 3A, which summarizes theoretical calculations that have been carried out to determine the temperature increase induced on and around a single gold nanorod in water, with dimensions defined by a long axis of 60 nm and a diameter of 15 nm, upon irradiation with an 800 nm laser beam operating at a power density of 880 kW cm−2.29 This irradiation leads to an increase in the temperature of the Au nanorods and their close environment (up to 60 nm from the rod surface in this particular case) above 40 °C, which clearly exceeds the LCST of pNIPAM.10 It is interesting to note that illumination with a laser operating at a wavelength close to the longitudinal plasmon band position leads both to an instantaneous increase of the absorbance at the low wavelength edge of the vis-NIR spectrum and to a significant red-shift and an increase in intensity of the longitudinal plasmon band, in agreement with the results obtained during bulk heating (Fig. 2 and 3B). This optical effect is observed as long as the laser illumination of the sample is maintained. The reason behind these observations is that the fast temperature increase of the gold nanorods upon laser irradiation is rapidly transferred into the pNIPAM network, therefore driving microgel collapse and a simultaneous increase in refractive index, which also leads to increased scattering. Therefore, this property of Au@pNIPAM colloids can be used to induce conformational changes in the nanoparticle coating, such as triggering deswelling, which can be exploited in applications such as drug delivery.
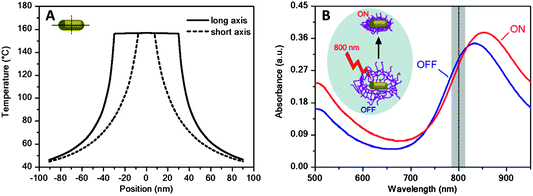 | ||
| Fig. 3 (A) Calculated cross-sectional temperature profile along the long (65 nm) and short (15 nm) axes of an Au nanorod in water, upon illumination with an 800 nm laser (power density 880 kW cm−2). (B) Vis-NIR extinction spectra of an Au nanorod@pNIPAM colloid before (blue line) and during (red line) irradiation with a laser beam. The vertical dashed line indicates the operating laser wavelength. The inset illustrates the induced collapse of the pNIPAM shell due to the optothermal heating of the Au NRs during irradiation. Reprinted with permission from ref. 29. | ||
Another interesting feature of the pNIPAM microgels is their tunable porosity, which allows dissolved ions or molecules to reach the gold core surface. This largely enhances the versatility of the composite system since the core size, shape and composition can be easily manipulated by in situ chemical reactions. For instance, through careful control of reaction conditions, growth of gold cores has been demonstrated via catalytic reduction of a gold salt on the nanoparticles surface, either preserving the spherical shape or evolving into a flower-like morphology (see Fig. 4A).13 A similar approach has also been employed for the reduction of silver ions, resulting in the formation of bimetallic cores with core@shell (Au@Ag) structure (see Fig. 4B).30 The most interesting aspect of this result is the possibility of tuning the optical response of the nanocomposite, according to the final size, shape and composition of the metal cores.
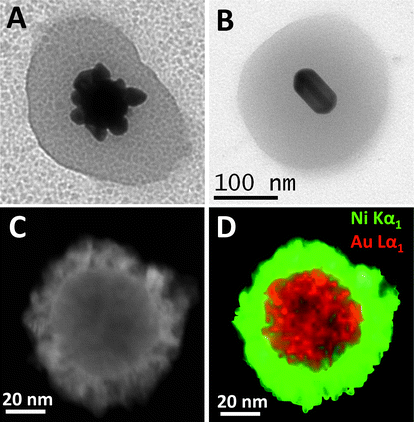 | ||
| Fig. 4 Detailed TEM images of the various structures that can be obtained through the selective growth of gold cores within Au@pNIPAM nanocomposites: (A) flower-like gold nanoparticle; (B) Au@Ag nanorod; (C) dark field scanning transmission electron microscopy (STEM) image of a single Au/Pt@Ni particle showing a typical core–shell structure; and (D) its corresponding STEM-XEDS RGB image (Au = red; Ni = green). Reprinted with permission from ref. 30 and 31. | ||
This accessibility of the metal core surface additionally allows the synthesis of composite colloids with multiple functionalities. For example, the addition of a magnetic functionality allows the manipulation of the composite particles by applying an external magnetic field. As a proof of concept, Ni shells were grown on the surface of gold spheres, previously encapsulated within porous pNIPAM microgels, so that a magnetic component could be added to the optically and thermally responsive Au@pNIPAM colloids.31 The growth of Ni on Au was reported to proceed only if a small amount of Pt was present on the particle surface, which would act as a catalyst for decomposition of a Ni–hydrazine complex.32 It should be pointed out that the Ni coating leads to a significant damping of the LSPR but a well-defined plasmon band could still be recorded, which was significantly red shifted as compared to the original one.
Applications
The unique properties of pNIPAM microgels have found numerous applications in different fields,33 but these can be even extended further if additional functionalities are incorporated within them, as described above. The combination of several materials with very different properties within the same nanoparticles opens up new possibilities in terms of applications, which have been demonstrated in important fields like catalysis, sensing or controlled release. Some of these potential applications are briefly addressed in what follows.Catalysis by metal nanoparticles has tremendously expanded during the last few decades. However, the nature of the metal catalyst largely depends on the target reaction to be catalyzed. For instance, gold and silver nanoparticles of different sizes have been used to catalyze electron transfer34 and oxidation reactions,35 but the same metals have also been recently found to exhibit catalytic effects on some important reactions, such as CO oxidation and desulfuration reactions.36 The encapsulation of metal nanoparticles within pNIPAM shells can be exploited not just to protect the particles from aggregation or to tune their optical response, but the polymer thermoresponsive behavior can be used to modulate the catalytic activity in a non-monotonous way, through variations in the porosity of the microgel when temperature is varied. Au@pNIPAM composite particles have been used as catalysts in the reduction of hexacyanoferrate(III) by borohydride ions in water.37 The colloidal stability provided by the pNIPAM shell makes these catalysts reusable for several cycles, with their catalytic properties almost unchanged. Besides the thermoresponsive behavior, the cross-linking density of pNIPAM can also be tuned, and both parameters allow us to control the diffusion of reactants toward the metal core surface and in turn the catalytic activity, as summarized in Fig. 5A. In the figure we can see that, regardless of shell composition, the observed rate constant (normalized by the gold surface in the Au@pNIPAM nanocomposites) increases with temperature according to Arrhenius law at temperatures below 20 °C (swollen state) and above 35 °C (collapse state) but it decreases between 20 and 35 °C due to the pore compression when the pNIPAM collapses when temperature is increased. Fig. 5A additionally shows the effect of cross-linking density (pre-determined during synthesis by the amount of cross-linker) on the catalytic activity. As one could intuitively expect, larger cross-linking densities (lower porosity) lead to lower observed rate constants.
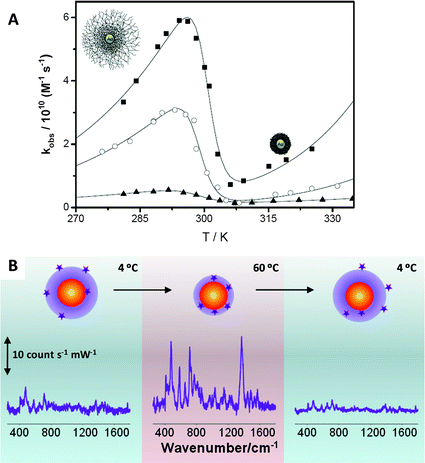 | ||
| Fig. 5 (A) Effect of the temperature on the catalytic efficiency normalized by the gold surface of Au@pNIPAM nanocomposites with different cross-linking densities: (■) 7%, (○) 10% and (▲) 17.5%. (B) Variation of the SERS (λex = 785 nm) intensity of 1-naphthol as a function of the solution temperature in a 4 to 60 to 4 °C heating–cooling cycle. Reprinted with permission from ref. 30 and 31. | ||
Another interesting application for Au(Ag)@pNIPAM microgel nanocomposites has been described in the field of SERS-based sensing. SERS (surface enhanced Raman scattering spectroscopy) is an optical spectroscopy technique in which the Raman scattering signal from a molecule gets enormously enhanced when it is located next to a nanostructured metal, under illumination at the LSPR wavelength. SERS has emerged as a powerful tool for the detection of a wide variety of analytes, ranging from metal ions to biomolecules, even at the single molecule level.38 Nevertheless, one of the main limitations of this technique is that molecules must be in close proximity to the metal surface to obtain a good SERS signal, which is often determined by the affinity of the analyte for Au and Ag surfaces. Although most molecules are intrinsically Raman active, only few present sufficient chemical affinity toward metals. Therefore, the development of new direct SERS sensing platforms for analytes with low or no metal affinity constitutes a very active SERS research area.39 We have recently demonstrated that Au(Ag)@pNIPAM nanocomposites might be useful for the detection of low metal-affinity molecules.30,40 The thermoresponsive behavior of the pNIPAM shell allows these systems to trap molecules and to bring them close enough to the metal surface for obtaining a detectable SERS signal. As a proof of concept 1-naphthol (1NOH) was used as a molecular probe with low metal affinity. When 1NOH was added to a Au@pNIPAM colloid (either swollen or collapsed), no SERS signal could be registered. However, detection was possible when the mixture of particles and 1NOH was heated above the LCST, as depicted in Fig. 5B. Before heating (4 °C), 1NOH could diffuse through the microgel network but it could also diffuse out, so that the number of molecules residing close to the gold core was insufficient to provide a meaningful SERS signal. However, the phase transition associated with the microgel collapse was sufficient to bring the molecular probes located within the pNIPAM shell into contact with the metal surface, so that a clear SERS signal could be registered. Finally, since 1NOH does not bind chemically to gold, a loss of signal was observed when the microgel was cooled down again below the LCST.
Another potential application, first reported by Kawano et al., is based on the light triggered release of molecules from pNIPAM coated gold nanorod nanocomposites.41 In their study the authors proposed the use of the optothermal conversion properties of gold nanorods to absorb NIR light to induce the selective collapse of the pNIPAM shell leading to the release of molecules previously loaded in it. Preliminary results on the light triggered release for in vivo applications were also reported.
Future perspectives
pNIPAM is probably the most well-known soft nanomaterial and has proven to be useful in various fields, especially in biomedical applications. Interestingly, the combination with metal nanoparticles in the form of core–shell nanocomposites with well-defined structure will open new possibilities in emerging fields such as catalysis, sensing or drug delivery. Nevertheless, pNIPAM presents some limitations or drawbacks, such as non-biodegradability and low biocompatibility, which need to be overcome. In this respect alternative thermosensitive polymers have been developed but combination with metal nanoparticles still remains to be demonstrated.The development of novel polymeric or composite systems capable to respond to several external stimuli in an intelligent way is also challenging. We expect that novel nanocomposites with a hierarchical organization of components, giving rise to systems where the stimuli and response signals can be separated by controlling the properties of the various constituting materials, will soon be reported, which will consequently lead to fascinating new possibilities.
Acknowledgements
The authors acknowledge funding from the Spanish Ministerio de Economía y Competitividad/FEDER (MAT2010-15374 and CTQ2010-18576), and the Xunta de Galicia/FEDER (10PXIB314218PR, 09TMT011314PR and INCITE09PXIB314259PR, INBIOMED).References
- A. Fernández-Barbero, I. J. Suárez, B. Sierra-Martín, A. Fernández-Nieves, F. Javier de las Nieves, M. Marquez, J. Rubio-Retama and E. López-Cabarcos, Adv. Colloid Interface Sci., 2009, 147–148, 88–108 CrossRef.
- M. Karg and T. Hellweg, J. Mater. Chem., 2009, 19, 8714–8727 RSC.
- Y. Lu and M. Ballauff, Prog. Polym. Sci., 2011, 336, 767–792 CrossRef.
- C. Burda, X. Chen, R. Narayanan and M. A. El-Sayed, Chem. Rev., 2004, 105, 1025–1102 CrossRef.
- M. Grzelczak, J. Pérez-Juste, P. Mulvaney and L. M. Liz-Marzán, Chem. Soc. Rev., 2008, 37, 1783–1791 RSC.
- K. Zhou and Y. Li, Angew. Chem., Int. Ed., 2012, 51, 602–613 CrossRef CAS.
- S. H. Ko, I. Park, H. Pan, C. P. Grigoropoulos, A. P. Pisano, C. K. Luscombe and J. M. J. Fréchet, Nano Lett., 2007, 7, 1869–1877 CrossRef CAS.
- K. Saha, S. S. Agasti, C. Kim, X. Li and V. M. Rotello, Chem. Rev., 2012, 112, 2739–2779 CrossRef CAS.
- M. Pelton, J. Aizpurua and G. Bryant, Laser Photonics Rev., 2008, 2, 136–159 CrossRef CAS.
- R. Pelton, Adv. Colloid Interface Sci., 2000, 85, 1–33 CrossRef CAS.
- O. J. Cayre, N. Chagneux and S. Biggs, Soft Matter, 2011, 7, 2211–2234 RSC.
- J. K. Oha, R. Drumrighta, D. J. Siegwart and K. Matyjaszewski, Prog. Polym. Sci., 2008, 33, 448–477 CrossRef.
- R. Contreras-Cáceres, A. Sánchez-Iglesias, M. Karg, I. Pastoriza-Santos, J. Pérez-Juste, J. Pacifico, T. Hellweg, A. Fernández-Barbero and L. M. Liz-Marzán, Adv. Mater., 2008, 20, 1666–1670 CrossRef.
- S. Gómez-Graña, F. Hubert, F. Testard, A. Guerrero-Martínez, I. Grillo, L. M. Liz-Marzán and O. Spalla, Langmuir, 2012, 28, 1453–1459 CrossRef.
- A. M. Alkilany, R. L. Frey, J. L. Ferry and C. J. Murphy, Langmuir, 2008, 24, 10235–10239 CrossRef CAS.
- R. Contreras-Cáceres, J. Pacifico, I. Pastoriza-Santos, J. Pérez-Juste, A. Fernández-Barbero and L. M. Liz-Marzán, Adv. Funct. Mater., 2009, 19, 3070–3076 CrossRef.
- M. Karg, S. Jaber, T. Hellweg and P. Mulvaney, Langmuir, 2011, 27, 820–827 CrossRef CAS.
- C. Fernández-López, C. Pérez-Balado, J. Pérez-Juste, I. Pastoriza-Santos, A. R. de Lera and L. M. Liz-Marzán, Soft Matter, 2012, 8, 4165–4170 RSC.
- A. Williams and T. Ibrahim, Chem. Rev., 1981, 81, 589–636 CrossRef CAS.
- A. P. R. Johnston, H. Mitomo, E. S. Read and F. Caruso, Langmuir, 2006, 22, 3251–3258 CrossRef CAS.
- J. F. Quinn, A. P. R. Johnston, G. K. Such, A. N. Zelikin and F. Caruso, Chem. Soc. Rev., 2007, 36, 707–718 RSC.
- L. Jierry, N. B. Ameur, J.-S. Thomann, B. Frisch, E. Gonthier, J.-C. Voegel, B. Senger, G. Decher, O. Felix, P. Schaaf, P. Mesini and F. Boulmedais, Macromolecules, 2010, 43, 3994–3997 CrossRef CAS.
- M. Karg, I. Pastoriza-Santos, J. Pérez-Juste, T. Hellweg and L. M. Liz-Marzán, Small, 2007, 3, 1222–1229 CrossRef CAS.
- R. Alvarez-Puebla, L. M. Liz-Marzán and F. J. García de Abajo, J. Phys. Chem. Lett., 2010, 1, 2428–2434 CrossRef CAS.
- H. Chen, X. Kou, Z. Yang, W. Ni and J. Wang, Langmuir, 2008, 24, 5233–5237 CrossRef CAS.
- S. Barbosa, A. Agrawal, I. Pastoriza-Santos, L. Rodríguez-Lorenzo, R. A. Alvarez-Puebla, A. Kornowski, H. Weller and L. M. Liz-Marzán, Langmuir, 2010, 26, 14943–14950 CrossRef CAS.
- J. Stehr, C. Hrelescu, R. A. Sperling, G. Raschke, M. Wunderlich, A. Nichtl, D. Heindl, K. Kurzinger, W. Parak, T. S. Klar and J. Feldmann, Nano Lett., 2008, 8, 619–623 CrossRef CAS.
- C. Hrelescu, J. Stehr, M. Ringler, R. A. Sperling, W. J. Parak, T. A. Klar and J. Feldmann, J. Phys. Chem. C, 2010, 114, 7401–7411 CAS.
- J. Rodríguez-Fernández, M. Fedoruk, C. Hrelescu, A. A. Lutich and J. Feldmann, Nanotechnology, 2011, 22, 245708 CrossRef.
- R. Contreras-Cáceres, I. Pastoriza-Santos, R. A. Alvarez-Puebla, J. Pérez-Juste, A. Fernández-Barbero and L. M. Liz-Marzán, Chem.–Eur. J., 2010, 16, 9462–9467 CrossRef.
- A. Sánchez-Iglesias, M. Grzelczak, B. Rodríguez-González, P. Guardia-Girós, I. Pastoriza-Santos, J. Pérez-Juste, M. Prato and L. M. Liz-Marzán, ACS Nano, 2009, 3, 3184–3190 CrossRef.
- M. Grzelczak, J. Pérez-Juste, B. Rodríguez-González, M. Spasova, I. Barsukov, M. Farle and L. M. Liz-Marzán, Chem. Mater., 2008, 20, 5399–5405 CrossRef CAS.
- Y. Guan and Y. Zhang, Soft Matter, 2011, 7, 6375–6384 RSC.
- S. Carregal-Romero, J. Pérez-Juste, P. Hervés, L. M. Liz-Marzán and P. Mulvaney, Langmuir, 2010, 26, 1271–1277 CrossRef CAS.
- J. Rodríguez-Fernández, J. Pérez-Juste, P. Mulvaney and L. M. Liz-Marzán, J. Phys. Chem. B, 2005, 109, 14257–14261 CrossRef.
- J. A. Rodriguez, P. Liu, Y. Takahashi, F. Viñes, L. Feria, E. Florez, K. Nakamura and F. Illas, Catal. Today, 2011, 166, 2–9 CrossRef CAS.
- S. Carregal-Romero, N. J. Buurma, J. Pérez-Juste, P. Hervés and L. M. Liz-Marzán, Chem. Mater., 2010, 22, 3051–3059 CrossRef CAS.
- K. Kneipp, H. Kneipp, I. Itzkan, R. R. Dasari and M. S. Feld, Chem. Rev., 1999, 99, 2957–2976 CrossRef CAS.
- R. A. Alvarez-Puebla and L. M. Liz-Marzán, Chem. Soc. Rev., 2012, 41, 43–51 RSC.
- R. A. Álvarez-Puebla, R. Contreras-Cáceres, I. Pastoriza-Santos, J. Pérez-Juste and L. M. Liz-Marzán, Angew. Chem., Int. Ed., 2009, 48, 138–143 CrossRef.
- T. Kawano, Y. Niidome, T. Mori, Y. Katayama and T. Niidome, Bioconjugate Chem., 2009, 20, 209–212 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2013 |