 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
1,5-, 2,6- and 9,10-distyrylanthracenes as luminescent organic semiconductors†
Afshin
Dadvand
ab,
Wei-Hsin
Sun
a,
Andrey G.
Moiseev
a,
Francine
Bélanger-Gariépy
c,
Federico
Rosei
b,
Hong
Meng‡
d and
Dmitrii F.
Perepichka
*a
aDepartment of Chemistry and Centre for Self-Assembled Chemical Structures, McGill University, 801 Sherbrooke Street West, Montreal, H3A 0B8, QC, Canada. E-mail: dmitrii.perepichka@mcgill.ca
bInstitut national de la recherche scientifique (INRS) and Centre for Self-Assembled Chemical Structures, 1650 Lionel-Boulet Boul., Varennes, J3X 1S2, QC, Canada
cDépartement de Chimie, Local A-634, Université de Montréal, 2900 Blvd Édouard-Montpetit, Pavillon Roger-Gaudry, Montréal, H3T 1J4, QC, Canada
dCentral Research and Development, Experimental Station, E. I. DuPont, Wilmington, DE 19880-0328, USA
First published on 6th March 2013
Abstract
We report the synthesis, molecular and crystal structures, optoelectronic properties and organic field-effect transistor (OFET) studies of three isomeric distyrylanthracene derivatives 2,6-DPSAnt, 1,5-DPSAnt and 9,10-DPSAnt. The analysis of the structure–property relationships reveals that π-conjugation in this series is defined by two competing factors: the co-planarity of the styryl substituents (highest in 2,6-DPSAnt) and the electronic communication through the anthracene core (strongest in 9,10-DPSAnt). The charge-transport properties of the compounds, on the other hand, are primarily controlled by the solid state packing which is more efficient for the linear 2,6-DPSAnt derivative. Accordingly, the highest field-effect mobility (up to 0.75 cm2 V−1 s−1) was measured for 2,6-DPSAnt. Unexpectedly high charge mobility of 1,5-DPSAnt (up to 0.15 cm2 V−1 s−1) was attributed to excellent thin-film morphology (nearly layer-by-layer growth) and favorable molecular packing in the films, different from that found in single crystals. Unfavorable molecular packing and poor film morphology observed for 9,10-DPSAnt predictably led to no transistor properties being observed for this isomer. Both 2,6-DPSAnt and 1,5-DPSAnt afforded light-emitting transistors with green light emission area localized near an electron-injecting electrode.
Introduction
The semiconducting conjugated organic molecules have received significant recent interest as they are the basis for a fundamental understanding of charge transport in the solid state and enable innovative technologies such as organic light-emitting diodes (OLEDs),1–3 organic field-effect transistors (OFETs)4,5 and organic photovoltaics (OPVs).6,7 The tunability of properties, multifunctionality and low-temperature processing are the three major advantages of molecular semiconductors (MSCs), pushing forward their use in optoelectronics. The combination of tunable light-emitting and charge-transport properties has been the basis of OLEDs – the most mature organic electronic technology. The short electrical channel of OLED devices does not demand high charge mobility in a semiconductor (μ ∼ 10−3 cm2 V−1 s−1) and a large number of OLED materials with highly efficient luminescence have been synthesized over the last two decades.8The design of high charge mobility and efficient solid state luminescence in the same molecule is more challenging and only a few materials have been shown to combine both properties. A specific technological application of such materials is organic light-emitting transistors (OLETs)9 which combine electrical switching and light emission function in a single device. In addition, the high current density achievable in OLETs10 could potentially lead to not yet realized organic injection lasers.11 Generally, achieving high charge mobility in MSCs requires a highly crystalline structure. However, many if not most semiconducting molecules exhibit strong quenching of the exciton in the crystalline state, empirically ascribed to the “π-aggregation” effect.12
Almost all high mobility MSCs are constructed from fused polycyclic aromatic and/or heteroaromatic (mostly, thiophene) rings.13 Their high charge carrier mobility is usually connected with their flat, symmetric and rigid molecular structure that leads to a low reorganization energy associated with charge injection and efficient intermolecular electron delocalization in the crystalline state. A classical example is pentacene, a benchmark p-type MSC with charge carrier mobility greater than 1 cm2 V−1 s−1.14 A number of other high-mobility MSCs based on the derivatives, homologues and heteroanalogues of pentacene have been reported.13,15–18 However, most of these polycyclic aromatic MSCs show either no luminescence or very weak luminescence in the solid state.
One of the reasons for luminescence quenching in the solid state is the singlet fission process that turns the singlets into two non-emissive triplet states.19 The thermodynamics of singlet fission requires the energy of the triplet state to be at or below half of that of the singlet. This condition is realized in tetracene and longer acenes. For shorter acene molecules, e.g. anthracene, singlet fission is not possible because of the higher triplet energy; a high PL quantum yield (PLQY, 64%) was reported for anthracene crystals.20 However, to the best of our knowledge, no working thin-film OFET devices have been realized with anthracene.21,22
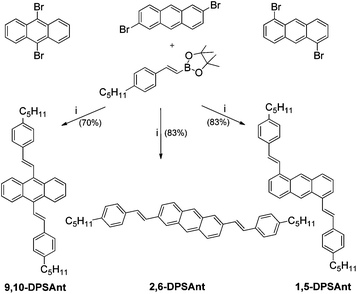
Substitution of anthracene in 2,6-positions with phenyl, thienyl, arylvinylene and arylethynyl groups has been used to prepare MSCs for practical OFET applications, leading to hole mobilities in the range of ∼0.1 to 1 cm2 V−1 s−1 in thin film transistors.23–28 Recently we reported a combination of high charge mobility (μ+ > 2 cm2 V−1 s−1) and strong solid state emission (70%) in crystals of 2-(4-hexylstyryl)anthracene and demonstrated its application in OLET devices.29 Much less is known about anthracene-based MSCs with substituents in other positions, although solution processed OFETs for tetralkoxy-substituted 9,10-distyrylanthracenes have been reported (and showed a relatively low charge mobility of ∼10−4 cm2 V−1 s−1).30 In the present work, we perform a detailed comparison of the structural, molecular and solid state optoelectronic properties of a series of three isomeric distyrylanthracenes 2,6-DPSAnt, 1,5-DPSAnt and 9,10-DPSAnt (Scheme 1) and study their performance in thin-film transistor devices.
Experimental part
General
Starting dibromoanthracenes were purchased from TCI America. 2,6-DPSAnt was synthesized as described earlier.25 PLQY in toluene solution were measured relative to 9,10-diphenylanthracene (95% in cyclohexane), for 2,6- and 1,5-DPSAnt), and relative to fluorescein (79% in ethanol), for 9,10-DPSAnt. Absolute PLQY of solid state (crystalline powders) samples were measured in the 6” integrating sphere from Labsphere Inc, using a Varian Cary Eclipse fluorometer. Fluorescence lifetimes were measured with an Edinburgh Instruments Mini Tau lifetime fluorometer exciting with an EPL 405 laser. Cyclic voltammetry measurements were performed in 0.1 M electrolyte solution (Bu4NPF6) in CH2Cl2 or PhCl using a Pt disk as a working electrode, Ag/AgCl as a reference electrode and Pt wire as an auxiliary electrode. Ferrocene (Fc) was added to the analyzed solution at the end of the measurement as an internal reference. All redox potentials are given relevant to the Fc/Fc+ couple. AFM characterization of molecular thin films was performed using a Bruker Multimode 8 instrument operated in the tapping mode. DFT calculations of the molecular structure (in the gas phase), molecular orbital energy, orbital splitting in the dimers32 and hole transfer reorganization energy35 were performed using the B3LYP functional and the 6-31G(d) basis set as implemented in Gaussian 09.31 Frequency analysis was performed for all optimized structures. The absorption/emission spectra and the structures of the relaxed excited state were analysed by time-dependent DFT at the same level of theory.4,4,5,5-Tetramethyl-2-[(E)-2-(4-pentylphenyl)ethenyl]-1,3,2-dioxaborolane
To a 250 mL Schlenk flask fitted with a magnetic bar and a nitrogen inlet were added 1-iodo-4-pentylbenzene (2.0 g, 7.3 mmol) and dry toluene (20 mL). Then, 2-ethenyl-4,4,5,5-tetramethyl-1,3,2-dioxaborolane (2.02 g, 13.1 mmol) and tributylamine (3.5 mL) were added. The resultant solution was degassed using three freeze–pump–thaw cycles. Pd(PPh3)4 (0.21 g, 0.18 mmol) was added in one portion, and the reaction mixture was heated up to 100 °C and kept at this temperature for 4 hours. After cooling down, it was diluted with dichloromethane and the resulting solution was washed with 10% HCl. The organic phase was separated, washed with brine and dried over MgSO4. Evaporation of the solvent under reduced pressure afforded a brownish oil which was purified by column chromatography (silica, hexane/EtOAc) to afford the pure product as a viscous orange oil (1.4 g, 64%).1H NMR (400 MHz, CDCl3): δ 7.39 (d, J = 8.0 Hz, 2H), 7.38 (d, J = 18.4 Hz, 1H), 7.15 (d, J = 8.0 Hz, 2H), 6.12 (d, 1H, J = 18.4 Hz, 1H), 2.59 (t, J = 7.2 Hz, 2H), 1.61 (m, 2H), 1.31 (m, 16H), 0.89 (m, 3H); 13C NMR (100 MHz, CDCl3) δ 149.6, 144.0, 135.0, 128.6, 127.0, 83.2, 35.7, 31.5, 31.0, 24.8, 22.6, 14.0; IR (neat) cm−1, ν 2977 (w), 2928 (m), 2857 (m), 1622 (s), 1567 (w), 1511 (m), 1458 (w), 1418 (w), 1380 (w), 1370 (w), 1347 (s), 1321 (s), 1271 (m), 1214 (s), 1142 (s), 995 (s), 969 (s), 900 (w), 846 (s), 811 (s), 752 (w), 679 (w), 644 (w), 558 (w); HRMS (APCI): calcld for C19H30O2B10 (M+ + H) 300.2370; found 300.2366.
9,10-Bis-[2-(4-pentylphenyl)vinyl]anthracene (9,10-DPPAnt)
To a 250 mL Schlenk flask fitted with a magnetic bar and a nitrogen inlet were added 9,10-dibromoanthracene (0.15 g, 0.45 mmol), 4,4,5,5-tetramethyl-2-[(1E)-2-(4-pentylphenyl)ethenyl]-1,3,2-dioxaborolane (0.40 g, 1.3 mmol), sodium carbonate (0.24 g, 2.2 mmol) solution in water (1.1 mL), aliquat 336 (0.092 g) and toluene (6 mL). The resultant solution was degassed by purging with nitrogen for 15 minutes. Then Pd(PPh3)4 (0.01 g, 0.009 mmol) was added in one portion, and the reaction mixture was heated up to 90 °C and kept at this temperature for three days. After cooling down to room temperature, the reaction mixture was diluted with dichloromethane (100 mL) and washed with water (3 × 50 mL). The organic phase was separated and dried over sodium sulphate. The solvent was removed under reduced pressure and the crude product was purified by column chromatography (silica, hexane) to afford a yellow powder (0.16 g, 70%). mp: 180–182 °C (from dichloromethane/ethanol).1H NMR (300 MHz, CDCl3) δ 8.40 (dd, J = 3, 6.9 Hz, 4H), 7.89 (d, J = 16.8 Hz, 2H), 7.62 (d, J = 8.1 Hz, 4H), 7.46 (dd, J = 3.0, 6.6 Hz, 4H), 7.28 (d, J = 8.1 Hz, 4H), 6.92 (d, J = 16.8 Hz, 2H), 2.68 (t, J = 7.2 Hz, 4H), 1.69 (m, 4H), 1.38 (m, 9H), 0.93 (t, J = 7.2 Hz, 6H); 13C NMR (75 MHz, CDCl3) δ 143.1, 137.3, 134.8, 132.8, 129.6, 128.9, 126.5, 125.1, 124.1, 35.8, 31.5, 31.2, 22.6, 14.1; IR (neat) ν cm−1 3005 (w), 2952 (m), 2926 (m), 2853 (w), 1510 (m), 1455 (m), 1436 (m), 1418 (m), 1369 (m), 1209 (m), 1117 (s), 971 (s), 846 (m), 757 (s), 725 (m), 660 (m), 653 (m), 607 (m), 564 (m), 551 (m), 519 (m), 506 (m); HRMS (APCI): calcld for C40H42 (M+ + 1) 523.3370; found 523.3363.
1,5-Bis-[2-(4-pentylphenyl)vinyl]anthracene (1,5-DPPAnt)
To a 100 mL two neck round-bottom flask fitted with a stop-cock adaptor, a magnetic bar, and an argon inlet were added 1,5-dibromoanthracene (0.224 g, 0.667 mmol), 4,4,5,5-tetramethyl-2-[(E)-2-(4-pentylphenyl)ethenyl]-1,3,2-dioxaborolane (0.601 g, 2.00 mmol), and toluene (7 mL). The resultant solution was degassed using three freeze–pump–thaw cycles. Then sodium carbonate (0.35 g, 3.3 mmol) solution in water (1.7 mL) and aliquat 336 (0.13 g) were added. The resultant solution was degassed with argon for 15 minutes. Then Pd(PPh3)4 (0.018 g, 0.015 mmol) was added in one portion, and the reaction mixture was heated up to 90 °C and kept at this temperature for 19 hours. After cooling down to room temperature, the reaction mixture was diluted with dichloromethane (100 mL) and washed with water (3 × 50 mL). The organic phase was separated and dried over magnesium sulphate. The solvent was removed under reduced pressure and the crude product was purified by column chromatography (silica, hexane/ethyl acetate) to yield a yellow crystalline product (0.29 g, 83%). mp: 183–185 °C (from chloroform/ethanol).1H NMR (300 MHz, CDCl3) δ 8.77 (s, 2H), 8.00 (J = 8.1 Hz, d, 2H), 7.98 (J = 16.2 Hz, d, 2H), 7.73 (J = 6.9 Hz, d, 2H), 7.60 (J = 8.1 Hz, d, 4H), 7.49 (J = 7.2 Hz, t, 2H), 7.26 (J = 8.1 Hz, d, 4H), 7.22 (J = 16.2 Hz, d, 2H), 2.67 (J = 7.2 Hz, t, 4H), 1.68 (J = 7.8, p, 4H), 1.37 (m, 8H), 0.93 (J = 6.6 Hz, t, 6H). 13C NMR (75 MHz, CDCl3) δ 142.9, 135.2, 135.1, 132.0, 131.9, 129.8, 128.9, 128.5, 126.7, 125.4, 125.0, 123.0, 35.8, 31.5, 31.2, 22.6, 14.1. IR (neat) ν cm−1 3026 (w), 2951 (m), 2915 (m), 2870 (m), 2854 (m), 1604 (w), 1511 (m), 1466 (m), 1422 (m), 1373 (m), 1354 (m), 1310 (m), 1275 (w), 1192 (m), 1125 (w), 962 (s), 946 (s), 908 (s), 876 (s), 857 (m), 843 (s), 809 (s), 743 (m), 727 (m), 701 (w), 582 (s), 548 (w), 501 (s); HRMS (ESI): calcld for C40H42 (M+) 522.3281; found 522.3283.
Thin-film transistor fabrication
Thin film OFETs were fabricated in top-contact configuration Au drain and source electrodes thermally evaporated through a shadow mask. Thin films of molecules were grown at room temperature by vacuum deposition (10−6 mbar, deposition rate 0.2–0.4 Å s−1) on HMDS treated SiO2/Si. SiO2 was thermally grown (200 nm thick) on heavily n-doped (Sb) Si (ρ ≈ 0.01 Ohm cm). The performance of the resulting OFETs was measured in a vacuum and using a Keithley 4200-SCS. A Toshiba IK 1000 charge coupled device (CCD) camera mounted on an optical microscope objective was used for EL imaging. The bias-modulated EL was recorded using a Hamamatsu (3137 QB) Si photodiode.Results and discussion
Synthesis
Disubstituted anthracenes 2,6-DPSAnt, 1,5-DPSAnt and 9,10-DPSAnt were synthesized by Suzuki coupling of corresponding dibromoanthracenes with 4-pentylphenylvinylene boronic ester (Scheme 1). Both 9,10-DPSAnt and 1,5-DPSAnt are highly soluble in common organic solvents and were purified by column chromatography followed by recrystallization. In contrast, 2,6-DPSAnt is highly insoluble and was purified by train sublimation.Thermogravimetric analysis (TGA) revealed high thermal stability of 2,6-DPSAnt, 1,5-DPSAnt, and 9,10-DPSAnt with onset decomposition temperatures (Tdec) of ∼355 °C, 335 °C, and 330 °C in air, respectively. TGA measurements under nitrogen showed no decomposition until evaporation of the compounds at 410 °C (2,6-DPSAnt), 360 °C (1,5-DPSAnt) and 350 °C (9,10-DPSAnt) (see ESI†). The differences in the evaporation temperature can likely be ascribed to stronger solid state intermolecular interactions in the linearly shaped 2,6-DPSAnt (see below). Similarly, this derivative showed a much higher melting point (318 °C) than 1,5-DPSAnt (183 °C) and 9,10-DPSAnt (180 °C).
Molecular and solid state structure
The effects of the position of substituents on planarity and solid-state packing of distyrylanthracenes were studied by X-ray crystallography and by DFT calculations at the B3LYP/6-31G(d) level of theory. Single crystals of 1,5-DPSAnt and 9,10-DPSAnt were prepared at room temperature by slow evaporation of chloroform and ethanol solutions, respectively. The X-ray structure of 2,6-DPSAnt (for vacuum-grown crystals) was reported previously (CCDC 621869).25 The crystal data and structure refinement parameters for all three isomers are summarized in Table 1.| Compound | ||||
|---|---|---|---|---|
| 2,6-DPSAnt 25 | 1,5-DPSAnt | 9,10-DPSAnt | ||
| Calcd. density (g cm−3) | 1.21 | 1.17 | 1.14 | |
| Crystal system | Monoclinic | Triclinic | Triclinic | |
| Space group | C2/c |
P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) |
P |
|
| Cell parameters (Å, °) | a | 67.66(6) | 6.7671(3) | 6.5844(3) |
| b | 7.230(5) | 10.0846(5) | 9.2194(3) | |
| c | 5.848(5) | 12.0321(6) | 13.5405(15) | |
| α | 90 | 98.990(2) | 100.0975(16) | |
| β | 91.395(11) | 106.294(2) | 99.8398(18) | |
| γ | 90 | 104.765(3) | 105.7088(16) | |
| Volume (Å3) | 2860(4) | 738.80(6) | 758.05(5) | |
| Z | 4 | 2 | 2 | |
| Temperature (K) | 293 | 100 | 100 | |
In all three molecules, the vinyl group is rotated out of the plane of aromatic rings (Fig. 1). This twist is small for 2,6-DPSAnt (15° between anthracene and vinylene planes, 11° between phenyl and vinylene planes), and is not expected to significantly disrupt the conjugation between anthracene and styryl groups. Considering that DFT calculations predict a planar structure in the gas phase, the observed twist must be due to crystal packing forces. In 1,5-DPSAnt, the additional steric interactions with hydrogens in 9,10-positions of the anthracene ring cause a larger twist (31° for anthracene/vinylene planes, 21° for phenyl/vinylene planes). The largest out-of-plane distortion was observed for 9,10-DPSAnt due to interaction with two pairs of peri-hydrogens (1,8 and 4,5); the twist of 60° between the anthracene and vinylene planes (12° between vinylene and phenyl planes) should drastically affect the conjugation with the styryl groups.
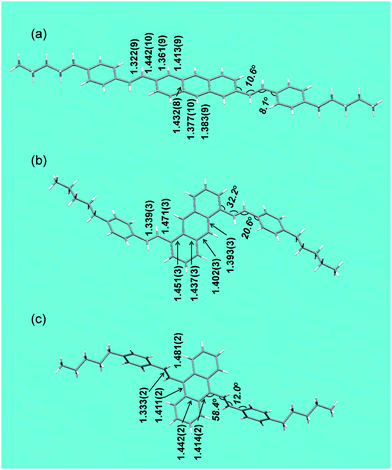 | ||
| Fig. 1 Molecular structures of 2,6-DPSAnt (a), 1,5-DPSAnt (b), and 9,10-DPSAnt (c) obtained from single crystal X-ray analyses, showing the corresponding bond lengths (in Å) and torsional angles. | ||
Interestingly, in 2,6-DPSAnt, the solid state packing also causes a slight distortion of the anthracene ring: the largest out-of-plane shift observed for C-2 and C-6 is 0.04 Å. The anthracene core in the other two isomers is fully planar.
We analyzed the bond length alternation as a rough indicator of the efficiency of π-conjugation. The shortest exocyclic bonds are found for 2,6-DPSAnt (Cant–Cvyn = 1.442(10) Å), followed by 1,5-DPSAnt (Cant–Cvyn = 1.471(3) Å) and 9,10-DSAnt (Cant–Cvyn = 1.481(2) Å). This suggests that the most efficient electronic coupling of substituents occurs in the 2,6-DPSAnt isomer, as expected based on the lowest out-of-plane distortion displayed by this derivative. On the other hand, the conjugation within the anthracene ring appears to be more efficient in 9,10-DSAnt and 1,5-DPSAnt isomers. The largest bond length alternation of the conjugation pathway is 0.031 Å, 0.035 Å, and 0.055 Å for 9,10-, 1,5- and 2,6-disubstituted anthracenes, respectively (Fig. 1). The electronic properties of the three derivatives are likely defined by a competition between the out-of-plane rotation of the substituents and different efficiency of conjugation of the 2–6, 1–5 and 9–10 pathways. Accordingly, it is quite difficult to predict the effect of substitution patterns, e.g. on the HOMO–LUMO gap (see below).
The different molecular shape imposed by the position of styryl substituents leads to very different packing motifs of 2,6-, 1,5- and 9,10-DSAnt in the crystal (Fig. 2). Similar to most acenes, 2,6-DSAnt adopts a herringbone packing (in the monoclinic space group C1(2)/c1). A uniform intermolecular separation (the distance between centroids of anthracene rings is 4.65 Å in both directions, cf. 4.71 Å for pentacene) provides two-dimensional π-interactions. The corresponding shortest π⋯π contacts (between carbon atoms of non-coplanar molecules) are 3.64 Å.
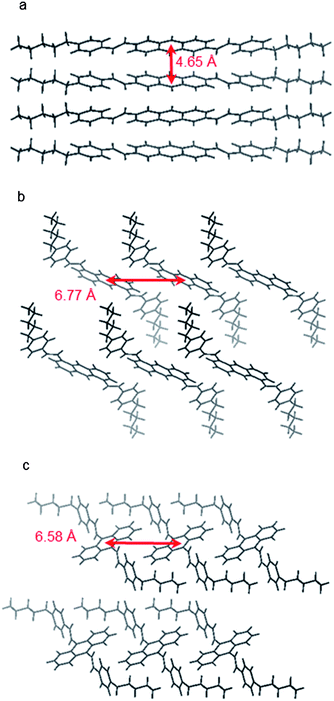 | ||
| Fig. 2 Crystal structures of (a) 2,6-DPSAnt, (b) 1,5-DPSAnt and (c) 9,10-DPSAnt. | ||
The position of substituents in 1,5-DSAnt and 9,10-DPSAnt hinders such herringbone packing and both isomers crystallize in the triclinic space group P. Within such a crystal lattice, the molecules form one-dimensional stacks and the anthracene cores of all molecules are co-parallel. However, the large slippage of the molecules within the stacks precludes any π–π interaction between the anthracene moieties. The distance between centroids of anthracene rings is 6.77 Å for 1,5-DSAnt and 6.58 Å for 9,10-DSAnt. The only π–π interactions observed in these crystals occur between the anthracene and styryl fragments (C⋯C contacts 3.5–3.7 Å). A similar packing arrangement was also observed in other substituted 9,10-(distyryl)anthracenes.32
The overall tighter packing of 2,6-DPSAnt is also apparent from comparison of the calculated density of the isomers (Table 1). The different solid state structures have a profound effect on the electronic communication between the isomeric molecules in the solid state. DFT calculations of the dimeric interactions within the experimentally observed crystal structures show that the orbital splitting33 in the closest-contact dimers decreases in the order 2,6-DSAnt, 1,5-DSAnt, 9,10-DSAnt (ΔHOMO = 0.41 eV, 0.11 and 0.03 eV, respectively, Table 2). Together with the calculated hole transfer reorganization energy λ+ (which increases along with the dihedral angle of the styryl substituents, Table 2) these trends predict that the highest hole mobility should be observed for the 2,6-DSAnt isomer.
| Compnd | E HOMO (eV) | E LUMO (eV) | E g (eV) | E optg (eV) | λ absmax (nm) | λ emmax (nm) | PLQY (%) | τ PL, nsec | Stokes shift (eV) | λ + (eV) | ΔHOMOd (eV) |
|---|---|---|---|---|---|---|---|---|---|---|---|
| a Estimated as ELUMO = [−4.8 − Eonsetredvs. Fc/Fc+] eV and EHOMO = [−4.8 − Eoxonsetvs. Fc/Fc+] eV.33 b 2,6-DPSAnt showed a biexponential decay with a small (6%) contribution of the second component τPL = 18.5 nsec.35 c DFT-calculated hole transfer reorganization energy.36 d Calculated splitting of the HOMO (HOMO − HOMO − 1) for the closest contact molecular dimers observed by X-ray crystallography. | |||||||||||
| 2,6-DPSAnt | −4.91(DFT) | −1.91(DFT) | 3.00(DFT) | 3.03(solut) | 395(solut) | 409 (solut) | 68(solut) | 3.1b | 0.11(solut) | 0.14 | 0.41 |
| −5.5(CV)a | −2.6 (CV)a | 2.9(CV)a | 2.58(solid) | 440(solid) | 546 (solid) | 14(solid) | 0.54(solid) | ||||
| 1,5-DPSAnt | −4.93(DFT) | −1.87(DFT) | 3.07(DFT) | 2.69(solut) | 412(solut) | 461 (solut) | 69(solut) | 3.9 | 0.32(solut) | 0.16 | 0.11 |
| −5.3(CV)a | 2.6(CV)a | 2.7(CV)a | 2.53(solid) | 433(solid) | 470 (solid) | 4–9(solid) | 0.27(solid) | ||||
| 9,10-DPSAnt | −4.82(DFT) | −1.85(DFT) | 2.97(DFT) | 2.65(solut) | 412(solut) | 581(solut) | 32(solut) | 4.3 | 0.81(solut) | 0.19 | 0.03 |
| −5.2 (CV)a | −2.7(CV)a | 2.5(CV)a | 2.47(solid) | 423(solid) | 531(solid) | 18–22(solid) | 0.65(solid) | ||||
Electrochemistry
The electrochemical properties of distyrylanthracene derivatives were studied in solution by cyclic voltammetry (CV, Fig. 3). The CV of 2,6-DPSAnt showed one reversible oxidation at Eox1/2 = 0.68 V and one reversible reduction at Ered1/2 = −2.28 V vs. Fc/Fc+. 1,5-DPSAnt displayed a similar reversible electrochemical behavior with Eox1/2 = 0.57 V and Ered1/2 = −2.27 V. The oxidation of 9,10-DPSAnt proceeds in two partially reversible waves at E1ox1/2 = 0.51 V and E2ox1/2 = 0.63 V; a single irreversible reduction is observed at Eredpc = −2.20 V. The HOMO–LUMO energies were determined from the onsets of the first oxidation and reduction potentials as EMO = [−4.8 − Eonsetvs. Fc/Fc+] eV (ref. 34) (Table 2). The electrochemical gaps are reduced in the range 2,6-DPSAnt (3.0 eV) → 1,5-DPSAnt (2.7 eV) → 9,10-DSAnt (2.5 eV).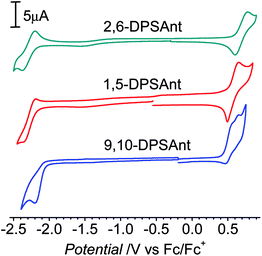 | ||
| Fig. 3 CV of 2,6-DPSAnt (in chlorobenzene, 85 °C), 1,5-DPSAnt and 9,10-DPSAnt (in CH2Cl2). | ||
Optical properties
The optical absorption/emission properties of distyrylanthracenes were studied in toluene solution and in the solid state (microcrystalline powders and thin films). In solution 2,6-DPSAnt exhibits an absorption band with a pronounced vibronic splitting (λmax = 341, 356, 374 and 395 nm; Fig. 4), suggesting a very rigid structure of the molecule. In contrast, the absorption spectra of 1,5-DPSAnt and 9,10-DPSAnt (λmax = 412 nm for both) are broad and featureless. This is attributed to the rotational degree of freedom of the styryl group, brought about by steric repulsion with peri-hydrogens of the anthracene ring. The optical band gaps of 2,6-, 1,5- and 9,10-DPSAnt determined from the onset of absorption are 3.03, 2.69 and 2.64 eV, respectively (Table 2 and ESI†). This red shift of absorption occurs notwithstanding the significant increase of twist of the styryl substituent from fully planar for 2,6-DPSAnt, to 34° in 1,5-DPSAnt to 52° in 9,10-DPSAnt (according to gas-phase DFT calculations). We conclude that electronic communication in the anthracene between the 9,10-positions is more efficient than that between 1,5- and, particularly, 2,6-positions, in agreement with the analysis of the bond-length alternation (see above). The same trend and very similar values of the HOMO–LUMO gap were obtained from electrochemical measurements (see above). However, DFT calculations, while giving a very similar value of the HOMO–LUMO gap (3.00 eV for 2,6-DPSAnt, Table 2), fail to reproduce the trend for the other two isomers. This might be due to an overestimation of the twist angle in gas-phase DFT calculations that increases the predicted gap for 1,5- and 9,10-DPSAnt as compared to solution-based experimental values.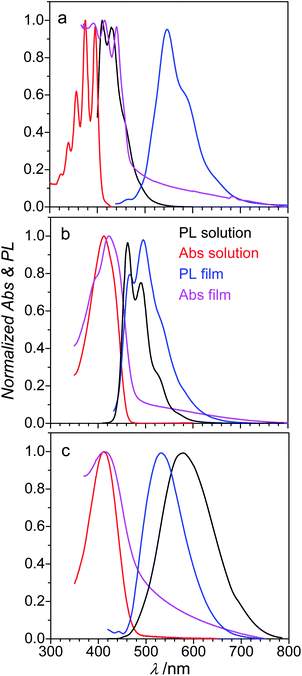 | ||
| Fig. 4 Absorbance and PL spectra of (a) 2,6-DPSAnt, (b) 1,5-DPSAnt and (c) 9,10-DPSAnt in toluene solution and in the solid state. | ||
The photoluminescence (PL) spectrum of 2,6-DPSAnt in solution shows a vibronically structured band (λmax = 409 nm, 428 nm) with a fairly small Stokes shift of 14 nm (0.11 eV). The PL spectra of 1,5-DPSAnt and 9,10-DPSAnt reveal much larger Stokes shift of 49 nm (0.32 eV) and 170 nm (0.81 eV), respectively. Such large reorganization of the excited state must be due to partial or full planarization of these molecules in the excited state. A clear vibronic structure of the emission of 1,5-DPSAnt (λmax = 461 nm, 491 nm and 530 nm) suggests a significant rigidification of the molecule in the excited state, expected for a planarized structure. In contrast, the emission of 9,10-DPSAnt remains featureless (λmax = 581 nm), implying that a substantial dihedral twist is maintained in the excited state.
The above conclusions are also supported by TD-DFT calculations (ESI†). For 2,6-DPSAnt, both the ground and the excited states are planar; for 1,5-DPSAnt, excitation is predicted to cause substantial planarization (anthracene/vinylene torsion angle decreases to 13°); however, for 9,10-DPSAnt, a non-negligible twist (35°) is maintained in the excited state. The calculated Stokes shifts are 0.29 eV, 0.51 eV and 0.55 eV for 2,6-, 1,5- and 9,10-DPSAnt, respectively.
In line with the rigid structure of their excited states, 2,6-DPSAnt and 1,5-DPSAnt showed the highest solution PLQY (∼70%) while the PLQY of 9,10-DPSAnt was lower (∼30%, Table 2). 9,10-DPSAnt possesses the longest luminescence lifetime in the series (4.3 ns, Table 2) that can likely be attributed to the shorter transition dipole moment of 9,10-DPSAnt brought about by its geometry.
Intermolecular interactions in the solid state have very different effects on the optical properties of the three isomers. The optical band-gap of 2,6-DPSAnt in the solid films is contracted vs. that in solution by 0.45 eV (to 2.58 eV), while this contraction for 1,5- and 9,10-DPSAnt is only 0.16 and 0.17 eV, respectively. This observation is in agreement with X-ray crystallographic analysis (see above) which showed that only 2,6-DPSAnt has direct π–π interactions between the anthracene cores in the solid state. Accordingly, the exciton delocalization in the solid state of 2,6-DPSAnt leads to a much larger Stokes shift in the solid state (0.54 eV) compared to that in solution (0.11 eV). In contrast, the Stokes shifts of 1,5- and 9,10-DPSAnt in the solid state (0.27 eV and 0.65 eV) are actually lower than those in solution since planarization of the excited states is hindered in the solid state. This effectively causes a blueshift of the PL band of 9,10-DPSAnt in the solid state compared to that in solution (Fig. 4).
The absolute PLQYs in the solid state were measured in the integrating sphere (Table 2). PLQY values were somewhat sensitive to the morphology of the sample, as also observed in anthracene.20 However, in all measurements the highest PLQY was observed for 9,10-DPSAnt (18–22%), followed by 2,6-DPSAnt (14%) and 1,5-DPSAnt (4–9%). The powder PLQYs are likely limited by the exciton quenching on surface defects and larger values are expected in single crystal samples.20,29 Strong aggregation-induced emission (AIE) was previously reported for 9,10-distyrylanthracene and its dialkoxy derivatives, with PLQY increasing from 0.4–0.7% in THF solution to 30–50% in aqueous THF and in the crystals.31 While the relatively high solid-state PLQY of 9,10-DPSAnt is in line with the AIE report,31 no net enhancement of PLQY for the solid vs. solution was observed in our case. Furthermore, only weak (∼2-fold) emission enhancement was observed upon dilution of THF solution of 9,10-DPSAnt with water (ESI†).
Thin-film device fabrication and studies
Thin films of 2,6-, 1,5- and 9,10-DPSAnt were prepared by slow (0.2–0.4 Å s−1) vacuum deposition on oxidized Si wafers (190 nm of SiO2) kept at room temperature. The morphologies of the formed films were analyzed by Atomic Force Microscopy (AFM), both for the early growth stage (sub-monolayers) and for the fully covered samples.The submonolayer films of 2,6-DPSAnt consist of islands of uniform height (∼3.0 nm) which implies the molecules are deposited in an upright configuration (molecular length 3.46 nm) with some tilt vs. surface normal (Fig. 5a and b). X-ray diffraction analysis of the films showed a series of (h00) peaks with a characteristic d-spacing of 3.4 nm which corresponds well with the unit cell found in the single crystal analysis (½a).25 However, the lateral size of these islands is relatively small (up to ∼0.1 μm, mean area 0.004 μm2) which leads to a large number of grain boundaries in fully covered films. This is in contrast to monosubstituted 2-(4-hexylstyryl)anthracene which produces very large islands in the same deposition conditions;29 the difference is likely attributable to higher molecular mobility of a smaller molecule.
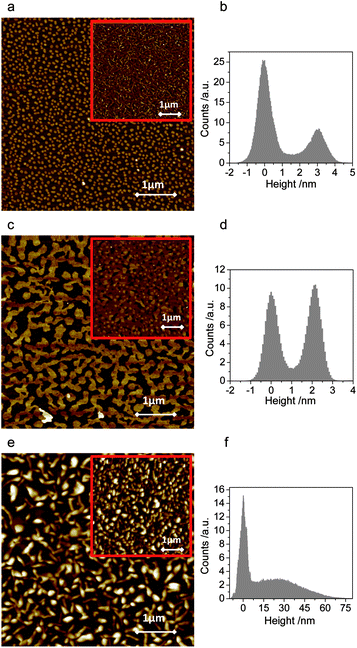 | ||
| Fig. 5 AFM images (5 μm × 5 μm) of submonolayer films of 2,6-DPSAnt (a), 1,5-DPSAnt (d) and 9,10-DPSAnt (e) grown on SiO2/Si. Corresponding height profiles are shown in (b, d and f). The insets show AFM images of fully covered substrates (nominal thickness of 35 nm for (a) and (c) and 100 nm for (e)). | ||
The films of 1,5-DPSAnt also grow in much larger islands (up to μm size, mean area 0.1 μm2) with good interconnection (Fig. 5c and d). At higher coverage, the islands continue to grow laterally to form an almost fully connected monolayer before the second layer starts growing. Such morphology formed at low nominal thickness is beneficial for high charge mobility in OFET applications. X-ray analysis of the films features a strong peak at 3.2° corresponding to a d-spacing of 2.75 nm, which is similar to the molecular length of 1,5-DPSAnt (2.94 nm). However, such a diffraction peak is not expected from the single-crystal X-ray data (see ESI†). Together with AFM data (monolayer thickness of ∼2.1 nm), this indicates that in vacuum-deposited films 1,5-DPSAnt adopts a layered structure with molecules standing upright on the surface – a different molecular arrangement from that found in solution-grown crystals.
In contrast, the growth mode of 9,10-DPSAnt is most detrimental for OFET applications. The nucleation of small grains is accompanied by 3D growth which leads to separate islands of large thickness (up to 75 nm) and poor interconnection (Fig. 5e and f). The full coverage of the surface required depositing films of very high nominal thicknesses (∼100 nm). We could not promote layer-by-layer growth of 9,10-DPSAnt through hexamethyldisilazane (HMDS) treatment of the substrate surface.
Top-contact OFET devices were prepared by evaporating gold electrodes on the ∼50 nm films of distyrylanthracenes. The measured output and transfer characteristics of 2,6-DPSAnt are shown in Fig. 6a and b. The devices showed hole mobility of 0.48 ± 0.11 cm2 V−1 s−1 (μ+max 0.75 cm2 V−1 s−1) and on–off ratio 105 to 107 in the saturated regime. There were no significant differences between the performance of devices prepared on bare Si/SiO2 and those on HMDS treated substrates. The relatively large negative threshold voltage VT = −18 ± 2 V can be ascribed to the low HOMO of 2,6-DPSAnt. These values are consistent with the earlier communication by Meng et al.25 reporting the hole mobility of 2,6-DPSAnt in top-contact OFETs in the range 0.1–1.28 cm2 V−1 s−1, depending on the temperature of the substrate during film deposition. Also, Klauk et al. reported hole mobility of non-alkylated 2,6-(di-β-styryl)-anthracene of up to 1.3 cm2 V−1 s−1 (in top-contact OFETs for films deposited at 80 °C).37 Very similar hole mobility (up to 1.3 cm2 V−1 s−1) was also measured for 2-(4-hexylstyryl)anthracene thin film OFET,29 which appears to be representative for 2(6)-substituted anthracenes with styryl groups.
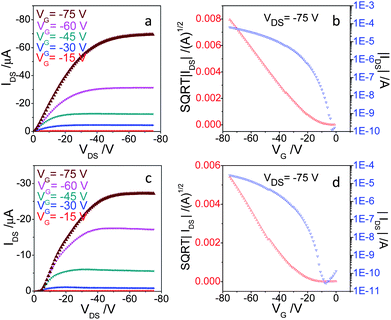 | ||
| Fig. 6 (a and c) Output and (b and d) transfer characteristics of OFETs based on 2,6-DPSAnt (top) and 1,5-DPSAnt (bottom) MSCs. | ||
OFETs fabricated with 1,5-DSAnt under identical conditions yield a somewhat lower but appreciable mobility in the range of 0.11 ± 0.05 cm2 V−1 s−1 (μ+max 0.15 cm2 V−1 s−1) with a threshold voltage of −23 ± 2 and on–off ratio 105 to 106. Such mobility was not expected based on the single crystal X-ray analysis data which show no orbital overlap between anthracene cores in 1,5-DSAnt. On the other hand, comparison of the X-ray diffraction pattern of thin films with that of single crystals clearly shows a different morphology of the former. In contrast, devices fabricated with 9,10-DPSAnt showed no transistor characteristics (i.e., only sub-nA current and no gate effect). This is attributed mainly to the unfavorable packing arrangements found in the crystal structure and poor film morphology of 9,10-DPSAnt.
The drain–source current flow in the transistors based on 2,6-DSAnt or 1,5-DPSAnt is accompanied by strong green electroluminescence readily observed even in single channel top-contact devices (Fig. 7a and b). Recording the current output and light emission as a function of the drain–source voltage (Vds) for different gate voltages (Vg) shows gate-modulated electroluminescence in p-channel driven devices (Fig. 7c); thus, the devices function as OLETs. Increasing the Vg reduces the turn-on voltage of the electroluminescence. For both materials, the electroluminescence zone is localized in the vicinity of the drain electrode which is consistent with the unipolar (p-type) transport characteristics of the devices. The asymmetric positions of the HOMO and LUMO of distyrylanthracenes (Table 2) with respect to the work function of Au (ca. −5 eV) creates an energy barrier for electron injection into the channel and confines the EL in the vicinity of the electron-injecting electrode.
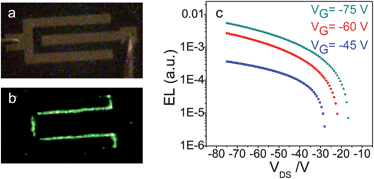 | ||
| Fig. 7 Optical images of top-contact OLET (W/L ∼ 2000/100 μm μm−1) before (a) and after (b) biasing, exhibiting a green electroluminescence from the device. (c) EL current characteristics of OLET based on a 2,6-DPSAnt. | ||
Conclusions
In summary, we studied the effect of positional isomerism of distyryl-substituted anthracenes on molecular packing in the solid state and their optoelectronic properties. Changing the position of styryl substituents from 2,6- to 1,5- and 9,10- leads to progressive increase of out-of-plane twist of the vinyl groups. Despite this fact, the lowest HOMO–LUMO gap, according to optical and electrochemical measurements, has been observed for the 9,10-DPSAnt which suggests a more efficient electronic communication between these positions. This can be rationalized by lower bond-length alternation in the anthracene core between the 1,5- and 9,10-positions compared to 2,6-positions, as observed by X-ray crystallography.The semiconducting properties of the three isomers were evaluated in thin-film transistors. In agreement with the lack of anthracene⋯anthracene contacts in single crystal structures and poor thin film morphology, no field-modulated conductance was observed in 9,10-DPSAnt based devices. However, both 2,6-DPSAnt and 1,5-DPSAnt showed good transistor characteristics with hole mobilities of up to 0.75 and 0.15 cm2 V−1 s−1, respectively. Such relatively high charge mobility was not expected for 1,5-DPSAnt based on its packing found by single crystal X-ray analysis. The molecules adopt a different arrangement in vacuum-grown thin films, which was confirmed by XRD and AFM studies. This emphasizes the role of polymorphism in complicating the predictions of charge mobility in thin films based on bulk crystallographic analysis.
All three isomers are highly emissive in the solid state and the transistors fabricated with 2,6-DPSAnt and 1,5-DPSAnt showed field-modulated intense green electroluminescence. The emission zone is localized near the electron-injecting drain electrode, in accordance with the unipolar (hole-transporting) characteristics of the devices.
The work shows that simple modification of the anthracene core allows access to new luminescent semiconducting materials with tunable properties. In particular, high solubility and surprisingly good charge mobility observed in 1,5-DPSAnt as well as enhanced electronic communication achieved through such substitution patterns (as compared to more explored 2,6-substition) make the 1,5-disubstituted anthracene an attractive building block for molecular and polymeric optoelectronic materials.
Author contributions
The manuscript was written with contributions from all authors. All authors have given approval to the final version of the manuscript.Acknowledgements
This work was supported by NSERC of Canada through Discovery Grants to D.F.P. and F.R. F.R. acknowledges partial salary support from the Canada Research Chairs program. D.F.P. and F.R. are also grateful to FRQNT for team grants. We acknowledge Prof. G. Hanan (University of Montreal) for access to life-time measurements.References
- S. R. Forrest, Nature, 2004, 428, 911 CrossRef CAS.
- S. B. Zhao and S. Wang, Chem. Soc. Rev., 2010, 39, 3142 RSC.
- M. C. Gather, A. Kohnen and K. Meerholz, Adv. Mater., 2011, 23, 233 CrossRef CAS.
- A. R. Murphy and J. M. Fréchet, Chem. Rev., 2007, 107, 1066 CrossRef CAS.
- A. N. Sokolov, B. C.-K. Tee, C. J. Bettinger, J. B.-H. Tok and Z. Bao, Acc. Chem. Res., 2012, 45, 361 CrossRef CAS.
- B. Walker, C. Kim and T.-Q. Nguyen, Chem. Mater., 2011, 23, 470 CrossRef CAS.
- Y. Sun, G. C. Welch, W. L. Leong, C. J. Takacs, G. C. Bazan and A. Heeger, Nat. Mater., 2012, 11, 44 CrossRef CAS.
- H. Meng and N. Herron, in Organic Light-Emitting Materials and Devices, ed. Z. R. Li and H. Meng, CRC Press, 2006 RSC; S.-B. Zhao and S. Wang, Chem. Soc. Rev., 2010, 39, 3142 RSC.
- A. Hepp, H. Heil, W. Weise, M. Ahles, R. Schmechel and H. von Seggern, Phys. Rev. Lett., 2003, 91, 157406 CrossRef.
- K. Sawabe, M. Imakawa, M. Nakano, T. Yamao, S. Hotta, Y. Iwasa and T. Takenobu, Adv. Mater., 2012, 24, 6141 CrossRef CAS.
- F. Dinelli, R. Capelli, M. A. Loi, M. Murgia, M. Muccini, A. Facchetti and T. J. Marks, Adv. Mater., 2006, 18, 1416 CrossRef CAS.
- I. F. Perepichka, D. F. Perepichka, H. Meng and F. Wudl, Adv. Mater., 2005, 17, 2281 CrossRef CAS; P. Taraneker, Q. Qiao, H. Jiang, K. S. Schanze and J. R. Reynolds, J. Am. Chem. Soc., 2007, 129, 8958 CrossRef.
- W. Wu, Y. Liu and D. Zhu, Chem. Soc. Rev., 2010, 39, 1489 RSC.
- H. Klauk, M. Halik, U. Zschieschang, G. Schmid, W. Radlik and W. Weber, J. Appl. Phys., 2002, 92, 5259 CrossRef CAS.
- C. Wang, H. Dong, H. Hu, Y. Liu and D. Zhu, Chem. Rev., 2012, 112, 2208 CrossRef CAS.
- M. J. Kang, I. Doi, H. Mori, E. Miyazaki, K. Takimiya, M. Ikeda and H. Kuwabara, Adv. Mater., 2011, 23, 1222 CrossRef CAS.
- A. N. Sokolov, S. Atahan-Evrenk, R. Mondal, H. B. Akkerman, R. S. Sanchez-Carrera, S. Granados-Focil, J. Schrier, S. C. B. Mannsfeld, A. Zoombelt, P. Zoombelt, Z. N. Bao and A. Aspuru-Guzik, Nat. Commun., 2011, 2, 437 CrossRef.
- A. Y. Amin, A. Khassanov, K. Reuter, T. Meyer-Friedrichsen and M. Halik, J. Am. Chem. Soc., 2012, 134, 16548 CrossRef CAS.
- M. B. Smith and J. Michl, Chem. Rev., 2010, 110, 6891 CrossRef CAS.
- R. Katoh, K. Suzuki, A. Furube, M. Kotani and K. Tokumaru, J. Phys. Chem. C, 2009, 113, 2961 CAS.
- A moderate hole mobility (μ+ = 2 × 10−2 cm2 V−1 s−1) was measured in anthracene single crystal transistors at low temperature: A. N. Aleshin, J. Y. Lee, S. W. Chu, J. S. Kim and Y. W. Park, Appl. Phys. Lett., 2004, 84, 5383 CrossRef CAS.
- Also, a very high hole and electron mobility (35–50 cm2 V−1 s−1) was reported for single crystals of anthracene based on low temperature time-of-flight (TOF) experiments: N. Karl and J. Martkanner, Mol. Cryst. Liq. Cryst. Sci. Technol., Sect. A, 2001, 355, 149 CrossRef CAS.
- H. Meng, F. P. Sun, M. B. Goldfinger, G. D. Jaycox, Z. G. Li, W. J. Marshall and G. S. Blackman, J. Am. Chem. Soc., 2005, 127, 2406 CrossRef CAS.
- J. A. Merlo, C. R. Newman, C. P. Gerlach, T. W. Kelley, D. V. Muyres, S. E. Fritz, M. F. Toney and C. D. Frisbie, J. Am. Chem. Soc., 2005, 127, 3997 CrossRef CAS.
- H. Meng, F. Sun, M. B. Goldfinger, F. Gao, D. J. Londono, W. J. Marshall, G. S. Blackman, K. D. Dobbs and D. E. Keys, J. Am. Chem. Soc., 2006, 128, 9304 CrossRef CAS.
- M. C. Um, J. Jang, J. P. Hong, D. Y. Yoon, S. H. Lee, J. J. Kim and J. I. Hong, J. Mater. Chem., 2008, 18, 2234 RSC.
- D. S. Chung, T. K. An, C. E. Park, H.-J. Yun, S.-K. Kwon and Y.-H. Kim, Appl. Phys. Lett., 2012, 101, 193304 CrossRef.
- H.-J. Yun, D. S. Chung, I. Kang, J. W. Park, Y.-H. Kim and S.-K. Kwon, J. Mater. Chem., 2012, 22, 24924 RSC.
- A. Dadvand, A. G. Moiseev, K. Sawabe, W. H. Sun, B. Djukic, I. Chung, T. Takenobu, F. Rosei and D. F. Perepichka, Angew. Chem., Int. Ed., 2012, 51, 3837 CrossRef CAS.
- F. Silvestri, A. Marrocchi, M. Seri, C. Kim, T. J. Marks, A. Facchetti and A. Taticchi, J. Am. Chem. Soc., 2010, 132, 6108 CrossRef CAS.
- M. J. Frisch, et al., Gaussian 09, Revision B.01, Wallingford CT, 2009 Search PubMed.
- J. He, B. Xu, F. Chen, H. Xia, K. Li, L. Ye and W. Tian, J. Phys. Chem. C, 2009, 113, 9892 CAS.
- J. L. Bredas, J. P. Calbert, D. A. da Silva and J. Cornil, Proc. Natl. Acad. Sci. U. S. A., 2002, 99, 5804 CrossRef CAS.
- Conversion of the electrochemical redox potentials into vacuum-scale orbital energies by referencing to ferrocene is somewhat contraversial. The recent literature rationalized the HOMO of ferrocene in the range of −4.76 to −5.2 eV: B. W. D'Andrade, S. Datta, S. R. Forrest, P. Djurovich, E. Polikarpov and M. E. Thompson, Org. Electron., 2005, 6, 11 CrossRef CAS; C. M. Cardona, W. Li, A. E. Kaifer, D. Stockdale and G. C. Bazan, Adv. Mater., 2011, 23, 2367 CrossRef The value of −4.8 eV, used in this paper, appears most often in the literature and is in good agreement with the (solid state) photoelectron spectroscopy data.
- G. Bartocci, U. Mazzucato, A. Spalletti and F. Elise, Spectrochim. Acta, Part A, 1990, 46, 413 CrossRef.
- N. E. Gruhn, D. A. da Silva Filho, T. G. Bill, M. Malagoli, V. Coropceanu, A. Kahn and J. L. Bredas, J. Am. Chem. Soc., 2002, 124, 7918 CrossRef CAS.
- H. Klauk, U. Zschieschang, R. T. Weitz, H. Meng, T. Sun, G. Nunes, D. E. Keys, C. R. Fincher and Z. Xiang, Adv. Mater., 2007, 19, 3882 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available: Details of DFT calculations; TGA data; details of fluorescence lifetime measurements; XRD of 1,5-DPSAnt films; a movie of operating OLET based on 2,6-DPSAnt. CCDC 923794 and 923795. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c3tc30247d |
| ‡ Current address: Institute of Advanced Materials, Nanjing University of Technology, 30 Puzhu Road(S), Nanjing, P. R. China. |
| This journal is © The Royal Society of Chemistry 2013 |