Chemometric determination of lipidic parameters in serum using ATR measurements of dry films of solvent extracts†
David
Perez-Guaita
a,
Angel
Sanchez-Illana
a,
Josep
Ventura-Gayete
b,
Salvador
Garrigues
*a and
Miguel
de la Guardia
a
aDepartment of Analytical Chemistry, University of Valencia, 50 Dr Moliner Street, Research Building, 46100 Burjassot, Valencia, Spain. E-mail: salvador.garrigues@uv.es
bUniversity Hospital Doctor Peset Aleixandre, Av. Gaspar Aguilar, 90, 46017 Valencia, Spain
First published on 25th October 2013
Abstract
Attenuated total reflectance (ATR) infrared spectroscopy of dried organic extracts of serum samples has been evaluated as a fast method for the determination of triglycerides, cholesterol, high density lipoprotein (HDL) and low density lipoprotein (LDL). After careful selection of solvents based on green parameters, serum samples were extracted using hexane–isopropanol and ethyl acetate–ethanol mixtures. Microscopy studies and comparison with standard spectra were performed in order to investigate whether the proposed methodology is suitable for the quantification of lipids in serum samples. The results of these preliminary studies confirmed that the variations in the IR spectra of sample extracts could be related quantitatively to variations in the concentrations of the target analytes. Then, ATR spectra of the dried sample extracts were obtained and direct measurement of the spectra were carried out and modelled using partial least squares (PLS) and reference concentrations. PLS models obtained from the extracts of the two mixtures were compared with those obtained from direct measurement of sera samples. The prediction errors obtained using the proposed approach were considerably (between 27 and 72%) better than those obtained by the direct measurements of sera. For triglycerides and cholesterol relative errors below 9% and 12% respectively were obtained with this method, which are comparable to the tolerance for the errors of the control analysis established at the hospital. For HDL and LDL, the errors found were between 18 and 20%. The incorporation of a preprocessing extraction step, involves time and solvent consumption. However, the results obtained provide evidence that the proposed method provides, in a few minutes and using simple instrumentation and with minimum cost, important information about the lipidic profile of patients sera at a good screening confidence level.
1. Introduction
The clinical analysis of blood and urine are almost indispensable for the diagnosis, prevention, and monitoring of several illness. Among them, the determination of lipids and related molecules such as triglycerides, cholesterol, high density lipoproteins (HDL) and low density lipoproteins (LDL) are crucial in the diagnosis and prevention of cardiovascular diseases.1 In addition, the concentration of the aforementioned analytes has been also recently related to depressive symptoms.2 Besides, lipidomics, i.e. the large-scale study of pathways and networks of lipids in biological systems,3 is an expanding research field which leads to important improvements in diagnosis and therapeutics.4Since the aforementioned analyses are often carried out using enzymatic methods through high-performance auto-analyzers, the associated cost can be too high for performing screening studies or monitoring analyses. In addition, samples must be transported to a fully-equipped laboratory, making the information available after a time delay, which may hamper timely diagnosis.5 Thus, in the last decades different analytical strategies have been applied for moving testing closer to the patient, so-called point-of-care testing (PoCT), which has several economic and clinical benefits.6
Fourier transformed infrared spectroscopy (FTIR) has been evidenced as one of the most promising techniques for obtaining concentration values of the main components, including the aforementioned lipids,7,8 in the analysis of several bio-fluids; such as serum,9 plasma,10 whole blood or urine.11 Since almost every bioorganic compound presents absorbance in the infrared region, infrared spectra provide a ‘snapshot’ of the main composition of the biological samples. However, the overlapping of the bands of the target analytes with those of several compounds contained in the samples makes mandatory the use of chemometrics, commonly partial least squares (PLS). For spectra acquisition, samples can be dried and measured by transmission12 or can be placed directly into an attenuated total reflectance (ATR) unit.11,13,14 ATR-FTIR permits a straightforward measurement of several parameters for the analysis of sera, which can be obtained in a few minutes and involves no or minimal pretreatment of samples or use of reagents. Besides, the equipment required is not complex and nowadays there are available compact and portable instruments suitable to be used for PoCT. Nevertheless, this technique also presents important drawbacks. First of all, the lack of sensitivity of the ATR technique implies that only analytes present at percentage levels in samples can be properly quantified.15 In addition, the protein bands overshadow most of the bands of the less concentrated compounds. There are also errors of precision and accuracy associated with reference data and finally, due to the high complexity and heterogeneity of the samples, several reference standards must be included in the calibration set in order to obtain a representative and robust calibration.9
Recent advances in ATR-FTIR have been focused on improving the prediction errors. E.g., quantum cascade lasers have been used as an infrared source in the determination of several clinical parameters in the plasma of critical patients, thus improving the signal-to-noise ratio of the measurements16 and compacting considerably the instrumentation needed. In addition, locally weighted regression-PLS has been used instead of PLS in order to improve the representativeness of the calibration set.17 Other approaches have been applied to eliminate the interference caused by the important contribution of proteins to the spectra. Proteins make up approximately 90% of the non-aqueous fraction of plasma and, because of that, proteins are easily determined using FTIR spectroscopy.18 However, the strong amide bands can hinder the determination of less concentrated molecules. Hence, serum creatinine was successfully quantified by using a laminar fluid diffusion interface which isolates small molecules as creatinine from proteins.19,20
To our knowledge, the analysis of serum components by FTIR after deproteinization with organic solvents has still not been investigated. From serum samples, organic solvents should extract the major lipids and related compounds, i.e. those involved in important clinical parameters (triglycerides, cholesterol and lipoproteins). Since lipids include a wide range of nonpolar and highly nonpolar molecules, normally a mixture of solvents is used for their extraction.21 Folch reagent is the most commonly used for this propose. Nevertheless, due to environmental issues,22 other mixtures of solvents such as hexane–isopropanol23 and ethyl acetate–ethanol24 have been proposed as alternatives.
Besides, drying samples or their organic components in an ATR crystal is an easy and rapid way of obtaining an infrared spectrum free from the contribution of solvent bands and thus with an enhanced signal-to-noise ratio. It has already been used for discriminating red wine cultivars25 and for detecting the presence of polyphenol cocoa metabolites in urine.26 However, due to the possible irreproducibility of the drying step and the low penetration depth of the beam in ATR measurements, the size and shape of the sample deposits obtained after a drying step should be investigated if this strategy is selected for performing a quantitative measurement.
The aim of this work has been to evaluate the use of ATR infrared spectra of serum organic extracts as a fast and PoC approach to determine the concentration of triglycerides, cholesterol, HDL and LDL. First, since the method is designed for the screening and control of several samples on a large scale, a careful selection of the solvents to be used for sample pretreatment was realized considering ‘green’ factors.22 The ATR spectra of dried sample extracts were obtained and modelled using PLS based on reference concentrations obtained by enzymatic methods. PLS models built from the extracts of the two solvent mixtures were compared with those obtained from direct measurements of sera samples. Results evidenced that the proposed approach is suitable as a fast tool for the determination of important lipidic parameters in sera, thus being interesting for PoCT.
2. Experimental
2.1 Chemicals and sample collection
Standards employed were cholesterol (purity > 99%), L-α-lysophosphatidylcholine (purity > 99%) and glyceryl trioleate (purity > 99%) from Sigma-Aldrich (Madrid, Spain). The organic solvents used in this study were purchased from Scharlau (Germany) and were of gradient HPLC grade.Serum samples with reference data of some of the studied parameters were obtained from the Hospital Dr Peset Alexandre (Valencia, Spain), being that the reason because we have not used a fixed number of samples to build all the models assayed. Reference concentrations of HDL, LDL, triglycerides and cholesterol were determined using an Abbot Architect c16000 auto-analyzer (Libertyville, IL, USA) as described elsewhere.9 The error of the reference data was considered as the tolerance in the validation analysis of controls established in the analysis protocol and performed daily at the hospital. The tolerance was two times the standard deviation (SD). However, since in most of the cases the values were inside the SD, a declared accuracy value of ±10% of the reported results, corresponding to the SD was considered. Table 1 summarizes the main descriptive statistics of the sample reference data used throughout this study.
| Analyte | Set | Hexane–isopropanol 3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 :1 |
Ethyl acetate–ethanol 3:1 |
Direct measurement | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Mean | SD | Min | Max | N | Mean | SD | Min | Max | N | Mean | SD | Min | Max | N | ||
| a All values are in mg dL−1. Cal and val indicate the sets used for calibration and validation respectively. SD, min, max and N indicate respectively standard deviation, minimum and maximum concentration value and the number of samples included on each set. | ||||||||||||||||
| HDL | Cal | 36 | 8 | 47 | 24 | 28 | 35 | 10 | 58 | 18 | 32 | 35 | 10 | 58 | 13 | 29 |
| Val | 34 | 11 | 58 | 15 | 21 | 34 | 9 | 50 | 15 | 24 | 36 | 8 | 50 | 21 | 16 | |
| LDL | Cal | 91 | 34 | 160 | 39 | 24 | 86 | 34 | 178 | 35 | 32 | 78 | 24 | 143 | 29 | 29 |
| Val | 82 | 28 | 159 | 46 | 21 | 86 | 30 | 160 | 39 | 24 | 77 | 28 | 124 | 35 | 14 | |
| Cholesterol | Cal | 158 | 41 | 247 | 83 | 28 | 148 | 44 | 259 | 53 | 38 | 139 | 28 | 200 | 87 | 29 |
| Val | 141 | 31 | 214 | 87 | 21 | 153 | 42 | 247 | 83 | 27 | 145 | 42 | 235 | 83 | 16 | |
| Triglycerides | Cal | 138 | 79 | 397 | 62 | 28 | 116 | 67 | 306 | 41 | 37 | 125 | 72 | 299 | 46 | 28 |
| Val | 137 | 84 | 345 | 46 | 21 | 153 | 90 | 397 | 65 | 26 | 124 | 61 | 227 | 41 | 13 | |
2.2 Direct ATR-FT-IR measurements
A Bruker IFS 66/v FTIR spectrometer (Bremen, Germany) equipped with a liquid nitrogen-refrigerated mercury–cadmium–telluride detector, a vacuum purge system, and a dry air purged sample compartment was used for the spectra acquisition. In all cases, an in-compartment ATR DuraSampleIR accessory with a nine reflection diamond/ZnSe DuraDisk from Smiths Detection Inc. (Warrington, UK) was adapted to the spectrophotometer. For the acquisition and handling of spectra OPUS software version 6.5 from Bruker was employed.Direct measurements were performed by depositing 150 μL of serum samples directly on the ATR cell and a spectrum was acquired averaging 300 scans, using a spectrum of the clean empty ATR cell as background. Spectra were acquired in the range between 600 and 4000 cm−1 with a resolution of 4 cm−1. A blank of Milli-Q water was obtained after each three samples measured and was subtracted from the samples in order to eliminate water contribution. After each measurement cleaning of the ATR cell was performed using soft cellulose and a 0.9% (w/v) sodium chloride aqueous solution in order to avoid protein precipitation and, finally the cell was rinsed with Milli-Q water.
2.3 Determination procedure involving extraction
80 μL of serum sample and 800 μL of the extraction solvent were mixed in an Eppendorf and homogenized in a vortex mixer for 5 seconds. The resultant slurry was then centrifuged at a speed of 4500g at 25 °C for 5 minutes. 2 μLof the organic extract were deposited carefully in the ATR crystal using a 5 μL Hamilton syringe #7005 (Bonaduz, GR, Switzerland) and the deposit was allowed to dry for 2 minutes. A spectrum of the resulting dry film was acquired using the same acquisition parameters considered for obtaining the direct spectra of serum samples. In this case after each measurement, the ATR crystal was cleaned using soft cellulose paper, the solvent used for the extraction and deionized water, until no bands were observed in the measured spectrum of the blank. A first evaluation of the most suitable solvent for the extraction was performed by using hexane and mixtures of ethyl acetate–ethanol 3:1 (v/v), hexane–isopropanol 3:1 (v/v), and chloroform–methanol 3:1 (v/v) (Folch reagent). Ethyl acetate–ethanol and hexane–isopropanol mixtures were selected for further studies.
2.4 Spectra obtained and PLS modeling
Since the volume of the samples was limited, not all the samples were analyzed using the assayed three methods (two solvents used for the extraction and the direct measurement), this is the reason why models were performed with a different number of samples. However, the reference values interval of the samples used for each method covered a wide range of concentrations for both, calibration and validation sets (see Table 1). For each measurement method, samples were randomly distributed between calibration and validation sets using the rand function available in Matlab. In some cases the rand function distributed the samples without covering the same range of reference concentrations for both sets, and the distribution was corrected exchanging manually a maximum of two samples. Approximately 30 samples were used for calibration and around 15–25 for validation, depending on the number of samples measured and the reference data available.PLS models were built using Matlab 7.7.0 (Mathworks Inc. Natik, MA, USA). Interval wavenumber ranges used for modeling were 810–1757 cm−1 for spectra obtained from direct measurements and 3161–2440 cm−1 and 1815–844 cm−1 for spectra obtained from the dried organic extracts. The root means square error of cross validation (RMSCV) calculated using the leave-one-out method was used for selecting the number of latent variables for performing the calibration for each model (see Tables 2–5). The spectra matrix was preprocessed by using a Savisky–Golatz 1st derivate (polynomial order 2 and filter width 15) and mean centering. Concentration data were mean centered as well.
| Parameter | Direct measurement | Hexane–isopropanol (3:1) |
Ethyl acetate–ethanol (3:1) |
|---|---|---|---|
| a RMSCV: root mean square error of cross validation, RMSEP: root mean square error of prediction, RRMSEP: relative root mean square error of prediction, RPD: residual predictive deviation. | |||
| RMSCV/mg dL−1 | 68.82 | 25.82 | 19.7 |
| RMSEP/mg dL−1 | 31.36 | 23.00 | 18.52 |
| RRMSEP (%) | 24.89 | 16.91 | 12.10 |
| RPD | 1.945 | 3.761 | 4.877 |
| Latent variables | 4 | 4 | 6 |
| Repeatability/mg dL−1 | 6.29 | 6.73 | |
| Relative repeatability (%) | 8.7 | 9.3 | |
For comparing the models obtained in the direct measurement to those obtained after protein precipitation and lipid extraction, the following parameters were considered: for evaluating the prediction capability of the models, root mean square error of prediction (RMSEP) and ratio of performance to deviation (RPD), defined as the ratio between RMSEP and the standard deviation of the prediction set reference values, were used. Besides, for quantifying the relative accuracy of the models, the relative root mean square error of prediction (RRMSEP) defined as the RMSEP divided by the average concentration value of the prediction set was used.
Since the step which involved the deposition and the drying of the organic extract was suspected to be an important source of imprecision, a study of the effect of these operations on the measurement repeatability and its influence on the precision of the predictions of analyte concentrations was performed. For this purpose, samples included in a new set of 5 samples that were not integrated in the calibration, nor in the validation set were extracted in triplicate using both assayed solvent mixtures and extracts were measured and predicted using the models obtained with the calibration set. The mean of the SD of the prediction results obtained for each sample was used for evaluating the precision of each measurement method.
2.5 Acquisition of the confocal images
Microscopy studies were performed in order to obtain information about the shape and size of the dry film produced after evaporation of the organic extracts of samples, using images of the dry films deposited on the ATR crystal. However, it was impossible to place the ATR unit in the focus of the microscope. To simulate the system, a steel ring with the same inner diameter as the ATR cell (4 mm) was pressed mechanically against a glass slide simulating the ATR unit. 2 μL of extracts were deposited on the crystal along the inner space of the O-ring. After drying, the O-ring was carefully removed. Images of the films of extracts of a sample obtained using hexane–isopropanol 3:1 and ethyl acetate–ethanol 3:1 were obtained. For comparison purposes images were also obtained of the dry film acquired by diluting the serum with Milli-Q water 1:10, at the same level employed to carry out extractions (1:10).
Images were obtained using an OLYMPUS (Barcelona, Spain) FV1000 confocal microscope operated in the reflection mode and equipped with a dry objective UPLSAPO (20×) with a numeric aperture of 0.75. The resolution of the measurements in the z-axis was 1.39 μm. Open source software ImageJ was used for handling the images.27
3. Results and discussion
3.1 FTIR spectra obtained and selection of solvents
Fig. 1 compares the spectra of untreated serum samples with those obtained from the extracts of samples using the two solvent mixtures selected for the PLS modeling, together with the spectra of three standards representing the analytes under study, i.e., cholesterol, triolein as a molecular model of triglycerides, and L-α-lysophosphatidylcholine as a typical phospholipid (one of the main family of components of HDL and LDL).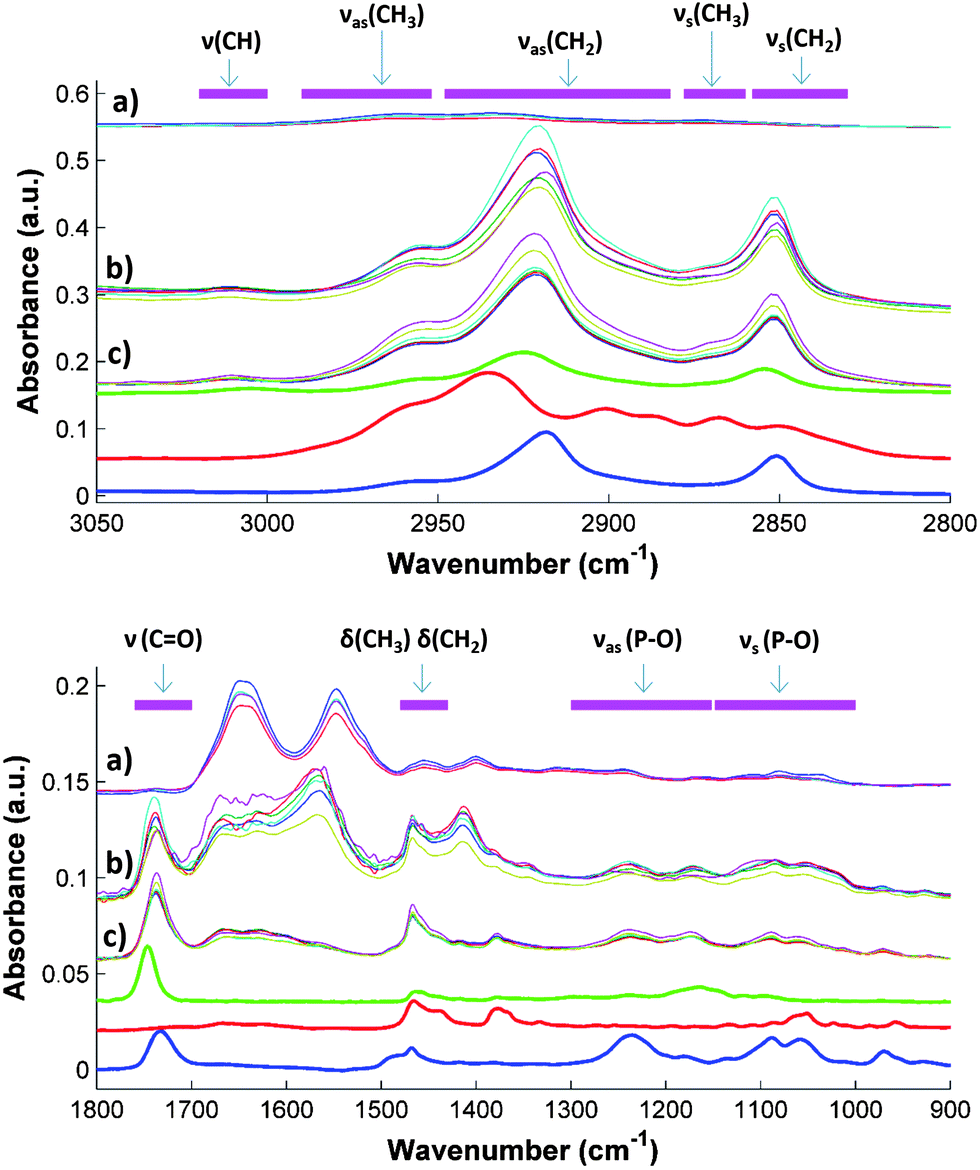 | ||
| Fig. 1 ATR-FTIR spectra of untreated and extracted serum samples: direct spectra after subtraction of a water blank (a), dried extracts of samples obtained using ethyl acetate–ethanol (b), extracts using hexane–isopropanol (c), spectra of L-α-lysophosphatidylcholine (blue), cholesterol (red) and triolein (green) standards. Horizontal magenta lines indicate the regions related to the absorption bands of the chemical bonds (see the text for additional information). NOTE: spectra were shifted for a clear comparison and untreated serum spectra were divided by 3. Standard spectra were obtained by drying two μL of a standard solution of 10 mg dL−1 in hexane–isopropanol on the ATR crystal. | ||
The type of molecules extracted was strongly related to the size, shape and position of the IR bands. Antisymmetric νas(P–O) and symmetric νs(P–O) stretching vibrations, characteristics of phospholipids, were found in the interval regions 900–1100 and 1100–1300 respectively.28 Regarding the alkyl groups, found in every lipid, the δas(CH3), δas(CH2), δs(CH3), δs(CH2) bending (scissoring) were found in the 1400–1450 cm−1 (ref. 10 and 29) range and the ν(CH3), δas(CH2), δs(CH3), δs(CH2) stretching vibrations between 2800 and 3100 cm−1. 10,29 Finally, the ν(C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O) stretching vibration of the phospholipids and the esters of fatty acids were found in the region between 1700 and 1760 cm−1.10,29
O) stretching vibration of the phospholipids and the esters of fatty acids were found in the region between 1700 and 1760 cm−1.10,29
A figure (SM1) available in the ESI† shows the ATR-FTIR spectra acquired from the different extracts of the same sample obtained using the solvents considered in the preliminary study. In SM1 there are also indicated the regions assigned to the major chemical groups found in the lipids present in sera.
A visual inspection of the spectra reveals that each solvent mixture extracted different amounts and types of lipids. Due to the low absorbance of the hexane extract, this solvent was discarded. Concerning solvent mixtures, ethyl acetate–ethanol 3:1 extracted the largest amount of lipids as indicated by the big absorption bands in the 2800–3200 cm−1 interval. Hence, this mixture was selected for further studies. Besides, the extracts obtained from the Folch reagent and the mixture composed of hexane–isopropanol 3:1 presented similar spectra, with an unidentified wide band at 1500–1700 cm−1. Since both mixtures presented similar behavior, only one was selected for performing the study. As the proposed method is intended for its large-scale use as a screening tool, mixtures were compared in terms of their green parameters (e.g. toxicity of the waste, safety for the operator, etc.30), using hazard statements available in the MERK index.31 Thus, the use of Folch reagent, which includes chloroform, a potentially carcinogen solvent, was excluded.23 An overview of the comparison of solvents employed for sample extraction is available in the ESI (SM2).†
Spectra obtained from the direct measurements were dominated by the protein amide bands, which hampered the specific bands of lipids. In the case of the C–H region (2800–3050 cm−1), water absorption was too high and the subtraction of the water blank was not effective enough to show clearly the C–H bands. In contrast, in the spectra of serum extracts strong differences between the samples can be seen in the absorption regions of the standards, providing evidence that changes in the spectra are caused by the different amount of cholesterol, triglycerides, HDL and LDL in the samples. For example, the maximum position and intensity of the CH2 and CH3 bands located in the 2800–3050 cm−1 range varied widely among the samples, specifically the maximum of the band assigned to the νas(CH2). Standard spectra provide evidence that the mentioned variation can be explained taking into account the different size and shape of the bands of the different components under study. Something similar occurs in the fingerprint region, where strong variations in the size and shape of the ν(CO) and the ν(P–O) bands were observed. In this region bands of triolein and L-α-lysophosphatidylcholine standard were also located. In short, the visual comparison of spectra shown in Fig. 2 reveals that the differences in the bands found in sample spectra are presumably due to differences in the concentration of the compounds under study.
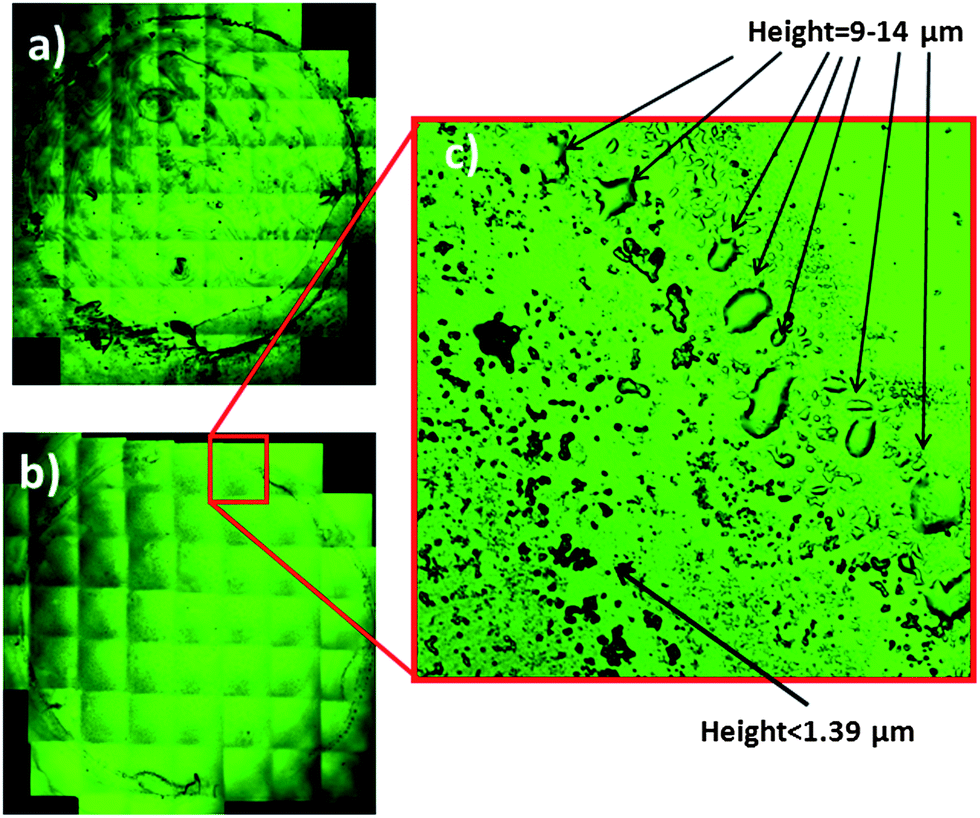 | ||
| Fig. 2 Confocal microscopy image of the dry film obtained by (a) drying 2 μL of a sample diluted 1:10 with deionized water and (b) drying an extract of the same sample obtained by diluting (1:10) in a mixture of ethyl acetate–ethanol. (c) Enlargement of a section of the crown formed in the proximity of the steel ring used for sample confinement (see the text for additional information). | ||
3.2 Size and shape of the dry films obtained after extraction
A confocal microscopy study of the topography of the deposits of the dry film extracts was performed in order to identify potential problems with the reproducibility of the spectra acquisition. This study was important, since (i) if the dry deposits obtained were not distributed regularly along the ATR crystal, the measurements will not be reproducible and (ii) because of the small penetration depth of the infrared radiation (0.5–2μm), only relative changes in the composition of the samples could be observed. The microscopy images of dry extracts obtained with ethyl acetate–ethanol 3:1 and those obtained for the same sample diluted with water and dried are shown in Fig. 2. For measuring the thickness of the deposits the number of reflection plans found was multiplied by the resolution in the z-axis of the apparatus (in this case 1.39 μm). Images obtained show that the dry film produced from water was an irregular shaped film which did not cover all of the crystal surface, and the thickness that could not be accurately measured because it was too high (>20 μm). This fact may be due to the high surface tension of water and the great amount of material deposited due to the presence of proteins. In contrast to this image, for the extract with the solvent mixture, a series of solid particles and little drops was found, which covered all of the crystal area delimited by the O-ring. In the extreme where the steel ring was placed (see enlargement) a little crown composed of big drops with a height between 9.73 and 13.9 μm was observed. Nevertheless, inside the crown, in the main part of the crystal the height of the drops was too low to be measured (<1.39 μm). Regarding the image obtained when the extract of hexane–isopropanol was dried, it a similar set of droplets with a height of less than 1.39 μm distributed along the crystal surface and a crown with big drops with a height around 10 μm were observed. Figures (SM 3–5) of the dried extracts are available in the ESI† (see also Fig. 3).
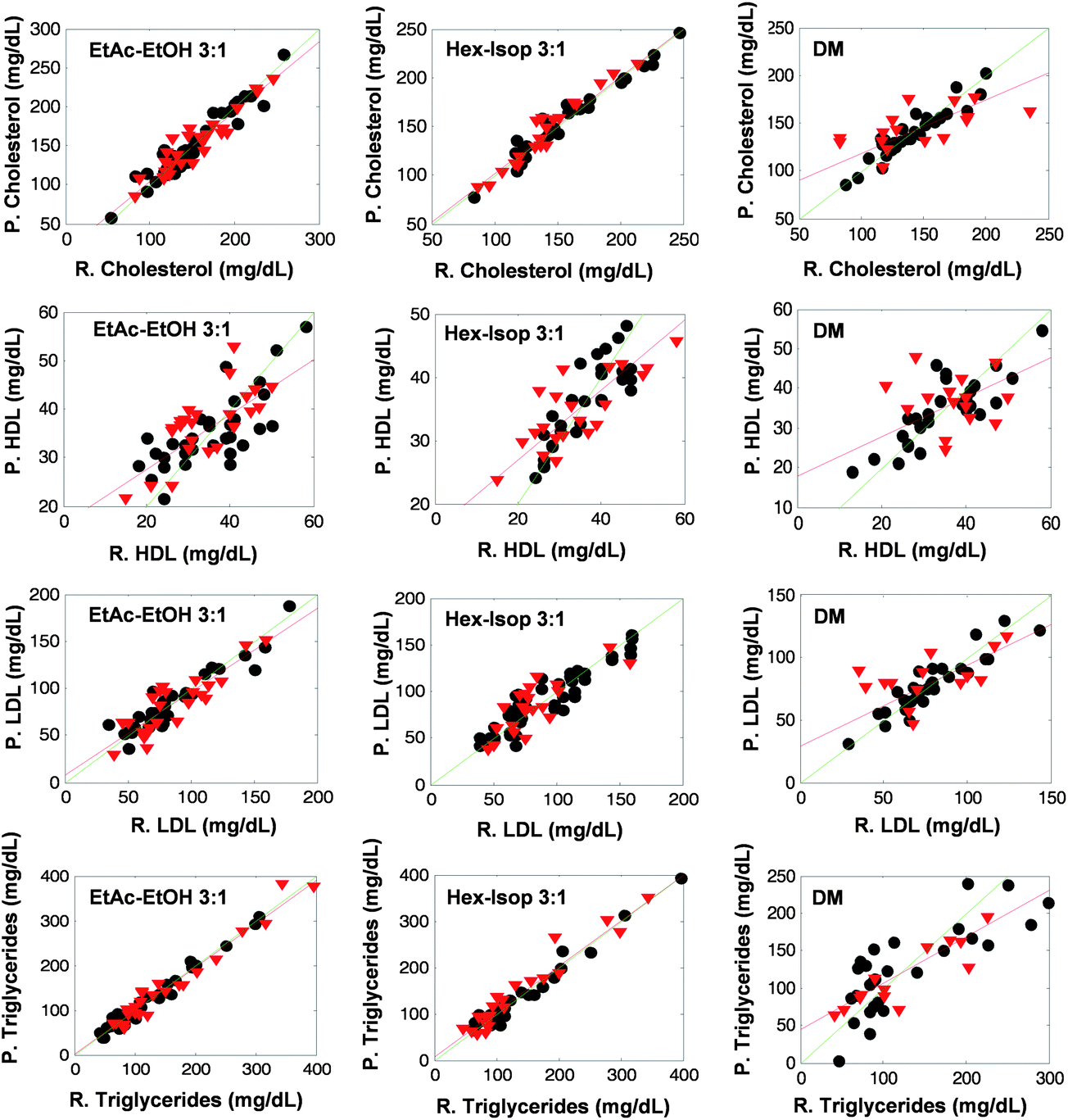 | ||
| Fig. 3 Predicted values vs. reference ones for cross validation (grey circles) and prediction (red triangles) obtained for all the models built through this study. For lipidic components determination in sera green line indicates a straight line with slope = 1 and intercept = 0 and red line represents the regression model for the prediction set. Note, EtAc–EtOH: ethyl acetate–ethanol, Hex–Isop: hexane–isopropanol, DM: direct measurement, P: predicted concentration value and R: reference concentration value. | ||
The main conclusion that can be obtained from this study is that irregular films were obtained by drying aqueous diluted sera. However, in the case of sample extracts obtained with organic solvents, the measurement area was covered uniformly by drops of regular size and their thickness in the main part of the crystal was smaller than the penetration depth of the beam, thus allowing the acquisition of quantitative information of the components present in the samples in a reproducible way.
3.3 PLS models
Tables 2–5 summarize the parameters considered in this study for the evaluation of the prediction capability of the PLS models built and the repeatability of the measurement step. In all the cases the worst prediction capability was found for direct measurement, the RMSEP of the models obtained from the spectra of extracts were always 27 to 72 per cent lower than those obtained by direct measurement. This fact is graphically denoted in Fig. 4, where the plots of reference values vs. predicted ones are shown. It can be seen that the sample concentration values for both, calibration and validation sets cover a wide range of concentrations. Besides, the regression line of the prediction samples (red lines) fitted better to a straight line with slope and intercept values equal to 1 and 0 respectively (green lines) for the models built through lipid extract measurements than in the case of models performed using direct measurements.| Parameter | Direct measurement | Hexane–isopropanol (3:1) |
Ethyl acetate–ethanol (3:1) |
|---|---|---|---|
| a RMSCV: root mean square error of cross validation, RMSEP: root mean square error of prediction, RRMSEP: relative root mean square error of prediction, RPD: residual predictive deviation. | |||
| RMSCV/mg dL−1 | 22.19 | 16.93 | 17.25 |
| RMSEP/mg dL−1 | 32.65 | 9.27 | 14.55 |
| RRMSEP (%) | 22.53 | 6.57 | 9.48 |
| RPD | 1.28 | 3.35 | 2.85 |
| Latent variables | 4 | 5 | 5 |
| Repeatability/mg dL−1 | 14.5 | 7.54 | |
| Relative repeatability (%) | 8.9 | 4.6 | |
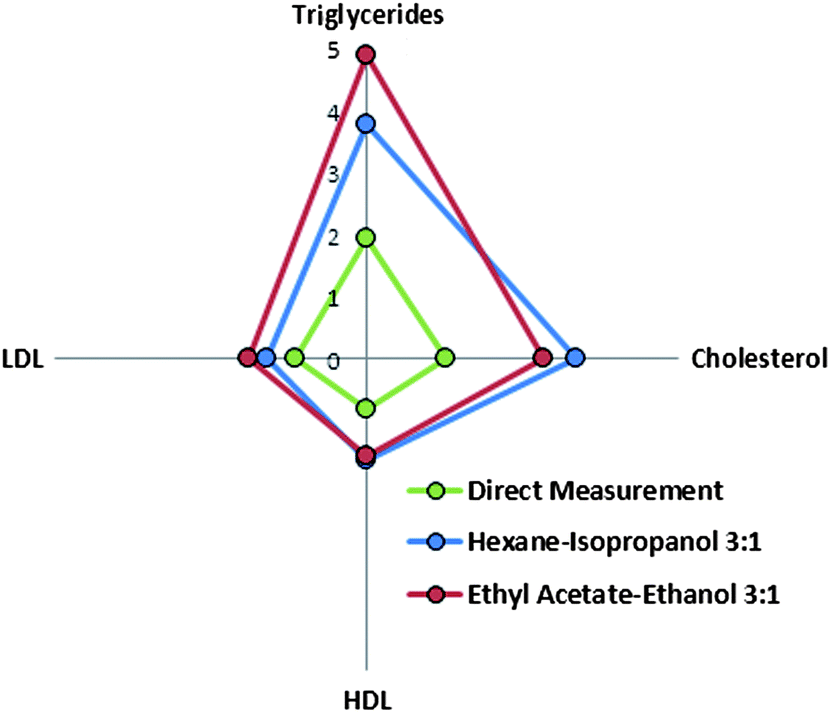 | ||
| Fig. 4 Radial plot of ratio of performance to deviation values obtained for all the models built for PLS-ATR-FTIR determination of lipidic compounds in sera. | ||
Nevertheless, the goodness of the validation was strongly dependent on the analyte considered. Fig. 4 represents the RPD calculated for the validation of the analytes under study. RPD values for cholesterol and triglycerides were found to be two times higher than those obtained for the lipoproteins. A similar profile of RPD can also be seen for models created using the spectra of extracts, in all the cases the RPD values obtained were better than those obtained by direct measurement. Comparing the two extraction procedures, the best prediction capability for triglycerides was found for the extraction of sera with ethyl acetate–ethanol mixture, with a RPD value 30% higher than the RPD obtained for the models obtained using the hexane–isopropanol mixture. In contrast, for cholesterol, the RPD obtained was 18% higher for the extraction with hexane–isopropanol than in the case of using ethyl acetate–ethanol mixture, thus providing evidence of the relative capability of each solvent mixture to extract well the different lipidic compounds present in sera.
3.4 Evaluation of the method as a screening tool and comparison with other methodologies based on IR
The accuracy of the determination of triglycerides and cholesterol in sera is strongly improved using the proposed approach based on PLS treatment of ATR-FTIR spectra of sample extracts dried on the ATR cell. Whereas the relative prediction errors obtained for the validation of models performed using direct measurement were found to be higher than 20%, the errors obtained from measurements made after solvent extraction were 16 and 12% for cholesterol and between 6.6 and 9.5% for triglycerides (see Tables 2–5). So, the incorporation of the extraction step produces an important enhancement of the accuracy of the prediction of the target analytes giving acceptable results for diagnosis, even when only a small number of samples was used for calibration. It has to be noted that the results for these analytes are comparable with the tolerance expressed as the SD of the controls measured daily in the hospital (10%) and thus it can be concluded that the accuracy of the developed approach is comparable to that of the costly instrumentation employed nowadays in our hospitals. In the case of HDL and LDL, relative accuracy errors obtained by direct ATR-FTIR measurements (around 30%) were also improved by employing measurements of extracts (between 18 and 20%). In this case those errors cannot be compared with the 10% of tolerance mentioned above. However the method developed could provide an effective screening tool and the fact that reference values of LDL were indirectly calculated using the reference data of HDL, triglycerides and cholesterol must be considered.| Parameter | Direct measurement | Hexane–isopropanol (3:1) |
Ethyl acetate–ethanol (3:1) |
|---|---|---|---|
| a RMSCV: root mean square error of cross validation, RMSEP: root mean square error of prediction, RRMSEP: relative root mean square error of prediction, RPD: residual predictive deviation. | |||
| RMSCV/mg dL−1 | 8.15 | 7.05 | 9.95 |
| RMSEP/mg dL−1 | 10.25 | 6.55 | 6.56 |
| RRMSEP (%) | 28.02 | 19.19 | 20.21 |
| RPD | 0.80 | 1.64 | 1.56 |
| Latent variables | 5 | 4 | 4 |
| Repeatability/mg dL−1 | 2.59 | 1.26 | |
| Relative repeatability (%) | 5.8 | 2.8 | |
The repeatability of the drying step was found to be an important source of irreproducibility. Relative standard deviations of the measurements of three replicates obtained by drying the same sample extracts (see the repeatability data in the last line of Tables 2–5) were higher than half of the relative accuracy in the case of triglycerides, cholesterol and HDL. Hence, an automation of the deposition of the samples could improve the prediction results. However, this automation also increases the complexity and cost of the instrumentation, thus limiting its application as a PoC methodology.
| Parameter | Direct measurement | Hexane–isopropanol (3:1) |
Ethyl acetate–ethanol (3:1) |
|---|---|---|---|
| a RMSCV: root mean square error of cross validation, RMSEP: root mean square error of prediction, RRMSEP: relative root mean square error of prediction, RPD: residual predictive deviation. | |||
| RMSCV/mg dL−1 | 20.11 | 15.58 | 16.00 |
| RMSEP/mg dL−1 | 24.64 | 17.13 | 15.82 |
| RRMSEP (%) | 32.00 | 20.90 | 18.38 |
| RPD | 1.139 | 1.607 | 1.912 |
| Latent variables | 5 | 4 | 4 |
| Repeatability/mg dL−1 | 14.51 | 7.6 | |
| Relative repeatability (%) | 14.7 | 7.7 | |
In the ESI†is available a table (SM6) which compares the errors obtained, number of samples used for calibration and number of latent variables selected in this study compared with those performed by Hosafçi et al.11 and Liu et al.7 It must be indicated that different approaches for obtaining the spectra were employed in each one of the three compared studies that were namely, direct measurement of whole blood in the case of Hoisafi et al., transmission measurement of dry films of sera in the case of Liu et al. and ATR measurement of organic extracts of sera in the case of the methodology proposed in this paper. Although the best prediction capability was obtained for the models built following the proposed methodology, it must be noticed that the validation sets used were different. However, it seems clear that the proposed methodology, using smaller calibration sets than previous ones and simpler PLS models (latent variables) is able to obtain better or comparable results than those obtained in previous works. This fact can be explained by the extraction step which eliminates the strong bands of proteins from the spectra. Nevertheless, this preprocessing step also implies the use of organic solvents and a preprocessing time.
4. Conclusions
The evaluation of the use of ATR-FTIR spectroscopy after sera extraction as a tool for the fast determination of triglycerides, cholesterol, HDL and LDL shows that prediction errors obtained using the proposed approach are considerably lower than those obtained by direct ATR measurements of sera. For triglycerides and cholesterol the method achieves accuracy prediction relative errors below 9% and 12% respectively, which are comparable to the tolerance of the reference methods. However, for HDL and LDL errors found were between 18 and 20%. The main drawback of this technique is that it requires a preprocessing extraction step which involves time and solvent utilization. However, in this work we have performed a careful selection of the solvent which minimizes the toxicity of the reagents and wastes. In addition, although the preconcentration step hampers the utilization of this methodology as a PoC tool the process can be miniaturized and the integration of the extraction step in the centrifugation of the blood for obtaining serum could also be studied. Thus, the developed method provides, in a few minutes and with minimum cost, important information about the lipidic profile of the serum of patients at a screening confidence level.Acknowledgements
Authors gratefully acknowledge the financial support of the Ministerio de Economía y Competitividad and FEDER (Projects CTQ2011-25743 and 2012-38635) and the Generalitat Valenciana (Project PROMETEO 2010-055). DPG acknowledges the “V Segles” grant provided by the University of Valencia. Furthermore the technical support from the Servicio de Microscopia and Antonio José Ibáñez González at the University of Valencia (Spain) for carrying out the microscope images measurements is kindly acknowledged.References
- W. S. Aronow, Clinical Lipidology, 2012, 7, 689–695 CrossRef CAS.
- C. Y. Fang, B. L. Egleston, K. P. Gabriel, V. J. Stevens, P. O. Kwiterovich, L. G. Snetselaar, M. L. Longacre and J. F. Dorgan, J. Behav. Med., 2012, 36, 143–152 CrossRef PubMed.
- M. R. Wenk, Nat. Rev. Drug Discovery, 2005, 4, 594–610 CrossRef CAS PubMed.
- X. Han and R. W. Gross, Mass Spectrom. Rev., 2005, 24, 367–412 CrossRef CAS PubMed.
- A. J. Tudos, G. J. Besselink and R. B. Schasfoort, Lab Chip, 2001, 1, 83–95 RSC.
- A. St John, Clin. Biochem. Rev., 2010, 31, 111–119 Search PubMed.
- K. Z. Liu, R. A. Shaw, A. Man, T. C. Dembinski and H. H. Mantsch, Clin. Chem., 2002, 48, 499–506 CAS.
- K. Z. Liu, A. Man, T. C. Dembinski and R. A. Shaw, Anal. Bioanal. Chem., 2006, 387, 1809–1814 CrossRef PubMed.
- D. Perez-Guaita, J. Ventura-Gayete, C. Pérez-Rambla, M. Sancho-Andreu, S. Garrigues and M. de la Guardia, Microchem. J., 2013, 106, 202–211 CrossRef CAS.
- G. Deleris and C. Petibois, Vib. Spectrosc., 2003, 32, 129–136 CrossRef CAS.
- G. Hosafçi, O. Klein, G. Oremek and W. Mantele, Anal. Bioanal. Chem., 2006, 387, 1815–1822 CrossRef PubMed.
- S. Low-Ying, R. A. Shaw, M. Leroux and H. H. Mantsch, Vib. Spectrosc., 2002, 28, 111–116 CrossRef CAS.
- G. Janatsch, J. D. Kruse-Jarres, R. Marbach and H. M. Heise, Anal. Chem., 1989, 61, 2016–2023 CrossRef CAS PubMed.
- A. Roth, F. Dornuf, O. Klein, D. Schneditz, H. Hafner-Giessauf and W. Mantele, Anal. Bioanal. Chem., 2012, 403, 391–399 CrossRef CAS PubMed.
- D. Rohleder, G. Kocherscheidt, K. Gerber, W. Kiefer, W. Kohler, J. Mocks and W. Petrich, J. Biomed. Opt., 2005, 10, 031108 CrossRef CAS PubMed.
- M. Brandstetter, T. Sumalowitsch, A. Genner, A. Posch, C. Herwig, A. Drolz, V. Fuhrmann, T. Perkmann and B. Lendl, Analyst, 2013, 138, 4022–4028 RSC , in press.
- D. Perez-Guaita, J. Kuligowski, G. Quintás, S. Garrigues and M. de la Guardia, Talanta, 2013, 107, 368–375 CrossRef CAS PubMed.
- D. Perez-Guaita, J. Ventura-Gayete, C. Pérez-Rambla, M. Sancho-Andreu, S. Garrigues and M. de la Guardia, Anal. Bioanal. Chem., 2012, 404, 649–656 CrossRef CAS PubMed.
- B. Schattka, M. Alexander, S. L. Ying, A. Man and R. A. Shaw, Anal. Chem., 2011, 83, 555–562 CrossRef CAS PubMed.
- R. A. Shaw, C. Rigatto, M. Reslerova, S. L. Ying, A. Man, B. Schattka, C. F. Battrell, J. Matthewson and C. Mansfield, Analyst, 2009, 134, 1224–1231 RSC.
- T. P. Ferraz, M. Fiúza, M. L. dos Santos, L. Pontes de Carvalho and N. Soares, J. Biochem. Biophys. Methods, 2004, 58, 187–193 CrossRef CAS PubMed.
- S. Armenta, S. Garrigues and M. de la Guardia, TrAC, Trends Anal. Chem., 2008, 27, 497–511 CrossRef CAS.
- A. Hara and N. Radin, Anal. Biochem., 1978, 90, 420–426 CrossRef CAS PubMed.
- J.-H. Lin, L.-Y. Liu, M.-H. Yang and M.-H. Lee, J. Agric. Food Chem., 2004, 52, 4984–4986 CrossRef CAS PubMed.
- A. Edelmann, J. Diewok, K. C. Schuster and B. Lendl, J. Agric. Food Chem., 2001, 49, 1139–1145 CrossRef CAS PubMed.
- C. Sánchez-González, W. Nigussie, R. Estruch, R. M. Lamuela-Raventós, M. Izquierdo-Pulido and S. de Lamo-Castellví, Analyst, 2012, 137, 3565–3570 RSC.
- C. A. Schneider, W. S. Rasband and K. W. Eliceiri, Nat. Methods, 2012, 9, 671–675 CrossRef CAS PubMed.
- F. M. Goñi and J. L. R. Arrondo, Faraday Discuss. Chem. Soc., 1986, 81, 117–126 RSC.
- C. Petibois, G. Cazorla, A. Cassaigne and G. Deleris, Clin. Chem., 2001, 47, 730–738 CAS.
- Handbook of Green Analytical Chemistry, ed. M. de la Guardia and S. Garrigues, Wiley, Chichester, 2012 Search PubMed.
- http://www.merckmillipore.com/ .
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c3an01057k |
| This journal is © The Royal Society of Chemistry 2014 |