Improved accuracy for label-free absolute quantification of proteome by combining the absolute protein expression profiling algorithm and summed tandem mass spectrometric total ion current†
Qi
Wu
ab,
Yichu
Shan
a,
Yanyan
Qu
ab,
Hao
Jiang
ab,
Huiming
Yuan
a,
Jianxi
Liu
ab,
Shen
Zhang
ab,
Zhen
Liang
a,
Lihua
Zhang
*a and
Yukui
Zhang
a
aNational Chromatographic Research and Analysis Center, Key Laboratory of Separation Science for Analytical Chemistry, Dalian Institute of Chemical Physics, Chinese Academy of Sciences, Dalian, 116023, China. E-mail: lihuazhang@dicp.ac.cn; Fax: +86 411 8437 9720; Tel: +86 411 8437 9720
bUniversity of Chinese Academy of Sciences, Beijing, China
First published on 17th October 2013
Abstract
Proteome scale absolute quantification is fundamental for the quantitative understanding of an organism. The unsatisfactory accuracy for protein abundance estimation of current algorithms has been partially improved by the Absolute Protein EXpression profiling (APEX) algorithm, which implements the prior expectations of peptides' appearances in the calculation of protein abundances. However, the abundance feature (AF) in APEX is the spectral count (SC); an AF suffers from a narrow dynamic range, thus, unsatisfactory accuracy. Therefore, we adopted another tandem mass spectrometric (MS/MS) level AF called Summed MS/MS Total ion current (SMT), which cumulates the MS/MS fragment intensities rather than simply counting the MS/MS spectra, to surmount this particular deficiency. The combination of APEX and SMT (abbreviated as APEX-SMT) is capable of improving the accuracy of absolute quantification by reducing the average relative deviation by ∼55–85% compared to that of APEX, through a series of tests on the Universal Proteomics Standard sample with a dynamic range of 5 orders of magnitude (UPS2). The algorithm could also be used for relative quantification. When applied to the relative quantification of a publicly available benchmark dataset, APEX-SMT could provide comparable accuracy to APEX. All these results suggest that APEX-SMT is a promising alternative to APEX for proteome quantification.
1. Introduction
Proteome scale absolute quantification plays a vital role in the comprehensive investigation of the metabolism of an organism and the mathematical modeling of systems biology, such as the rates of protein production and degradation, and the cost of a metabolic pathway.1 The ability to quantitate the proteomes could be helpful for investigating the life activities.2Although mass spectrometry (MS)-based absolute quantification of proteins has been achieved by spiking in known amounts of isotopically labeled peptides or proteins (e.g., Absolute Quantification of proteins, AQUA;3 Selected Reaction Monitoring or Multiple Reaction Monitoring assay, SRM/MRM;4 Protein Standard Absolute Quantification, PSAQ5), the proteome scale absolute quantification remains a privilege for label-free methods, which mainly focuses on development of new algorithms to meet the need for accurate and reproducible quantification. Many abundance features (AFs) in MS detection have been successfully utilized for the estimation of absolute quantities of all identified proteins in a sample,6 such as the precursor intensity (top 3 most abundant peptides,7 intensity-Based Absolute Quantification, iBAQ8), peptide coverage (Exponentially Modified Protein Abundance Index, emPAI9), tandem mass spectrometric (MS/MS) spectral count (SC) (Normalized Spectral Abundance Factor, NSAF10) and the newly accepted MS/MS fragment intensity (Spectral Index, SI;11 Summed MS/MS Total ion current, SMT12–14).
It is worth noting that the detectabilities and intensities of peptides on a certain liquid chromatography (LC)-MS platform could vary dramatically due to the different physicochemical properties of peptides. In other words, different proteins of equal amount could yield very diverse MS AFs.15 With such a phenomenon taken into consideration, the quantification accuracy might be improved. Lu et al. applied machine learning methods to reduce this discrepancy and developed a SC-based algorithm, named “Absolute Protein EXpression profiling” (APEX),16 which correlates well with other non-MS measurements, such as western blotting, two-dimensional gels, flow cytometry, and mRNA abundance validation, for both yeast and Escherichia coli proteomes. Different from precursor-based quantification, the information for MS/MS-based quantification is readily available. Therefore, algorithms based on SC have received massive adoption in the label-free quantification community.17 However, they suffer from narrow dynamic ranges, which could seriously jeopardize the quantification accuracy.14
Recent discoveries12–14,18 including our previous work find that summations of MS/MS fragment intensities are promising alternatives to broaden the quantitative dynamic ranges. Two major forms have been developed, abbreviated as SMT12–14 and SI.11 The former cumulates all fragment intensities of all MS/MS spectra assigned to a protein, while the latter cumulates only the intensities of matched fragments in all MS/MS spectra assigned to a protein. To the best of our knowledge, the combination of APEX and SMT (or SI) has not been reported yet. This combination has the potential to broaden the dynamic range of absolute quantification and preserve the merit of machine learning algorithms.
Herein, SC is replaced with SMT and SI in the APEX formula, resulting in two modified versions of the APEX algorithm, which are named as APEX-SMT and APEX-SI. Through a series of tests using the Universal Proteomics Standard sample with a known dynamic range (UPS2), we find that compared to APEX and APEX-SI, APEX-SMT could yield wider dynamic range and better accuracy. In the meantime, the benefit of machine learning methods is indeed preserved. Moreover, we find that any existing high quality dataset on a certain instrumental setup could be utilized for training. Furthermore, through testing the algorithms on a publicly available benchmark dataset,19 we find that APEX-SMT could offer comparable accuracy to APEX in relative quantification as well.
2. Materials and methods
2.1 Algorithm design
The APEX algorithm, given by formula (1), first corrects SCi of protein i with the summation of peptides' detectabilities (Oi) and protein identification probability (pi), and then normalizes across the sample (totally N proteins), generating the proportion of each protein relative to the total sample abundance. The absolute abundance of every individual identified protein could be calculated through multiplying the proportion by the estimated total protein abundance (C) in the sample. The reason APEX is termed as an absolute quantification measurement is that it does not require at least two samples to be compared with each other, which is a prerequisite in relative quantification.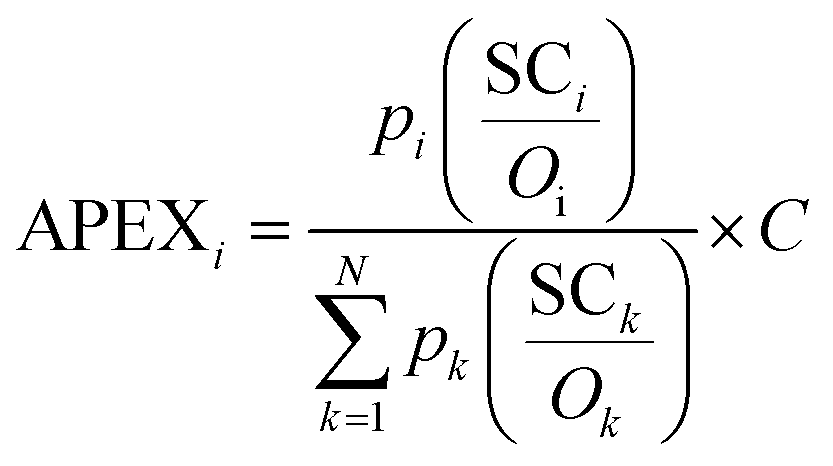 | (1) |
The calculations of SC, SI and SMT are shown in formulae (2)–(4), which are similar to our previous strategy,12 except that SC, SI and SMT of shared peptides are proportionally divided into the corresponding proteins according to the weight value reported by ProteinProphet, to avoid the multiple counting effect when dealing with shared peptides.20
 | (2) |
 | (3) |
 | (4) |
Replacing SC in the APEX formula with SI and SMT would generate two modified versions of APEX, which are named as APEX-SI and APEX-SMT, as defined in formulae (5) and (6):
 | (5) |
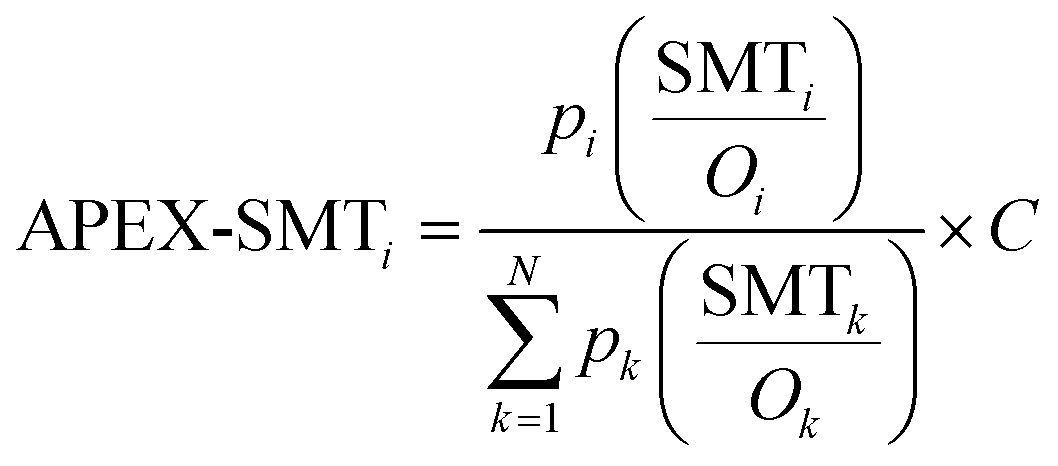 | (6) |
Furthermore, the training-free algorithms (normalized AF) were used as comparisons, where AF referred to SC, SI or SMT. They are in the manner of formula (7):
 | (7) |
2.2 Datasets
Two datasets were used in this study. The first one was termed as the Absolute Quantification Evaluation (AQE) dataset, which was generated in our own laboratory and used for absolute quantification evaluation. In the AQE dataset, yeast and rat brain digests were used for training, while UPS2 (Proteomics Dynamic Range Standard, Sigma) digests were used for absolute quantification. UPS2 consists of 48 standard human proteins, with molar quantities spanning 5 orders of magnitude (6 individual molar levels). Each molar level contains 8 proteins with different properties, including the molecular mass, isoelectric point and hydrophobicity. All three samples received 5 replicate runs on both LTQ XL and Orbitrap Velos. All raw files and databases in use can be downloaded from http://ProteomeCommons.org tranche service (hash: shGb+RfiTGPBtKOnBAut5Rar5JcczeEeosBouV5Lh8Ixj/NvlXc5dsxcb6AmMeCPqXhNcOVEYvYQf6dJYmZ/Zw6v+IkAAAAAAAATtA==). The details of sample preparation and LC-MS/MS analysis are given in the ESI.† The second one was the study 6 Orbitrap@O86 dataset19 from the Clinical Proteomic Technology Assessment for Cancer (CPTAC) network. It was downloaded from the http://ProteomeCommons.org tranche service (hash: NGX3cBUAZXSWvc+6XFNIdVhpLPJTO87lzAxUQmwwR2KHUwWDrdFwV1dso3bvxf7HeXZ4C/juqwEUIz4boC9H3HcLrxEAAAAAAAAmDw==) and used for relative quantification evaluation. This dataset is comprised of triplicate runs of five samples on Orbitrap, namely sample A–E. Each sample contains constant yeast digests (60 ng μL−1) with different amounts of UPS1 spike-ins (0.24, 0.74, 2.2, 6.7, and 20 fmol μL−1, from A–E). UPS1 is similar to UPS2, except that the 48 proteins were equimolarly mixed. Every two adjacent samples provide 3-fold changes for the UPS1 mixture under a large and constant background of yeast digests. Since the amounts of UPS1 proteins in samples A and B are little, they are regarded as yeast digests and used for training, while UPS1 spike-ins in samples D and E are utilized for relative quantification evaluation. Sample C is discarded.2.3 Database search and post-processing
The data analysis pipeline of the AQE dataset is shown in Fig. 1. The pipeline was accomplished through combining some Java source code from APEX Quantitative Proteomics Tool21 and some written in-house except the database search and post-processing of search results. The first step after collecting RAW files was database search and post-processing of search results, then the search results were further filtered by PeptideProphet and ProteinProphet (both are components of the Trans-Proteomic Pipeline,22 version 4.6, http://tools.proteomecenter.org/wiki/index.php?title=Software:TPP) to produce interpreted results in pep.xml and prot.xml formats. These XML files were then delivered to the APEX, APEX-SI and APEX-SMT calculation pipelines (Fig. 1). For multiple runs in any training data, they were processed altogether to generate one pair of result files (one pep.xml file and one prot.xml file). For multiple runs in any quantification data, they were processed separately to generate multiple pairs of result files. The false discovery rate (FDR) control was accomplished through q-value calculation for each peptide to spectrum match (PSM) according to decoy identifications,23,24 and PSMs with q-value ≤ 0.01 were considered acceptable. Detailed procedures of database search were listed as follows.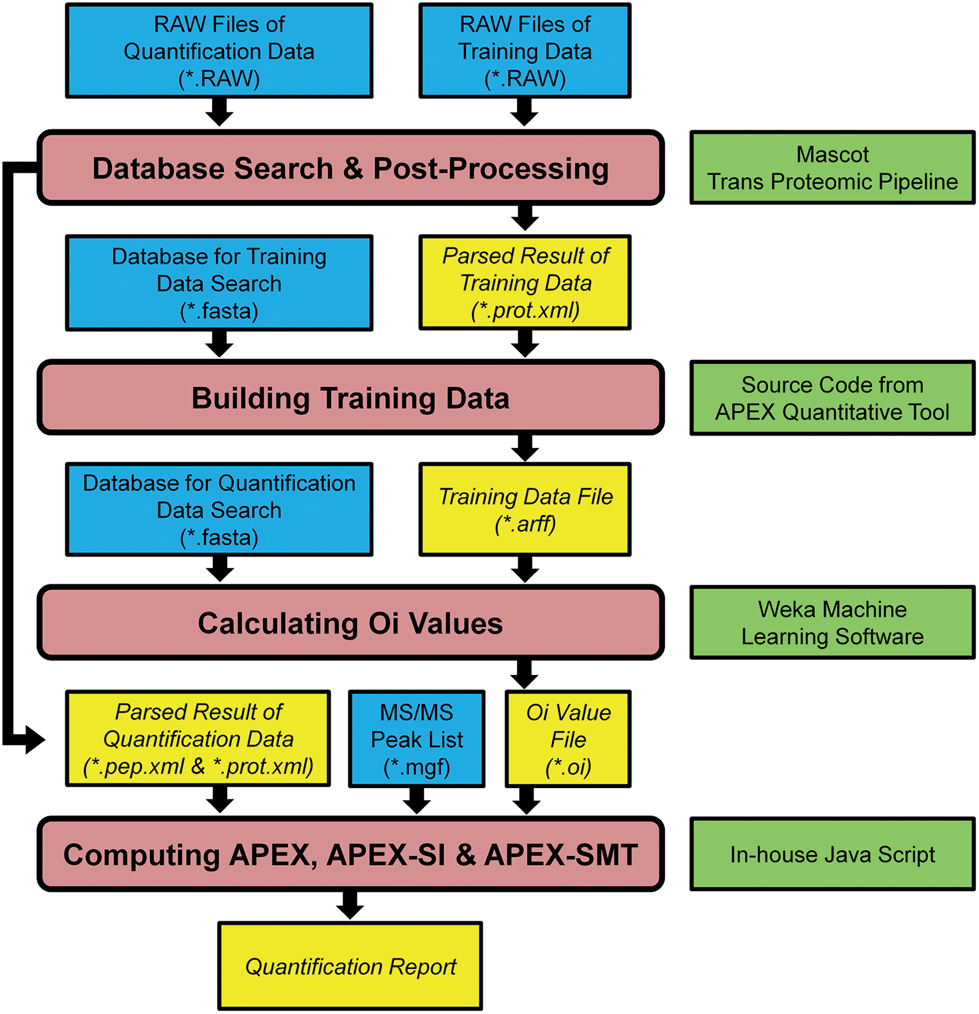 | ||
| Fig. 1 Pipeline of APEX, APEX-SI and APEX-SMT implementation. Four primary processes are depicted in rounded rectangles while files and software are depicted in rectangles. The files generated during the processes are shown in italic, and the software required is shown on the right side rectangle of the corresponding process. The whole processes except database search and post-processing are wrapped in a Java code package consisting of some source code from ref. 21 and some written in-house. | ||
RAW files, no matter generated in our own lab or downloaded from Proteome Commons, were first converted to the mzML format using msconvert.exe, then to the mgf format using mzxml2search.exe with default parameters, except altering mass precision to 5 and intensity precision to 1. Msconvert.exe and mzxml2search.exe are both components of TPP. The database searches were performed with an in-house installation of Mascot25 (version 2.3.02). The databases used are all in the target-reverse forms. The introduction of reversed sequences is used for FDR calculations.
The databases used are as follows: the yeast sequences in the Swissprot repository (downloaded from http://www.uniprot.org on August 31st, 2012), concatenated with their reversed forms (7786 target sequences and 7786 reverse sequences, named as SP_yeast_20120831_plus_RE.fasta), are used for searching yeast data in the AQE dataset and samples A and B in the Orbitrap@O86 dataset; the rat sequences in the International Protein Index (IPI) database (version 3.87), concatenated with their reversed forms (39931 target sequences and 39931 reverse sequences, named as ipi.RAT.v3.87_plus_RE.fasta), are used for searching rat data in the AQE dataset; the 48 UPS2 sequences, concatenated with their own reversed forms and the reversed forms of yeast sequences (48 target sequences and 7834 reverse sequences, named as UPS2_plus_RE_plus_yeastREonly.fasta), are used for searching UPS2 data in the AQE dataset; the 48 UPS1 sequences (notably, UPS1 and UPS2 have 46 identical sequences, while each has 2 unique sequences), concatenated with the yeast sequences and the reversed forms of both UPS1 and yeast sequences (7834 target sequences and 7834 reverse sequences, named as UPS1_plus_yeast_20120831_plus_RE.fasta), are used for searching samples D and E in the Orbitrap@O86 dataset.
The database search parameters are dependent on the sample preparation procedures and the resolution of the mass spectrometer in use. The search parameters for LTQ XL data are as follows: precursor and fragment mass tolerances are 2 Da and 1 Da respectively; only 2+, 3+ and 4+ tryptic peptides with at most two missed cleavage sites are considered; carbamidomethylation of cysteine is set as fixed modification while oxidation of methionine is set as variable modification. The search parameters for Orbitrap Velos data are similar to those for LTQ XL, except the followings: precursor and fragment mass tolerances are 10 ppm and 0.5 Da respectively; observed precursor masses are permitted to differ from the peptide monoisotopic mass by 1 or 2 Da in case that a non-monoisotopic peak is chosen for fragmentation.
2.4 Construction of training data
The arff (attribute-relation file format) files used for training were generated according to protocols proposed by John et al.21 and Vogel et al.26 First, confidently identified proteins were chosen based on their probabilities and SCs reported by ProteinProphet. Then they were in silico digested, and the resulting peptides were filtered according to several predefined filters such as length, molecular weight and FDR. 35 calculated physicochemical properties of each peptide, together with their detectabilities retrieved from parsed search results, constructed the arff file. For a graphical view of the process to construct the training data, please refer to the ESI (Fig. S1A†).2.5 Calculation of Oi values
The training process was performed with the Weka software package (http://www.cs.waikato.ac.nz/ml/weka/), and the optimized machine learning algorithm reported by the original APEX study16 was utilized. The algorithm is a cost-sensitive classifier based upon bagging with a random forest. The performance of this algorithm was evaluated with a 10-fold cross-validation. Once the model was established, it was used to predict the probabilities of observing the peptides on the LC-MS/MS system. The Oi value of a protein was calculated by summing all probabilities of peptides assigned to that particular protein. The Oi file contains Oi values of all proteins in the database for searching quantification data. For a graphical view of the process to calculate the Oi value, please refer to the ESI (Fig. S1B†).2.6 Computation of APEX, APEX-SI and APEX-SMT
SC, SI and SMT, together with protein probability values, the Oi file and the MS/MS peak list file, were imported into the formulae (1), (5) and (6) to obtain the final absolute quantification results.3. Results and discussion
3.1 Choice of machine learning algorithms
Since the emphasis of this study was not to optimize the training process, the training was performed entirely on the basis of the previously optimized parameters,21 which include three optimal machine learning algorithms, including Random Forest, RIpple DOwn Rule learner (RIDOR) and J4.8 Decision Trees. All of them are cost-sensitive classifiers based upon bagging. They were tested with our five arff files (2 from LTQ XL, 2 from Orbitrap Velos, and 1 from Orbitrap XL). The results indicate that Random Forest does perform the best as reported (ESI, Table S1†). Therefore, Random Forest is our final choice for the machine learning algorithm. All parameters used for training and estimators generated during the 10-fold cross-validation process in different datasets are summarized in Table 1. As can be seen, the model performances are better than empirical thresholds from the original study16 and the concrete protocol description,26 ensuring the high-quality of the established models and the trustworthiness of the training results generated from these models. When the models were built, Oi files and quantification reports could be sequentially produced according to the procedures described in Sections 2.5 and 2.6.| Dataset | AQE dataset | Orbitrap@O86 dataset | Empirical thresholds or (previously reported values) from ref. 16 and 26 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Instrument | LTQ XL | Orbitrap Velos | Orbitrap XL | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Data source for training | Yeast | Rat | Yeast | Rat | Yeast | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| a This value was from ref. 16 since it was not reported in ref. 26 all other thresholds or reported values were from ref. 26. | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Parameters used for training | Protein probability | Equals 1 | Equals 1 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| SC threshold | ≥50 | ≥70 | ≥30 | Depends on the system setup | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Number of proteins entering the training process | 105 | 64 | 105 | 92 | 74 | ∼30–150 (89) | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Fraction of observed peptides | 0.13 | 0.08 | 0.13 | 0.10 | 0.06 | Bigger the better (0.09) | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Machine learning algorithm in use | A cost-sensitive classifier based upon bagging with a random forest (weka.classifiers.meta.CostSensitiveClassifier -cost-matrix “[cost matrix]” -S 1 -W weka.classifiers.meta.Bagging -- -P 100 -S 1 -I 10 -W weka.classifiers.trees.RandomForest -- -I 10 -K 5 -S 1) | A cost-sensitive classifier based upon bagging with a random forest | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Estimators generated during the 10-fold cross-validation | Percentage of correctly classified instances | 92.3% | 94.3% | 91.5% | 92.4% | 95.7% | Bigger the better, (86%)a | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| F-measure of the observed peptides | 0.713 | 0.672 | 0.691 | 0.654 | 0.712 | >0.5, bigger the better (0.61) | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Recall of the observed peptides | 0.758 | 0.748 | 0.731 | 0.731 | 0.840 | Bigger the better (0.63) | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
3.2 Technical reproducibility of APEX, APEX-SI and APEX-SMT
The good technical reproducibility of the algorithm in use is a prerequisite for both absolute quantification and relative quantification. Therefore, the technical reproducibility of APEX, APEX-SI and APEX-SMT was tested using the AQE dataset. Among the five injections, a three out of five protocol was used to determine the confidently identified proteins. Totally 22 UPS2 proteins are identified on LTQ XL (22 in all 5 runs), with all 8 proteins at the two highest molar levels (500 and 50 fmol) and 6 proteins at 5 fmol. On Orbitrap Velos, totally 30 proteins are identified (29 in 5 runs and 1 (P01008, 500 amol) in 4 runs), with all 8 proteins at the three highest molar levels (500, 50 and 5 fmol) and 6 proteins at 500 amol. The average RSDs for the calculated quantities of APEX, APEX-SI and APEX-SMT of all identified UPS2 proteins are summarized in Table 2. Besides, those obtained with normalized AFs (formula (7)) are also included. APEX-SI shows obvious higher RSDs than the others, indicating that the fragment matching process for SI calculation does adversely affect its reproducibility. SMT, in contrast, is much more stable than SI among injections, because it contains the intact information of MS/MS spectra. A large SC would definitely result in a large SMT but not necessarily in a large SI. However, cumulating fragment intensities from one MS/MS spectrum surely introduces more variation than simply counting “1”, and that's why the stability of SMT is not as good as SC. The following conclusions could be made from Table 2. Firstly, training by different organisms scarcely affects the technical reproducibility. Secondly, RSDs of APEX-SMT are higher than APEX by approximately 7% for LTQ XL and 5% for Orbitrap Velos, indicating slightly worse but still comparable technical reproducibility.| Algorithm | LTQ XL (22 proteins) | Orbitrap Velos (30 proteins) | ||||
|---|---|---|---|---|---|---|
| Training by yeast | Training by rat | Normalized AF | Training by yeast | Training by rat | Normalized AF | |
| APEX (normalized SC) | 18.5 | 18.6 | 18.7 | 24.8 | 24.8 | 25.1 |
| APEX-SI (normalized SI) | 42.8 | 42.3 | 46.3 | 51.4 | 51.9 | 59.0 |
| APEX-SMT (normalized SMT) | 25.3 | 25.3 | 25.9 | 30.3 | 30.3 | 30.1 |
3.3 Evaluation of absolute quantification on the AQE dataset
The accuracy of APEX, APEX-SI and APEX-SMT in absolute quantification was evaluated with the AQE dataset. The average value of the calculated quantities in five replicates for each protein is deemed as its final calculated quantity. After logarithmic transformation, the relationships between the injected amount and the average value for the final calculated quantities of proteins in every molar level were plotted, and standard deviations were plotted as error bars, as shown in Fig. 2. Two parameters were designed to evaluate the variance and uniformity between injected and calculated quantities, namely VAR and UNI, and shown as formulae (8) and (9),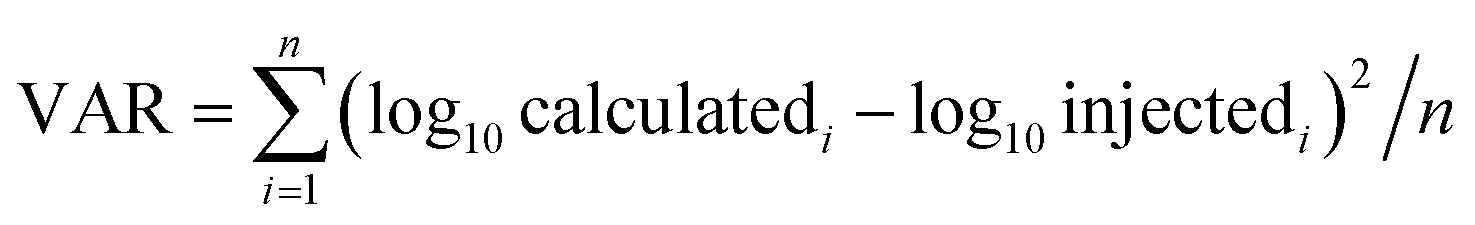 | (8) |
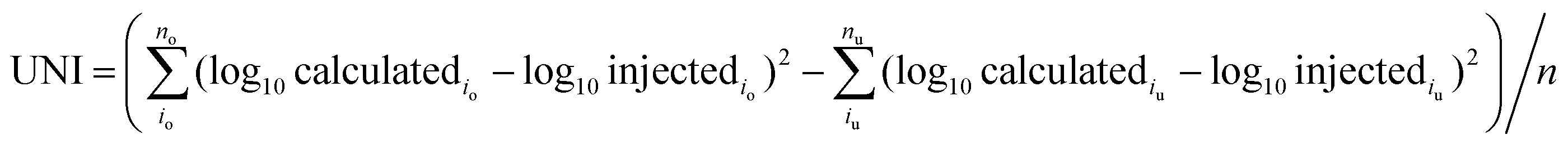 | (9) |
 | ||
| Fig. 2 Relationships between injected amounts and calculated quantities for the AQE dataset. (A–C), LTQ XL, yeast-training; (D–F), LTQ XL, rat-training; (G–I), LTQ XL, training free; (J–L), Orbitrap Velos, yeast-training; (M–O), Orbitrap Velos, rat-training; (P–R), Orbitrap Velos, training free. After logarithmic transformation, the average value for the final calculated quantities of proteins in every molar level was plotted as a black dot, and standard deviation (s.d.) was plotted as an error bar. In some cases, the s.d. is larger than the average value, leading to the inability to calculate the log10 (average-s.d.). The lower limit of the error bar is extended unlimitedly to the y minus direction to represent this situation. VAR and UNI, as described in formulae (8) and (9), together with the linear fitting equation, are shown in the lower right corner of each individual figure. VAR represents the total deviation regardless of underestimation or overestimation while UNI represents the uniformity of the deviation; both are closer to 1 the better. As for the linear fitting equation, the slope and R2 closer to 1 and the intercept closer to 0 indicate better accuracy. | ||
VAR, UNI and the linear fitting equation were printed in the lower right corner of each individual figure. VAR represents the total deviation, regardless of overestimation or underestimation, and is surely a positive number. UNI represents the uniformity of the deviation, so it can be either positive or negative. Positive UNI indicates overall overestimation while negative one indicates overall underestimation. For both the closer to 0 the better. As pointed out by Spinelli et al.,18 the linearity (Pearson's R2) of the linear fitting alone cannot reflect the accuracy comprehensively; the slope and intercept of the linear fitting equation matter as well. The slope closer to 1, intercept closer to 0 and R2 closer to 1 indicate better accuracy.
From Fig. 2A–F and J–O, we could see a sharp reduction of VAR and UNI for APEX-SMT compared to those of the corresponding APEX and APEX-SI on both instruments. Also, the slope of APEX-SMT is closer to 1 than APEX and APEX-SI, and the intercept of APEX-SMT is closer to 0 than APEX and APEX-SI. Even the linearity (R2) of APEX-SMT is better than APEX and APEX-SI on LTQ XL (the linearity of APEX-SMT is slightly worse than APEX and APEX-SI on Orbitrap Velos due to the obvious overestimation on the 500 amol molar level). The same trend is observed for the normalized AF algorithms (Fig. 2G–I, P–R). All these results suggest that APEX-SMT surmounts corresponding APEX or APEX-SI in accuracy considering all the estimators discussed above no matter on which instrument. The major problem of APEX and APEX-SI is the evident overestimation. In other words, the dynamic ranges of APEX and APEX-SI are too narrow to truly reflect protein concentrations of 3 orders of magnitude. In contrast, the true dynamic range of the sample could be approximately reflected by APEX-SMT. The comparison among the three algorithms could be concluded as follows. APEX-SMT shows its superiority over APEX-SI in every aspect, including better reproducibility among injections and much better ability to reflect the protein concentrations. The only drawback of APEX-SMT is its slightly worse reproducibility than APEX, which is unavoidable due to the slightly worse reproducibility of SMT itself.
The normalized AF algorithms slightly outperform corresponding training algorithms on Orbitrap Velos (Fig. 2J–R). However, all estimators are based on the average calculated quantity in each molar level; the deviation among individual proteins in each molar level remains untested. Therefore, we calculated the largest fold change among individual calculated quantities in every molar level (the largest calculated quantity divided by the smallest), and summarized the result in Table 3, from which the effect of machine learning algorithms is validated. The fold changes are significantly reduced after training at every molar level on both instruments (with 2 exceptions at 500 amol on Orbitrap). The fold changes could be up to ∼40 on LTQ and ∼110 on Orbitrap without training, while after training, they could be reduced within 10 on LTQ and ∼20 on Orbitrap. Although the average calculated quantities correlate well with injected amounts using either APEX-SMT or normalized SMT, the quantification accuracy of any individual protein could not be guaranteed by the latter. It confirms our hypothesis that the dynamic ranges are intrinsic to AFs themselves; meanwhile the machine learning algorithm does its job in reducing the discrepancy of calculated quantities of different proteins of equal amount. In other words, the combination of APEX and SMT does perverse the advantages of both.
| Molar level | LTQ XL | Orbitrap Velos | ||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Trained by yeast | Trained by rat | Training free | Trained by yeast | Trained by rat | Training free | |||||||||||||
| APEX | APEX-SI | APEX-SMT | APEX | APEX-SI | APEX-SMT | Normalized SC | Normalized SI | Normalized SMT | APEX | APEX-SI | APEX-SMT | APEX | APEX-SI | APEX-SMT | Normalized SC | Normalized SI | Normalized SMT | |
| 500 fmol | 2.2 | 3.3 | 7.5 | 2.2 | 2.5 | 8.6 | 15.3 | 15.0 | 38.9 | 4.1 | 4.6 | 18.2 | 4.2 | 4.2 | 18.6 | 24.1 | 27.8 | 106.3 |
| 50 fmol | 2.7 | 4.8 | 3.7 | 3.1 | 4.2 | 3.7 | 6.9 | 26.9 | 8.8 | 2.5 | 6.5 | 11.5 | 2.4 | 7.7 | 10.7 | 17.0 | 21.7 | 32.1 |
| 5 fmol | 3.6 | 2.9 | 3.7 | 4.3 | 3.4 | 4.3 | 5.3 | 5.8 | 6.0 | 2.7 | 6.5 | 6.6 | 2.9 | 6.8 | 6.9 | 8.3 | 8.8 | 8.1 |
| 500 amol | — | — | — | — | — | — | — | — | — | 9.6 | 22.2 | 8.1 | 9.9 | 18.7 | 7.8 | 4.2 | 29.2 | 4.0 |
We continued to conduct the comparison among APEX, APEX-SI and APEX-SMT using the yeast-training data by calculating the absolute value of relative deviation (|(calculated-injected)|/injected) for every individual protein through dividing the absolute difference between the calculated and the injected value by the injected value. The average deviations for the 22 proteins identified by LTQ are 101% (APEX), 144% (APEX-SI) and 45% (APEX-SMT), while those for the 30 proteins identified by Orbitrap are 1134% (APEX), 787% (APEX-SI) and 146% (APEX-SMT). The major deviations on Orbitrap come from the lowest molar level (500 amol). After excluding the 6 proteins at 500 amol, the deviations of the remaining 24 proteins could be reduced to 383% (APEX), 276% (APEX-SI) and 59% (APEX-SMT). The average deviations of APEX-SMT are reduced by 55% (LTQ)-85% (Orbitrap Velos) compared to APEX. Similar results were obtained from the rat-training data (Tables S2 and S3†). All these results demonstrate that APEX-SMT could obviously improve the quantification accuracy compared to APEX and APEX-SI.
Our study shows that no bias is observed along the way from training to reporting the quantification result, no matter which training organism (yeast or rat) is in use. Therefore, we could come to the conclusion that any high-quality real sample dataset on an LC-MS/MS system could be used for training on that particular system setup, allowing researchers to retroactively make use of the existing data in their own labs for either training or quantification.
3.4 Evaluation of relative quantification on the Orbitrap@O86 dataset
Once the absolute quantities of proteins are determined, their relative expression changes in different samples could be calculated through comparing the absolute quantity of the same protein in these samples. Since APEX has been proven to be capable of the sensitive detection of differentially expressed proteins,16 we compared the performance of relative quantification of APEX with that of APEX-SMT and APEX-SI on the Orbitrap@O86 dataset. We did a series of tests on APEX, APEX-SI and APEX-SMT following our previous procedure12 on the same testing dataset.19 Briefly, 9 binary sets of UPS1 protein ratios were exhaustively calculated with the 3 respective replicates of samples D and E (similar to what Colaert et al.27 did), and then the ratios were logarithmically transformed and plotted by MATLAB (MathWorks, version 2010b) ksdensity function with default parameters, as shown in Fig. 3A. A vertical line was used to indicate the expected log2 ratio (log2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 3 = 1.5850). From Fig. 3A, we could see that the distributions of APEX-SI and APEX-SMT are almost centered on the expected value while that of APEX has an obvious shift to the underestimation side. This phenomenon indicates the unsatisfactory overall accuracy for APEX, consistent with the narrow dynamic range of SC. The width of distribution for APEX-SI, APEX-SMT and APEX is in the descending order, demonstrating the goodness of precision for the three algorithms in the same order. The slightly better precision of APEX-SMT compared to APEX-SI could be attributed to the better reproducibility of SMT compared to SI. To take both accuracy and precision into consideration, 1.5–6 (0.5–2 times of the actual fold change) was empirically set as the acceptable range, thus, the area under the density distribution plot in this range (0.5850 to 2.5850 on log2 scale) could represent the probability of the ratios falling in this range. The probabilities of getting an acceptable result by the three algorithms are 56.83% (APEX), 52.38% (APEX-SI) and 55.34% (APEX-SMT). APEX-SMT is better than APEX-SI and comparable to APEX.
3 = 1.5850). From Fig. 3A, we could see that the distributions of APEX-SI and APEX-SMT are almost centered on the expected value while that of APEX has an obvious shift to the underestimation side. This phenomenon indicates the unsatisfactory overall accuracy for APEX, consistent with the narrow dynamic range of SC. The width of distribution for APEX-SI, APEX-SMT and APEX is in the descending order, demonstrating the goodness of precision for the three algorithms in the same order. The slightly better precision of APEX-SMT compared to APEX-SI could be attributed to the better reproducibility of SMT compared to SI. To take both accuracy and precision into consideration, 1.5–6 (0.5–2 times of the actual fold change) was empirically set as the acceptable range, thus, the area under the density distribution plot in this range (0.5850 to 2.5850 on log2 scale) could represent the probability of the ratios falling in this range. The probabilities of getting an acceptable result by the three algorithms are 56.83% (APEX), 52.38% (APEX-SI) and 55.34% (APEX-SMT). APEX-SMT is better than APEX-SI and comparable to APEX.
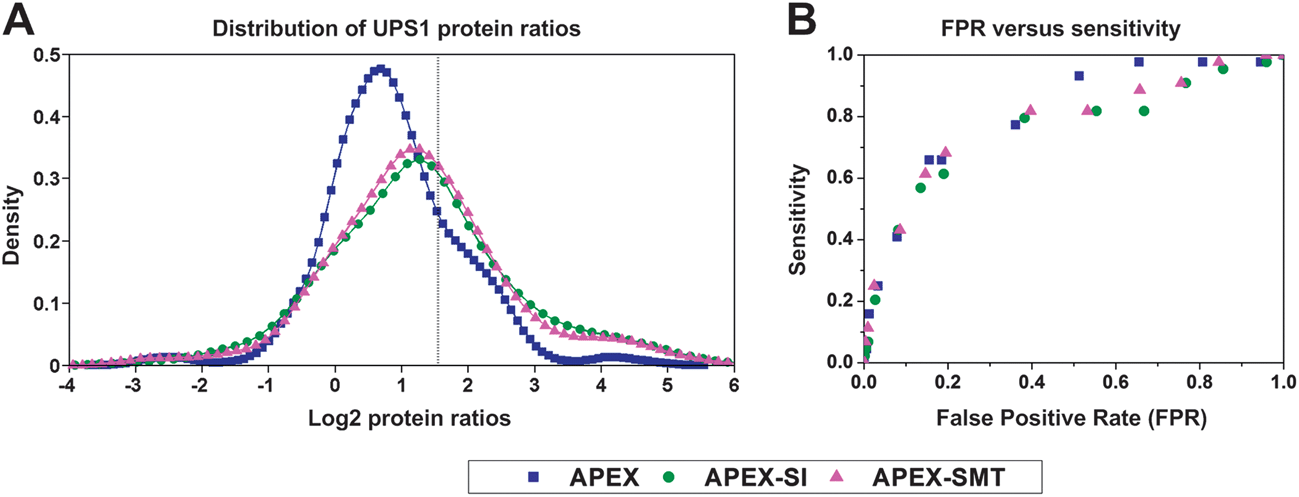 | ||
| Fig. 3 Relative quantification evaluation on the Orbitrap@O86 dataset. (A) Density plot of UPS1 protein ratios calculated for samples D and E in an exhaustive manner by APEX, APEX-SI and APEX-SMT, with a vertical line indicating the expected log2 value of 1.5850 (log23 = 1.5850). (B) FPR versus sensitivity plot for the three algorithms. FPR = the number of detected significantly changed yeast proteins/1074, Sensitivity = the number of detected significantly changed UPS1 proteins/44. 14 ascending p values were set at 0.001, 0.005, 0.01, 0.02, 0.05, 0.08, 0.1, 0.2, 0.3, 0.4, 0.5, 0.6, 0.7, and 0.8. FPR and sensitivity are both saturated when p ≥ 0.8 for any algorithm, meaning they are both equal to 1. | ||
The significant differentially expressed proteins were detected by the Power Law Global Error Model (PLGEM)28 (version 1.24.0) under an R environment (version 2.13.0) at 14 ascending p values (p = 0.001, 0.005, 0.01, 0.02, 0.05, 0.08, 0.1, 0.2, 0.3, 0.4, 0.5, 0.6, 0.7, 0.8). For details of PLGEM settings and performance evaluation, please refer to the ESI (Fig. S2†). The significant differentially expressed proteins' list contains both UPS1 proteins and yeast proteins. The significant changes of UPS1 proteins are defined as “true positive” changes while those of yeast proteins are defined as “false positive” changes. Therefore, the false positive rate (FPR) could be defined as the proportion of false positive yeast proteins in all identified yeast proteins (1074), while the sensitivity could be defined as the proportion of true positive UPS1 proteins in all identified UPS1 proteins (44). FPR versus sensitivity was plotted, as shown in Fig. 3B. Note that almost all statistical models including PLGEM have a high demand on the reproducibility of the AF to discriminate the true significantly changed proteins from the false ones. We are not surprised to observe that the performance of APEX-SI is worse than the other two in the whole FPR range. APEX and APEX-SMT perform almost the same in the low FPR range (≤0.4). These results indicate the similar ability of APEX-SMT in detecting differentially expressed proteins compared to that of APEX in the reasonable FPR range. All these results demonstrate the reliability of APEX-SMT to be applied in relative quantification.
4. Conclusion
A big obstacle in computational absolute quantification of proteomes in a label-free manner is the disparity in MS AFs of equal amount of peptides/proteins. The probability of observing a peptide, predicted by machine learning algorithms and implemented in the APEX algorithm, provides a fast and easy way to largely alleviate this barrier. This makes the choice of abundance features even more important. MS/MS level AFs are much easier to calculate and less dependent on the resolution of mass spectrometers than MS level AFs. Through a series of tests on a comprehensive homemade dataset for absolute quantification evaluation, the results show that APEX-SMT could significantly improve the quantification accuracy compared to APEX. When evaluated with a publicly available benchmark dataset for relative quantification, APEX-SMT could also provide comparable accuracy to APEX. All these results suggest that the APEX-SMT algorithm is a promising alternative to APEX to enhance the accuracy of label-free quantification of proteomes.The appropriate incorporation of MS level AFs, such as the precursor intensity or area under the curve (AUC), preserves the potential to further improve the quantification accuracy. This study could help us to open up more studies regarding the combination of the computational methods to correct the mass spectrometric bias of disparate proteins and the novel types of reproducible and accurate AFs. Hopefully, researchers could ultimately get an accurate perspective of biological and metabolic processes.
Acknowledgements
The authors want to thank Dr David C. Trudgian (Central Proteomics Facility, Sir William Dunn School of Pathology, University of Oxford) for providing some Perl scripts for reference. They are grateful for the financial support from the National Basic Research Program of China (Grant 2012CB910603), National Natural Science Foundation (Grant 20935004), The Creative Research Group Project by NSFC (Grant 21021004), and The Analytical Method Innovation Program of MOST (Grant 2012IM030900).References
- L. Arike, K. Valgepea, L. Peil, R. Nahku, K. Adamberg and R. Vilu, J. Proteomics, 2012, 75, 5437–5448 CrossRef CAS PubMed.
- M. Baker and L. DeFrancesco, Nat. Biotechnol., 2011, 29, 221–227 CrossRef CAS PubMed.
- S. A. Gerber, J. Rush, O. Stemman, M. W. Kirschner and S. P. Gygi, Proc. Natl. Acad. Sci. U. S. A., 2003, 100, 6940–6945 CrossRef CAS PubMed.
- T. Yoneyama, S. Ohtsuki, M. Ono, K. Ohmine, Y. Uchida, T. Yamada, M. Tachikawa and T. Terasaki, J. Proteome Res., 2013, 12, 753–762 CrossRef CAS PubMed.
- V. Brun, A. Dupuis, A. Adrait, M. Marcellin, D. Thomas, M. Court, F. Vandenesch and J. Garin, Mol. Cell. Proteomics, 2007, 6, 2139–2149 CAS.
- Q. Wu, H. M. Yuan, L. H. Zhang and Y. K. Zhang, Anal. Chim. Acta, 2012, 731, 1–10 CrossRef CAS PubMed.
- J. C. Silva, M. V. Gorenstein, G. Z. Li, J. P. C. Vissers and S. J. Geromanos, Mol. Cell. Proteomics, 2006, 5, 144–156 CAS.
- B. Schwanhausser, D. Busse, N. Li, G. Dittmar, J. Schuchhardt, J. Wolf, W. Chen and M. Selbach, Nature, 2011, 473, 337–342 CrossRef PubMed.
- Y. Ishihama, Y. Oda, T. Tabata, T. Sato, T. Nagasu, J. Rappsilber and M. Mann, Mol. Cell. Proteomics, 2005, 4, 1265–1272 CAS.
- L. Florens, M. J. Carozza, S. K. Swanson, M. Fournier, M. K. Coleman, J. L. Workman and M. P. Washburn, Methods, 2006, 40, 303–311 CrossRef CAS PubMed.
- N. M. Griffin, J. Yu, F. Long, P. Oh, S. Shore, Y. Li, J. A. Koziol and J. E. Schnitzer, Nat. Biotechnol., 2010, 28, 83–89 CrossRef CAS PubMed.
- Q. Wu, Q. Zhao, Z. Liang, Y. Y. Qu, L. H. Zhang and Y. K. Zhang, Analyst, 2012, 137, 3146–3153 RSC.
- N. Colaert, K. Gevaert and L. Martens, J. Proteome Res., 2011, 10, 3183–3189 CrossRef CAS PubMed.
- J. M. Asara, H. R. Christofk, L. M. Freimark and L. C. Cantley, Proteomics, 2008, 8, 994–999 CrossRef CAS PubMed.
- P. Mallick, M. Schirle, S. S. Chen, M. R. Flory, H. Lee, D. Martin, J. Ranish, B. Raught, R. Schmitt, T. Werner, B. Kuster and R. Aebersold, Nat. Biotechnol., 2007, 25, 125–131 CrossRef CAS PubMed.
- P. Lu, C. Vogel, R. Wang, X. Yao and E. M. Marcotte, Nat. Biotechnol., 2007, 25, 117–124 CrossRef CAS PubMed.
- D. H. Lundgren, S. I. Hwang, L. F. Wu and D. K. Han, Expert Rev. Proteomics, 2010, 7, 39–53 CrossRef CAS PubMed.
- K. J. Spinelli, J. E. Klimek, P. A. Wilmarth, J. B. Shin, D. Choi, L. L. David and P. G. Gillespie, Proc. Natl. Acad. Sci. U. S. A., 2012, 109, E268–E277 CrossRef CAS PubMed.
- P. A. Rudnick, K. R. Clauser, L. E. Kilpatrick, D. V. Tchekhovskoi, P. Neta, N. Blonder, D. D. Billheimer, R. K. Blackman, D. M. Bunk, H. L. Cardasis, A. J. L. Ham, J. D. Jaffe, C. R. Kinsinger, M. Mesri, T. A. Neubert, B. Schilling, D. L. Tabb, T. J. Tegeler, L. Vega-Montoto, A. M. Variyath, M. Wang, P. Wang, J. R. Whiteaker, L. J. Zimmerman, S. A. Carr, S. J. Fisher, B. W. Gibson, A. G. Paulovich, F. E. Regnier, H. Rodriguez, C. Spiegelman, P. Tempst, D. C. Liebler and S. E. Stein, Mol. Cell. Proteomics, 2010, 9, 225–241 CAS.
- Y. Zhang, Z. Wen, M. P. Washburn and L. Florens, Anal. Chem., 2010, 82, 2272–2281 CrossRef CAS PubMed.
- J. Braisted, S. Kuntumalla, C. Vogel, E. Marcotte, A. Rodrigues, R. Wang, S. T. Huang, E. Ferlanti, A. Saeed, R. Fleischmann, S. Peterson and R. Pieper, BMC Bioinf., 2008, 9, 529 CrossRef PubMed.
- A. Keller, J. Eng, N. Zhang, X. Li and R. Aebersold, Mol. Syst. Biol., 2005, 1–17 CrossRef CAS PubMed.
- L. Käll, J. D. Storey, M. J. MacCoss and W. S. Noble, J. Proteome Res., 2007, 7, 29–34 CrossRef PubMed.
- D. C. Trudgian, G. Ridlova, R. Fischer, M. M. Mackeen, N. Ternette, O. Acuto, B. M. Kessler and B. Thomas, Proteomics, 2011, 11, 2790–2797 CrossRef CAS PubMed.
- D. N. Perkins, D. J. C. Pappin, D. M. Creasy and J. S. Cottrell, Electrophoresis, 1999, 20, 3551–3567 CrossRef CAS.
- C. Vogel and E. M. Marcotte, Nat. Protoc., 2008, 3, 1444–1451 CrossRef CAS PubMed.
- N. Colaert, J. Vandekerckhove, K. Gevaert and L. Martens, Proteomics, 2011, 11, 1110–1113 CrossRef CAS PubMed.
- N. Pavelka, M. L. Fournier, S. K. Swanson, M. Pelizzola, P. Ricciardi-Castagnoli, L. Florens and M. P. Washburn, Mol. Cell. Proteomics, 2008, 7, 631–644 CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c3an01738a |
| This journal is © The Royal Society of Chemistry 2014 |